orthologs
Orthologs are derived from a common ancestor gene and have some degree of sequence similarity
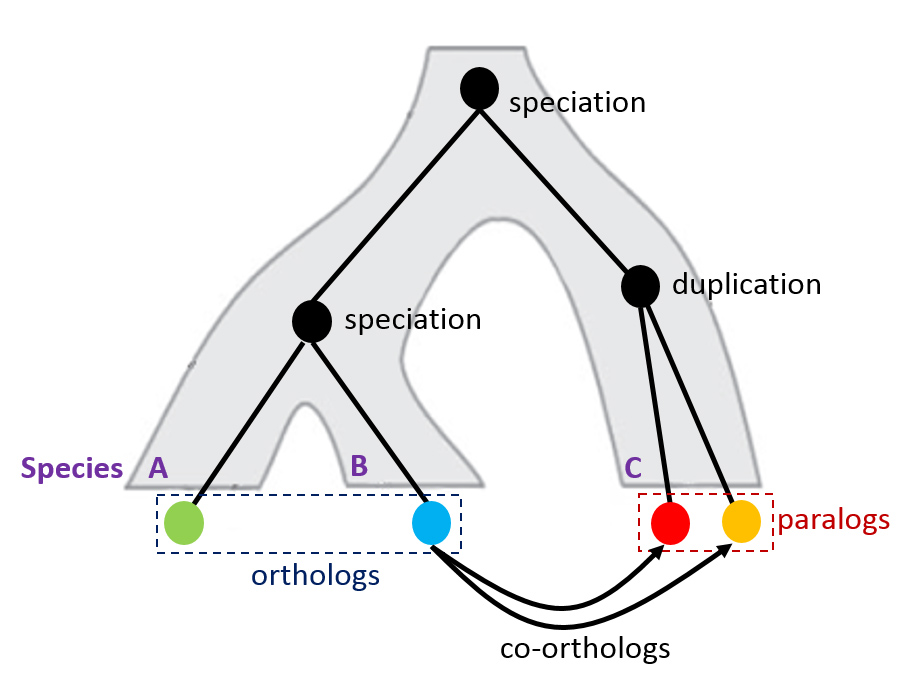
Image credit: https://www3.beacon-center.org/blog/2015/03/23/beacon-researchers-at-work-same-behavior-same-genes/
orthologs
Orthologs are derived from a common ancestor gene and have some degree of sequence similarity
Image credit: https://www3.beacon-center.org/blog/2015/03/23/beacon-researchers-at-work-same-behavior-same-genes/
20. Y. Zheng, C. Lorenzo, P. A. Beal, Nucleic Acids Res. 45, 3369–3377 (2017)
The authors showed that ADAR proteins were able to deaminate adenosines not only in RNA duplexes but also in RNA-DNA heteroduplexes.
While this work was done in vitro, it may help understand the functions of ADAR in the cell, specifically the link between ADAR impairment and the autoimmune disease Aicardi-Goutieres Syndrome (AGS). Also it may be used as a new tool for DNA editing techniques.
10. O. O. Abudayyeh et al., Science 353, aaf5573 (2016).
A large group of scientists from several labs conducted an extensive study on the protein C2c2 (now known as Cas13a). They demonstrated that C2c2 was an RNA-guided RNA nuclease and predicted that it would be an important tool for RNA targeting.
Moreover, they showed that in vitro C2c2 once activated by the target binding could cleave not only the target but also the surrounding mRNA molecules. The authors termed this effect "the collateral cleavage."
Extension of our rational design approach, such as combining promising mutations and directed evolution, could further increase the specificity and efficiency of the system, while unbiased screening approaches could identify additional residues for improving REPAIR activity and specificity.
It may also be possible to reduce the size of Cas13b as was done with the Cas9 protein. Or use an ortholog of a different size, as was also done with Cas9.
Smaller Cas13b variants would have lower chance of interfering with cellular processes, making it an even more useful tool for studying RNA biology.
The transient nature of REPAIR-mediated edits will likely be useful for treating diseases caused by temporary changes in cell state, such as local inflammation, and could also be used to treat disease by modifying the function of proteins involved in disease-related signal transduction.
Compared to DNA editing, RNA editing is temporary and can be reversed more easily. This means that RNA editing with REPAIR can be used not only to correct inherited mutations that cause disease, but disease-causing mutations that arise later in life. While DNA editing could treat a mutation that occurred later in life, the treatment would be permanent. RNA editing could treat something temporarily, so it could be used for temporary cell growth or inhibition of imflammation in response to an infection.
The programmability of RNA and DNA editing by simple base pairing rules makes it much easier to develop than alternative therapies such as drug inhibitors that are targeted to specific enzymes. Finding an inhibitor that is specific enough without causing side effects is difficult. The REPAIR system is a potential solution to this precision problem.
the REPAIR system directly deaminates target adenosines to inosines and does not rely on endogenous repair pathways to generate desired editing outcomes.
This is an advantage over the traditional CRISPR/Cas9-mediated genome editing. CRISPR/Cas9 uses the cell's machinery to make changes, so it is efficient only in actively dividing cells.
Moreover, we can compare the REPAIR system to DNA cytidine base-editors (BE). The BE system has been modified to prevent cellular DNA repair processes that are triggered by Uracil (U) bases in DNA. The BE system, in any form, still relies on DNA repair machinery to propagate the edit. RNA editing does not.
Check out the schematic below for more information on the BE editing paths.

can target either the sense or antisense strand
Cas9 (as the targeting unit of the DNA base editor) can be directed against the sense or antisense strand. The gRNA pairs with the strand, causing a short sequence of the opposite strand to be displaced. Then, the "free" fragment is accessible for the cytidine deaminase that acts on single-stranded DNA.
This means that DNA base editors can be used to correct mutations in either strand. By contrast, RNA editors can only work with the mRNA which limits their editing ability two times.
