g likelihood or Bayesian probabilistic phylogene
If you have a molecular data partition, you can just use total evidence approach and the standard 1-parameter Markov model.
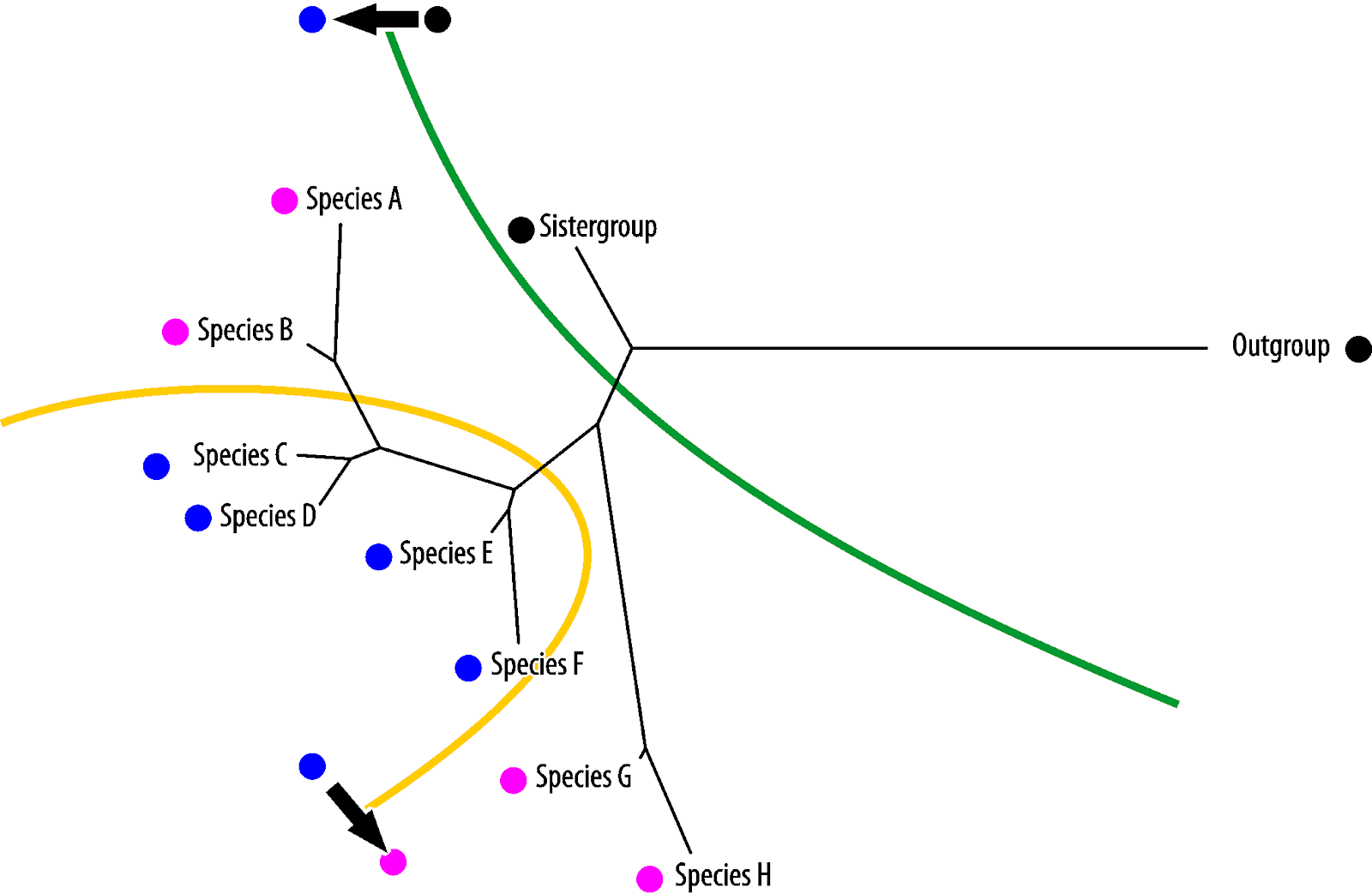
Potential synapomorphies will be compatible with the molecular tree and considered not likely to change. Potential homoiologies and symplesiomorphies are partly ("semi-")compatible with the molecular tree and, hence, considered less likely to change than highly homoplastic traits with (random) convergence.
Just try out a couple of datasets, and infer the (Bio)NJ and ML trees and then compare the result with the strict consensus network (not tree) of all equally parsimonious trees and the Bayesian tree sample.