In contrast, REPAIR is not constrained by PAM, PFS, and other motifs, meaning it can act on a broader range of sequences (but can only act on transcripts).
The lack of motif for ADAR editing, in contrast with previous literature, is likely due to the increased local concentration of REPAIR at the target site owing to dCas13b binding.
Previously, it has been shown that the ADAR proteins prefer certain nucleotides at the 5' and 3' positions next to the target sequence (reference 21). However, this is not a strict preference, and some mutants are more strict than others.
In addition, previous work has shown that when ADAR2 is site-specifically directed at a target, its local concentration is increased and less favorable reactions sometimes occur.
The REPAIR system offers many advantages compared with other nucleic acid–editing tools.
REPAIR stands for RNA Editing for Programmable A to I Replacement, i.e. it is a system that precisely edits single bases in nucleic acids. Because it is a base editor, the REPAIR system is mainly compared to other base editors and not to all possible genome editing tools.
To learn more about base editing systems, read more here.
Deeper sequencing and novel inosine enrichment methods could further refine our understanding of REPAIR specificity in the future.
In this work, the authors used next-generation sequencing to detect editing events. They compared the obtained sequences (reads) with the genomic sequence. If the genome contained A but the read had G at the same position, they considered that an editing event.
However, this approach may overestimate the number of edits. First, the sequencing itself generates errors that can be mistakenly attributed to ADAR activity. Second, the gene may have many versions in a genome that differ at some positions, including A>G differences. This leads to some ambiguity in data interpretation.
To make up for these problems, several approaches have been suggested to detect inosine modification directly, including chemical modification and antibody-based enrichment. Post publication of this paper, newer techniques have been introduced that could be used for chemical modification.
it is unlikely to do so in this case because Cas13b does not bind DNA efficiently and because REPAIR is cytoplasmically localized.
ADAR activity on DNA-RNA heteroduplexes might be a problem for precise RNA editing by REPAIR. Fortunately, Cas13b predominantly binds RNA and the REPAIR system is sequestered from the DNA in the cytoplasm as a result of the nuclear export sequence (NES) fused to Cas13b.
fidelity
How specifically an enzyme carries out its function. We can say that Cas13b has high fidelity because it produced very few off-targets in the knockdown experiment (see Figures 1E, 1F, and 1G).
Compared with REPAIRv1, REPAIRv2 exhibited increased specificity, with a reduction from 18,385 to 20 transcriptome-wide off-targets with high-coverage sequencing (125x coverage, 10 ng of REPAIR vector transfected)
To more rigorously compare the off-target activity of two systems, the authors performed sequencing with higher coverage.
Recall that earlier they used 12.5x coverage. Here, they used 125x coverage.
Why is this important? Cellular genes are expressed at different levels which leads to a different number of individual mRNA molecules. The more abundant a particular molecule is, the easier it is to detect it at a given coverage. When you increase the coverage, you have a chance to catch molecules that are less abundant in the cell.
This is exactly what happened in the experiment with the REPAIRv1. At 125x coverage, the authors detected off-targets in the majority of transcripts (18385 of around 20000 protein-coding genes in our genome). By contrast, the REPAIRv2 system was astonishingly more specific and produced off-targets 1000 times less frequently.
We further explored motifs surrounding off-targets for the various specificity mutants
Inspired by other explorations into 3' and 5' motifs, the authors looked at transcripts with off-target effects—specifically at two nucleotides surrounding the edited adenosine.
A majority of mutants either significantly improved the luciferase activity for the targeting guide or increased the ratio of targeting to nontargeting guide activity, which we termed the specificity score
The authors looked at two characteristics of the modified protein variants.
First, they tested whether the mutant had changed its editing activity. This was calculated by looking at the restoration of the Cluc luciferase signal. While the authors didn't necessarily want increased editing activity, they wanted to avoid a loss of editing activity.
However, catalytic activity sometimes leads to more off-target effects. Therefore, they authors calculated the ratio between the Cluc signal in targeting and non-targeting conditions, i.e. the specificity score. The higher the score, the more specific a mutant variant was.
structure-guided protein engineering
When researchers want to modify a protein to improve a particular feature, they can use the knowledge of the protein's 3D structure to identify and modify key amino acids in the sequence.
The overlap in off-targets between the targeting and nontargeting conditions and between REPAIRv1 and BoxB conditions suggests that ADAR2DD drives off-targets independent of dCas13 targeting
In summary, the authors found two pieces of evidence supporting the hypothesis that the ADAR2 deaminase domain is the main source of off-targets:
First, they found overlap between the off-target effects of REPAIRv1 under different targeting conditions and ADAR2(DD).
Second, they saw overlap in the off-target effects between REPAIR and ADAR2(DD) in a completely different targeting system. This offers strong support that ADAR2 drives off-target effects regardless of the targeting module.
Given the high number of overlapping off-targets between the targeting and nontargeting guide conditions, we reasoned that the off-targets may arise from ADARDD.
The authors found a lot of overlap between the off-target effects they saw in the targeting and non-targeting conditions, meaning that the REPAIRv1 complex was modifying adenosines even when the gRNA did not guide it. This suggests that ADAR is increasing off-target editing effects independent of dCas13b.
coverage
When you sequence a genome, you do it in pieces rather than in a single, continuous stretch. This is similar to cutting the genome up and then putting it back together again, like a puzzle. Each base may be read multiple times and be a part of multiple sequences—comparing pieces to see where they overlap is how the full genome is reconstructed. The number of times a base is read is called the "coverage," and higher coverage leads to a more accurate sequence.

adeno-associated viral (AAV) vectors
Adeno-associated virus (AAV) is a human virus that is present in 80% - 90% of the adult population but does not cause any disease. This virus has been extensively used as a biomolecular tool because it is small and has low risk of genome integration and causing unwanted mutations.
Delivering the REPAIRv1 system to diseased cells is a prerequisite for therapeutic use, and we therefore sought to design REPAIRv1 constructs that could be packaged into therapeutically relevant viral vectors
There are two main approaches for the inserting of exogenous genetic material into cells: non-viral and viral.
Non-viral techniques such as transfection with DNA-liposomes can be used for almost any construct, and are relatively easy to use. Some therapeutic companies are looking at lipid nanoparticles for ribonucleoprotein (RNP) delivery to the liver. The authors of the paper used liposome transfection in all of the preceding experiments they describe here. However, this method is inappropriate for in vivo (live) delivery because of its non-specificity and high toxicity.
An alternative approach is to use viral delivery. This strategy utilizes the natural ability of viruses to infect cells and integrate viral DNA into cellular DNA. A part of the viral genome is replaced with a desired sequence, and then cells are infected with the modified virus. When viruses insert their DNA into the cellular DNA, the desired sequence is added. Viruses are very specific and use only certain molecules to enter the cell, so this method is useful for controlling the type of cell that receives exogenous DNA. However, precautions must be taken to make sure the virus does not go out of control and that the cell's immune response does not destroy the virus before it delivers the DNA.
the ClinVar database
ClinVar is an archive that collects data about the relationship between gene variants and phenotypes. It contains more than 400,000 records.
ClinVar is free to use—try looking up information about AVPR2. Search for AVPR2 to find out more about the 878G>A mutation that the authors looked at.
Fanconi anemia
Fanconi anemia (FA) is a rare genetic condition resulted from nonfunctional DNA repair mechanisms.
Because DNA repair is vital for every cell in the body, all organs are affected when repair mechanisms don't function properly. Organs that contain frequently dividing cells (such as skin) are the most affected. People with Fanconi anemia have bone marrow defects, organ abnormalities, and an increased risk of some cancers.
FANCC
FANCC codes for a protein involved in the Fanconi anemia (FA) disease pathway.
This pathway is activated when the cell DNA gets damaged. The FANCC protein is a part of a complex responsible for recognizing this DNA damage and activating repair mechanisms.
PPIB
Stands for, Peptidylprolyl Isomerase B (also known as Cyclophilin B), which codes for a protein that regulates protein folding in the cytoplasmic reticulum. Some mutations of PPIB result in impaired bone development.
we generated an RNA-editing reporter on Cluc by introducing a nonsense mutation [W85X (UGG→UAG)],
To create the reporter, the researchers "broke" the gene for the Cluc luciferase by introducing a mutation in the UGG codon, changing it to UAG (a nonsense mutation, which signals the ribosome to stop translation). Since this codon was positioned in the beginning of the Cluc transcript, no luciferase was synthesized in the cell.
The A (adenosine) in the UAG codon was the target for RNA editing. Both Cas13b-mediated RNA recognition and ADAR-mediated editing were required to remove the stop codon at the beginning of the Cluc transcript, which would restore Cluc expression.
This means that Cluc luminescence would only be seen where editing (both targeting and cleavage) was successful.
We next characterized the interference specificity of PspCas13b and LwaCas13a across the mRNA fraction of the transcriptome.
The next question was to understand the specificity of Cas13 in the whole cellular transcriptome (the portion of the genome that's transcribed). In the previous experiments, the researchers looked at the expression of only one target (unmodified or modified). Here, they narrowed their focus from the entire genome to just the transcriptome.
To do that, the authors transfected the cells with Cas13, LwaCas13a or PspCas13b, a gRNA to target Gluc, and a plasmid containing the gene for Gluc.
The control cells got an irrelevant gRNA instead of the gRNA targeting Gluc. As an additional control for comparison, the authors used shRNA-mediated knockdown of Gluc in a parallel cell culture.
After 48 hours the researchers collected the cells, extracted mRNA, and determined the sequences and the number of copies for each transcript.
We transfected HEK293FT cells with either LwaCas13a or PspCas13b, a fixed guide RNA targeting the unmodified target sequence, and the mismatched target library corresponding to the appropriate system.
The cells were transfected with a nuclease, a gRNA for the non-mutated target site and a whole library with all possible plasmid variants. After 48 hours the researchers collected the cells, extracted RNA, and determined which mutated sequences from the library were left uncleaved.
The transcript with the unmodified sequence was depleted most efficiently so that its level was the lowest after the cleavage. The levels of all other sequences with substitutions decreased to a lesser extent or did not decrease at all. The better Cas13 cut the sequence, the higher the depletion of this sequence was.
The authors then compared the sequences by their "depletion scores."
These results indicate that Cas13a and Cas13b display similar sequence constraints and sensitivities against mismatches.
The researchers found that Cas13a and Cas13b had the same sequence requirements for successful target recognition and cleavage. Neither nuclease was affected by changes to the PFS sequence, but both of them functioned less well when there were changes in the middle of the target sequence.
Another consequence of these findings is that, because changes to PFS sequences (which are at the edge of target sequences) don't affect Cas13 activity, off-target mRNA molecules that differ from the target only in PFS sequences might be mistakenly destroyed by Cas13.
Fortunately, because of its length (more than 30-nt), the spacer is highly specific and binds unique sequences.
Sequencing showed that almost all PFS combinations allowed robust knockdown
Substitutions in the PFS motifs did not affect how well Cas13a and Cas13b found and cut the target sequences. As a result, sequences with such substitutions were depleted as successfully as the control sequence, which was unmodified.
found that PspCas13b had consistently increased levels of knockdown relative to LwaCas13a (average of 92.3% for PspCas13b versus 40.1% knockdown for LwaCas13a)
As you can see on the figures 1C and 1D, the blue line corresponding to the PspCas13b nuclease goes far beyond the red line which corresponds to the LwaCas13a nuclease.
The signal of the Gluc luciferase that resulted from the PspCas13b knockdown was around 10% for all gRNA molecules along the transcript. This means that the average knockdown efficiency was 90%. The signal of Gluc in the case with LwaCas13a was not as consistent, but on average dropped by about 40%.