Note that if you apply TNT's iterative character weighting procedure, what you effectively do is sorting the random convergences from parallelisms/ characters that are more compatible with the preferred tree.
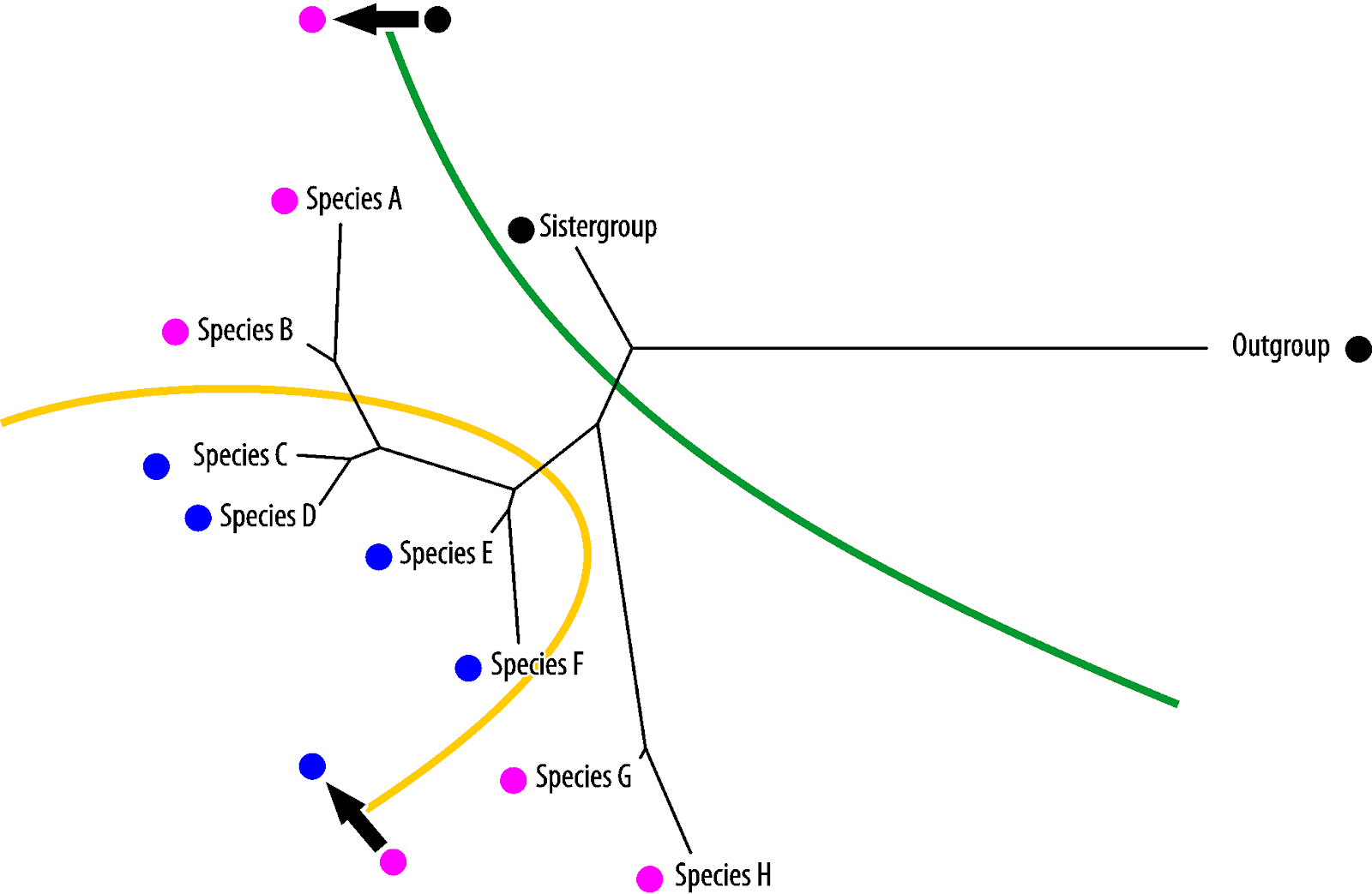 Vice versa, blue is the symplesiomorphy, pink the homoiology found only in the most derived (in absolute evolutionary terms) taxa
Vice versa, blue is the symplesiomorphy, pink the homoiology found only in the most derived (in absolute evolutionary terms) taxa