To more rigorously define the activity of PspCas13b and LwaCas13a, we designed position-matched guides tiling along both Gluc and Cluc transcripts and assayed their activity using our luciferase reporter assay.
To figure out which parts of the Gluc or Cluc RNA molecules were the best targets for Cas13, the authors generated a series of gRNA guides where each guide was shifted one to several nucleotides relative to the previous one. This is called tiling.
In this way, the guides together could cover the whole sequence or a part of a sequence that a researcher was interested in. See figure 4A and 4C or figure 5A for a visual of how the guides were "tiled."
Therefore, we tested the interference activity of the seven selected Cas13 orthologs C-terminally fused to one of six different localization tags without msfGFP.
The authors took six of the best-performing orthologs from the previous part of the study and replaced the msfGFP domain at the C-terminus of each ortholog with different localization sequences. They then tested Gluc knockdown in the same way they previously tested LwaCas13a.
We selected the top five Cas13b orthologs and the top two Cas13a orthologs for further engineering
Several Cas13a and b orthologs decreased Gluc luciferase signal in up to 10-30% of the control cells. Some Cas13b nucleases demonstrated even greater effectiveness than the previously characterized Cas13a from Leptotrichia wadei.
We transfected human embryonic kidney (HEK) 293FT cells with Cas13-expression, guide RNA, and reporter plasmids and then quantified levels of Cas13 expression and the targeted Gluc 48 hours later
To directly compare the effectiveness of Cas13a, b, and c orthologs, the authors transfected cells with two luciferases, Cas13 and two different gRNAs targeting Gluc luciferase.
They measured Gluc luciferase activity. Reduced Gluc luciferase activity indicated interference from the Cas13 ortholog and successful targeting by the gRNA.
They determined the expression of Cas13 to see whether Gluc knockdown was dependent on the quantity of Cas13 rather than the specific orthology.
hydrolytic deamination of adenosine to inosine, a nucleobase that is functionally equivalent to guanosine in translation and splicing
ADAR enzymes bind to adenosine and convert them to inosine by deamination, or the removal of an amino group from the molecule. In RNA, inosine is structurally similar to guanine (G), so it can form a base pair with cytidine (C). ![] (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2953425/bin/nihms234588f1.jpg)
Source: Nishikura, K. (2010). Functions and Regulation of RNA Editing by ADAR Deaminases. Annual Review of Biochemistry 79, 321–349. http://doi.org/10.1146/annurev-biochem-060208-105251
We recently reported that Cas13a enzymes can be adapted as tools for nucleic acid detection (14)
The authors developed a technique called SHERLOCK (Specific High-Sensitivity Enzymatic Reporter UnLOCKing) and successfully used it to detect Zika and dengue virus RNA, pathogenic bacterial DNA, and mutations in human DNA.
Both RNA and DNA molecules can be used as a starting material. First, they are converted to DNA and amplified by in vitro transcription into RNA. Next, Cas13 finds target RNA sequences and emits a signal that researchers use to detect the presence of certain RNA molecules.
See the creators of SHERLOCK talk about how it works and potential applications in this video.
Here, we describe the development of a precise and flexible RNA base editing technology using the type VI CRISPR-associated RNA-guided ribonuclease (RNase) Cas13
In this article, the authors describe how they created a system that can edit RNA molecules. They used a Cas13 protein fused to an adenine deaminase. The Cas13 protein recognized specific sequences on an RNA molecule, and the adenine deaminase edited bases, which can convert A to I (which is functionally read as a G).
The authors improved the targeting specificity (accuracy) and editing rate (precision) by Cas13 and deaminase mutagenesis, and determined the sequences for which this system is most effective. They showed one application of this technology by correcting a series of disease-causing mutations at the cellular level.
frame shifts
Genes are transcribed into proteins by codons, which are made up of sequences of three amino acids. Mutations that occur in numbers divisible by three will not affect the reading frame, but those not divisible by three will cause a shift in the reading frame. Generally, a mutation in-frame will not disrupt the protein function and can be handled by the cell. A frame-shift mutation will completely inactivate a protein and can be very detrimental to cellular function in some cases.
For example:
Original sequence: THE CAT WAS RED
Frame shift mutation: ATH ECA TWA SRE D
Non-frame shift mutation: THE BIG CAT WAS RED
Cpf1
Cpf1 is a nuclease that is analogous to Cas9.
Cpf1 differs from Cas9 in a number of ways. The most important one is that when Cpf1 cuts DNA, it leaves overhangs. It also requires different PAM sequences, which are short sequences that help the system distinguish self DNA from non-self DNA.
Cfp1 has the potential to be more accurate than Cas9, and can sometimes be used when there are no sequences that Cas9 can use as a target.
Precise nucleic acid–editing technologies
These techniques allow researchers to modify a chosen nucleic acid sequence.
The most widely used technologies are TALENs (transcription activator-like effector nucleases), ZFNs (zinc finger nucleases) and CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9).
CRISPR is the most popular since it can be programmed to new sequences using a guide RNA, whereas other tools must be engineered at the protein level, which is difficult and time consuming. The focus of this paper is a variation of CRISPR that uses Cas13.
Current editing tools, based on programmable nucleases
Check out this interview with author Feng Zhang about the development and potential uses of CRISPR technology. Dr. Zheng also compares Cas9 and Cpf1 nucleases.
To see an animation for how Cas9 works, check out this video.
endogenous editing of transcripts
RNA editing happens in the cell after DNA has been transcribed (RNA molecules have been synthesized from a using a DNA sequence as a template). This editing is usually insertion, deletion, or substitution of bases. The most common type of RNA editing is the substitution of adenosine to inosine, which functionally reads as guanosine (facilitated by the ADAR family of enzymes). Essentially, this reads as an A to G base change.
cloned
Cloning means to insert a gene, a gene fragment, or an accessory sequence into a vector. Vectors are vehicles that carry DNA into a cell and exist as a plasmid in the nucleus,
When the vector is multiplied in a cell or in a cell-free system, many copies of this fragment (clones) are generated.
To investigate PFS constraints on REPAIRv1, we designed a plasmid library that carryies a series of four randomized nucleotides at the 5′ end of a target site on the Cluc transcript
Though the authors had already characterized PFS preferences for Cas13b, they needed to check that the fusion of Cas13b with ADAR did not change its targeting efficiency and specificity. The researchers also wanted to confirm that PFS would work when generating RNA edits. This is important as DNA base editors are limited by the PAM of Cas9 and Cpf1 making RNA more powerful since you can target anywhere in the transcriptome. Therefore, it was important to check PFS constraints again.
small enough to fit within the packaging limit of AAV vectors
The wild type Cas9 is also a relatively big protein at 1368 amino acids. It is too long for many applications, including packaging into viral particles. Recently, scientists have developed a way to dramatically reduce the Cas9 size while retaining its DNA binding properties.
Smaller orthologs of Cas9, like saCas9, can also be used. Some therapeutic companies are now using this for viral delivery.
We mutated residues in ADAR2DD(E488Q) previously determined to contact the duplex region of the target RNA (Fig. 6A) (19).
The researchers mutated amino acids in ADAR involved with binding to the RNA target or catalytic deamination to test whether they affected deamination.
28. K. A. Lehmann, B. L. Bass, Biochemistry 39, 12875–12884 (2000)
The authors of this work investigated and described the activity of the human ADAR1 and ADAR2 deaminases.
They showed that ADAR proteins had certain sequence preferences, including the nucleotides around the target and the position of the target relative to the RNA duplex ends. Additionally, they determined that ADARs did not edit all accessible adenosines. That implied these proteins possessed some specificity with unknown mechanism.
22. S. K. Wong, S. Sato, D. W. Lazinski, RNA 7, 846–858 (2001).
This paper demonstrated that ADAR deaminases preferentially edit adenosine bases that are mismatched with a cytidine.
21. A. Kuttan, B. L. Bass, Proc. Natl. Acad. Sci. U.S.A. 109, E3295–E3304 (2012).
The authors of the paper mutated the ADAR2 protein and, by screening of multiple mutants, identified the E488Q variant that could edit adenosine in all possible triplet targets.
16. K. Nishikura, Annu. Rev. Biochem. 79, 321–349 (2010).
A comprehensive review on research into ADAR proteins.
15. O. O. Abudayyeh et al., Nature 550, 280–284 (2017).
The authors of this study showed that the Cas13a protein was comparable to RNAi in its efficiency at knocking down RNA, but has superior specificity.
They also generated a catalytically dead Cas13a and demonstrated that it retained RNA-binding properties. This dCas13a was successfully used for RNA tracking.
Finally, the authors did not observe cleavage at sites surrounding the target (collateral cleavage) with Cas13a in mammalian cells. This was extremely important as it suggests the nuclease can be safely used to target specific sites without major adverse effects.
14. J. S. Gootenberg et al., Science 356, 438–442 (2017).
The authors of the paper presented a new application of the CRISPR-based tools as diagnostic devices. They created a technique called SHERLOCK (Specific High-Sensitivity Enzymatic Reporter UnLOCKing) which was able to detect very small amounts of disease-causing viruses and bacteria, as well as mutations in human DNA.
9. Y. B. Kim et al., Nat. Biotechnol. 35, 371–376 (2017).
The same group of researchers that developed the first DNA base editor (7) improved the complex's specificity and expanded its ability to target.
They improved specificity by making the targeting window smaller, and expanded its ability to target different sequences by fusing the complex to Cas9 proteins that use different PAM sequences.
7. A. C. Komor, Y. B. Kim, M. S. Packer, J. A. Zuris, D. R. Liu, Nature 533, 420–424 (2016)
The authors of the study suggested an alternative strategy to genome editing that doesn't use DNA breaks and donor templates. They created a DNA base-editor complex composed of a mutant Cas9 and a cytidine deaminase APOBEC1. This complex was able to convert all C-G pairs into T-A within a narrow targeting window. However, it required additional modules to control the editing.
8. K. Nishida et al., Science 353, aaf8729 (2016).
This group of scientists created a DNA base editor consisting of Cas9 and the cytidine deaminase PmCDA1. Similar to other work (7), this editor modified all cytidines in a target window and required an additional protein module for the control of editing.
Cas13 has no targeting sequence constraints
An important advantage of Cas13 over Cas9 is that it does not have a PAM or PFS that is required for it to target a specific site. To expand the number of sequences that Cas9 can target, researchers must use Cas9 proteins from different species (which have different associated PAM sequences). This can complicate the development and testing of gene editing systems that use Cas9.
the exact target site can be encoded in the guide by placing a cytidine within the guide
In terms of precision, i.e., on-target editing, we can compare REPAIR to other base editors.
For instance, cytidine editors contain the cytidine deaminase APOBEC1 and convert C-G pairs to A-T pairs. The activity of APOBEC1, directed by Cas9, is not restricted to only one C but extends for several bases away from the PAM sequence. Recent work has demonstrated that the editing window of APOBEC may be reduced to 1-2 nucleotides by modifying of the deaminase.
Similarly, the precision of ADAR2 may be increased by placing a C across from the target A, creating a mismatch that is ideal for ADAR2 editing.
Although it is possible that ADAR could deaminate adenosine bases on the DNA strand in RNA-DNA heteroduplexes
The authors of the paper looked at the crystal structure of the ADAR deaminase with the RNA duplex and predicted that ADAR might also operate on RNA-DNA duplexes. Later, they experimentally confirmed this prediction.
For guides targeting either KRAS or PPIB, we found that REPAIRv2 had no detectable off-target edits, unlike REPAIRv1, and could effectively edit the on-target adenosine at efficiencies of 27.1% (KRAS) or 13% (PPIB)
REPAIRv2 showed a moderate decrease in on-target activity, but at the same time it had substantially lower off-target editing.
There is a trade-off between on-target activity and off-target numbers. Depending on how an editor is being used, it may be more useful to have high activity and low specificity or lower activity and high specificity.
There is a middle ground where an editor is less active (but still useful) but more accurate.
Furthermore, we speculated that different transcriptome states could also potentially alter the number of off-targeting events.
Different cell types specialized to performing particular functions require different proteins for their work. Therefore, they have distinct transcriptomes.
Moreover, a cell's transcriptome changes depending on where it is in the cell cycle and the conditions and needs of the surrounding environment.
As a result, off-targets of the REPAIR system may vary from one cell type to another, reflecting changes in the presence and abundance of individual transcripts.
REPAIRv1 off-target edits were predicted to result in numerous variants, including 1000 missense base changes (fig. S13C), with 93 events in genes related to cancer processes
Earlier the authors only counted off-targets and did not look at the effect of adenosine modification on the transcript function. Here, they determined the position of the modification and the possible impact on the amino acid choice if the edit was in the protein coding region.
We selected a subset of these mutants (Fig. 6B) for transcriptome-wide specificity profiling by next-generation sequencing.
The recovery of the Cluc signal is an easy way to get the first look at the activity of the mutants. However, a single transcript cannot reflect changes in the whole transcriptome.
we tested 17 single mutants with both targeting and nontargeting guides, under the assumption that background luciferase restoration in the nontargeting condition would be indicative of broader off-target activity.
It would be too expensive to test all the mutants by RNA sequencing to find whole-transcriptome off-targets. Instead, the authors looked at a single transcript for Cluc luciferase.
The authors assumed that if luciferase activity was restored even non-targeting guide RNAs, it would mean that ADAR2 increased off-target activity.
We analyzed the editing efficiency of these two systems compared with REPAIRv1 and found that the BoxB-ADAR2 and full-length ADAR2 systems demonstrated 50 and 34.5% editing rates, respectively, compared with the 89% editing rate achieved by REPAIRv1
The authors demonstrated that the constructs with mutated ADAR had enhanced editing activity at both on-target and off-target sites. They also observed that the construct with the full-length ADAR protein was less efficient at editing and had fewer off-target effects.
There was a high degree of overlap in the off-target editing events between ADARDD(E488Q) and all REPAIRv1 off-target edits, supporting the hypothesis that REPAIR off-target edits are driven by dCas13b-independent ADARDD(E488Q) editing events
This conclusion was drawn from the observation that ADAR deaminase domain alone could produce a substantial number of off-targets found under the condition when the full REPAIRv1 system was applied.
Nevertheless, figure S8C shows that ADAR activity cannot account for all off-targets and there are likely other sources of off-target effects. Comparing the off-target numbers for four different conditions shows that the REPAIRv1 construct has a greater level of irrelevant editing than the ADAR domain alone.
To reduce the size, we tested a variety of N-terminal and C-terminal truncations of dCas13
AAVs can accommodate up to 4.7kb, meaning dCas13b and the extra regulatory sequences are too big to fit in one virus. To solve this problem, the authors truncated (shortened) dCas13b by removing sections of the protein with unnecessary functions.
The dCas13b nuclease is composed of different domains (parts), some of which are used for RNA binding and some which are used for cleavage. The RNA cleavage is mediated by two well-defined HEPN domains which are located close to the N- and C-terminus of the protein. We are still unsure which domains mediate binding.
However, the REPAIRv1 system needs only the RNA binding ability of dCas13b. Therefore, the researchers were able to remove sections of dCas13b containing HEPN while retaining its binding function.
We found that all C-terminal truncations tested were still functional and able to restore luciferase signal (fig. S7)
The REPAIR construct with the largest C-terminal truncation (removal of the end) of dCas13b demonstrated the same level of RNA editing as the original REPAIRv1. This meant that the researchers were able to shorten dCas13b enough to fit into an AAV, but not destroy its ability to bind RNA.
AAV vectors have a packaging limit of 4.7 kb
Almost the entire AAV genome can be replaced with a desired construct of max length of 4.7kb. This limits how much genetic material the virus can carry, so some essential viral genes are carried in an additional construct or constructs (the "helper plasmid" in the picture below).

Learn more about AAV at Addgene.
We then tested the ability of REPAIRv1 to correct 34 different disease-relevant G→A mutations
The researchers chose 34 disease causing mutations to test. Each mutation was a G to A substitution that changed the amino acid sequence of the protein and disrupted normal function.
Using guide RNAs containing 50-nt spacers
The authors chose three 50-nt spacers to target two genes carrying disease-relevant mutations. As was determined at the previous step, the spacers of 50-nt length showed higher rates of editing but more off-target effects than 30-nt spacers.
AVPR2
AVPR2 is a gene that codes for a protein called vasopressin V2 receptor. AVPR2 binds the hormone vasopressin and contributes to the regulation of water in the body.
To demonstrate the broad applicability of the REPAIRv1 system for RNA editing in mammalian cells, we designed REPAIRv1 guides against two disease-relevant mutations
After the authors had generated successful REPAIRv1-mediated editing of mRNA for an exogenous gene (i.e., a gene introduced from outside the organism), they chose two endogenous genes (i.e., native to the organism) to further explore the power of REPAIRv1.
They tested Cas13b nuclease activity in the same way as they did with the exogenous gene.
we modified the linker between dCas13b and ADAR2DD(E488Q)
The dCas13b and ADAR2 protein parts are joined together via a stretch of amino acids called a linker. There are more than a thousand linker variants in different multi-domain proteins. The sequence of a linker can influence protein folding and stability, as well as functional properties of individual domains. Therefore, the choice of linker is an important step in protein design.
The authors tested linkers of different length and flexibility and found that shorter and more flexible variants produced the best results.
We also observed that 50-nt spacers had an increased propensity for editing at nontargeted adenosines within the sequencing window, likely because of increased regions of duplexed RNA
The longer, 50-nt spacers showed higher rates of editing but also more off-target effects. This suggests a tradeoff between the efficiency of editing and the specificity.
To validate that restoration of luciferase activity was due to bona fide editing events, we directly measured REPAIRv1-mediated editing of Cluc transcripts via reverse transcription and targeted next-generation sequencing.
The ADAR deaminase can introduce changes not only into the target adenosine but also in the surrounding adenosine bases. To check how specific the dCas13-ADAR2 protein was in targeting the correct adenosine base, the researchers sequenced edited Cluc transcripts and determined all positions with A to I substitutions.
Further, they used tiling gRNA molecules in order to determine the influence of the spacer (PFS) length and the mismatch distance on the off-target editing.
position-matched guides
The authors directly compared the RNA knockdown efficiency of two technologies, Cas13 cleavage and RNA interference (RNAi). Both technologies require guide RNA (gRNA) molecules for targeted recognition. Cas13 is directed by a gRNA, while RNAi complex uses a molecule termed shRNA.
Different parts of an RNA molecule can be more or less accessible to gRNAs. Therefore, gRNA and shRNA target sequences have to be selected such that they are close to each other, i.e. position-matched, so that a more fair comparison between Cas13 and RNAi can be made.
Cas13b from Prevotella sp. P5-125 (PspCas13b) and Cas13b from Porphyromonas gulae (PguCas13b) C-terminally fused to the HIV Rev nuclear export sequence (NES), and Cas13b from Riemerella anatipestifer (RanCas13b) C-terminally fused to the mitogen-activated protein kinase NES
Why did the authors test several NESs from different proteins?
Over 200 NESs from different proteins have been described. Each NES is around 10 amino acids long and has a unique structure. The variation between NESs means that they have different effects on export efficiency and protein stability in different environments.
We hypothesized that Cas13 activity could be affected by subcellular localization, as we previously reported for optimization of LwaCas13a
The RNA molecule is transcribed in the nucleus and then exported to the cytoplasm. It can be recognized and cleaved by Cas13 at any moment along this way.
Previously, the authors used nuclear localization sequences (NLS) and nuclear export sequences (NES) to determine at what point during the transcription and export process Cas13 is most efficient. They tested knockdown efficiency of LwaCas13a with either NLS or NES tags.
They found that constructs with a nuclear localization sequence (NLS) were most efficient. However, this observation cannot be generalized to all Cas13 orthologs, which must be tested individually.
We found that dCas13b-ADAR1DD(E1008Q) required longer guides to repair the Cluc reporter, whereas dCas13b-ADAR2DD(E488Q) was functional with all guide lengths tested
The authors found that the the length of the guide RNA affects how efficiently the RNA-editing protein complex finds the target sequence.
In order to find the best construct, it is important to test different targeting conditions, testing as many target sequences as possible while also considering the resources required to test each one.
We introduced a mismatched cytidine opposite the target adenosine, which has been previously reported to increase deamination frequency,
Previous work has shown that ADAR works better when an RNA molecule contains a mismatch at a target site. This may be because it makes it easier for the deaminase to flip the target base, which has to happen for editing. Panel B shows what base flipping looks like:
Zheng, Y., Lorenzo, C., & Beal, P. A. (2017). DNA editing in DNA/RNA hybrids by adenosine deaminases that act on RNA. Nucleic Acids Research, 45(6), 3369–3377. http://doi.org/10.1093/nar/gkx050
To engineer a PspCas13b lacking nuclease activity (dPspCas13b, referred to as dCas13b hereafter), we mutated conserved catalytic residues in the HEPN domains and observed loss of luciferase RNA knockdown
The authors hypothesized that even without nuclease activity, Cas13b would still be able to recognize target molecules directed by a gRNA.
Mutations of the key part of the protein responsible for RNA cleavage eliminated Cas13b's catalytic ability. The next step was to test whether the mutated Cas13b was still able to find the target sequences, even if it couldn't cleave them.
containing hyperactivating mutations in order to enhance catalytic activity [ADAR1DD(E1008Q) (27) or ADAR2DD(E488Q)] (21)
Previously, ADAR2 was randomly mutated, and protein variants with improved activity were selected. The protein with a substitution at the 488 position showed the highest activity.
Later, another group tried the same thing with ADAR1. The group compared protein sequences and found the corresponding amino acid position on ADAR1 and performed the same substitution, which resulted in the same increase in activity. See the image below for a comparison of ADAR1 and ADAR2:
Tomaselli, S., Bonamassa, B., Alisi, A., Nobili, V., Locatelli, F., & Gallo, A. (2013). ADAR Enzyme and miRNA Story: A Nucleotide that Can Make the Difference. International Journal of Molecular Sciences, 14(11), 22796–22816. http://doi.org/10.3390/ijms141122796
off-targets
Off-target effects occur when the nuclease introduces changes to irrelevant sequences because of their similarity to the target sequence. High frequency of off-target effects are undesirable because it corresponds to low specificity, making it hard to control the nuclease activity.
We found that LwaCas13a and PspCas13b had a central region that was relatively intolerant to single mismatches
The sequences with substitutions in the middle part (between 12 and 26 nucleotides) had the lowest depletion scores. A lower depletion score means that the nuclease was not as successful at cleaving the RNA sequence (i.e. depleting expression).
To characterize the interference specificities of PspCas13b and LwaCas13a, we designed a plasmid library of luciferase targets containing single mismatches and double mismatches throughout the target sequence and the three flanking 5′ and 3′ base pairs
To figure out how specifically the Cas13 orthologs would target RNA, they created a "library" (collection) of sequences with one or two mutations in the gRNA target site for the Gluc gene. The idea was to see how different a sequence could be and still be recognized by the nuclease.
Remember that the goal is to find a highly specific nuclease. This means that ideally the nuclease would not recognize sequences with mutations.
KRAS
KRAS is a protein which participates in intracellular signal transduction. Importantly, it controls cell proliferation. When mutated, it becomes constitutively active (always turned on) and contributes to the development of several cancers.
without msfGFP
The authors ran the assay without msfGFP to determine if any of the orthologs do not require stabilization domains. This is important because orthologs that do not require these domains could be used when an experimental construct needs to be small.
For each Cas13 ortholog, we designed PFS-compatible guide RNAs, using the Cas13b PFS motifs derived from an ampicillin interference assay
Cas13a prefers to recognize target sequences when a spacer (target sequence) is surrounded by specific PFS motifs.
To determine which PFS sequences Cas13b orthologs prefer, the researchers used an ampicillin interference assay. They transformed bacterial cells with two plasmids: One contained a Cas13b ortholog while the second plasmid included an ampicillin resistance gene and a gRNA against this gene with randomly generated 5' and 3' PFS around the target sequence. If the PFS led to the Cas13b ortholog targeting the correct gene, bacterial cells would die because they would lose ampicillin resistance when the gene was cleaved.
The authors collected and analyzed cells that survived to determine which PFS sequences were irrelevant for targeting. They then subtracted these PFSs from the starting pool to determine which PFSs are preferred by Cas13b orthologs.
To assay interference in mammalian cells, we designed a dual-reporter construct expressing the independent Gaussia (Gluc) and Cypridina (Cluc) luciferases under separate promoters, allowing one luciferase to function as a measure of Cas13 interference activity and the other to serve as an internal control.
To monitor the effect of Cas13 nuclease activity, the researchers constructed a vector that contained genes for two different luciferases. These luciferases use different substrates (source materials) to generate light.
One luciferase was used to measure Cas13 interference, and the other was used as a control. In cells where Cas13 did not interfere, both luciferases would emit light. In cells with interference, however, activity from one of the luciferases would decrease.
nuclear localization signal
Localization signals make sure proteins go to the right place in a cell. These signals are in the form of sequences that are recognized by different parts of the cell.
codon-optimized
Typically, a single amino acid is coded by many different codons. Different species sometimes use different codons for the same amino acid, or produce different numbers of amino acids from the same codon.
As a result, when a researcher introduces a gene for a protein from one species into another, the amount of protein made is usually small. To increase the amount of protein produced, it is important to use codons for that particular species. This is done by introducing synonymous mutations in the gene. Synonymous mutations change a DNA sequence but result in the same amino acid.
We sought to identify a more robust RNA-targeting CRISPR system by characterizing a genetically diverse set of Cas13 family members
Previously, the authors tested 15 orthologs of the Cas13a nuclease for their ability to knock down RNA. In this study, they expanded their search to other members of the Cas13 family, including a, b, and c variants.
ADAR1 and ADAR2
In addition to the functional ADAR1 and ADAR2 enzymes, there is a catalytically inactive ADAR3 enzyme in the human genome. Although it cannot deaminate RNA, ADAR3 might still regulate gene expression, for example by inhibiting its two functional homologs (ADAR1 and ADAR2).
it required a monomeric superfolder green fluorescent protein (msfGFP) stabilization domain for efficient knockdown
Superfolder GFP is a protein derived from GFP, a "reporter" that can be added to genes to see where and when they are expressed in an organism. GFP lights up when it is exposed to a specific wavelength of light.
Superfolder GFP has several mutations that improve its folding properties and stability in comparison to regular GFP. In this particular experiment, the researchers fused a msfGFP (Superfolder GFP) domain to Cas13 to serve as a reporter and to increase the stability of the nuclease.
To learn more about GFP, check out this Science in the Classroom resource.
Although previous approaches have engineered targeted ADAR fusions via RNA guides (23–26), the specificity of these approaches has not been reported, and their respective targeting mechanisms rely on RNA-RNA hybridization without the assistance of protein partners that may enhance target recognition and stringency.
Several groups have built programmable RNA-editing techniques based on the ADAR deaminase. The groups used different approaches to direct the editor to target mRNAs and demonstrated that these systems are efficient.
However, none of the groups have extensively studied how accurately these systems find target sequences, or the effects systems have when they hit the wrong target. This is important, because a gene editor must be highly accurate (specific) to be useful for most applications.
repetitive regions
Stretches of repeated sequences of nucleic acids (DNA or RNA). Most repetitive regions do not occur in protein-coding genes. However, they are important for regulation.
The ADAR catalytic domain is capable of deaminating target adenosines without any protein cofactors in vitro
This is very important because it means that ADAR catalytic domain can be used independently of any other cofactors (which help enzymes catalyze reactions). As a result, it can still catalyze the deamination reaction when fused to another protein.
host machineries
Systems of enzymes and accessory molecules that carry out cellular functions.
However, the potential targeting sites of DNA base editors are limited by the requirement of Cas9 for a protospacer adjacent motif (PAM) at the editing site
For CRISPR/Cas9 to work, there must be a special sequence (called a PAM sequence) next to the target site. Cas9 uses this PAM sequence to target a certain part of the DNA molecule.
Different versions of Cas9 can use different PAM sequences. Researchers have experimented with different natural and artificial Cas9 enzymes to expand the range of sequences we can edit. However, there are still some sequences that we cannot edit because we do not have a version of Cas9 (or another enzyme) that can target them.
The distribution of edits on transcripts was heavily skewed for REPAIRv1, with highly edited genes having more than 60 edits
These transcripts are the most abundant in the cell and are therefore easily accessible for the deaminase. Multiple edits along the transcript may imply that the editing complex is able to stay bound to the mRNA long enough to modify not only the target A but also the surrounding adenosines.
X-linked nephrogenic diabetes insipidus
Nephrogenic diabetes insipidus (NDI) is a congenital disease characterized by the body's inability to make concentrated urine. It can be caused by mutations in two different genes, including AVPR2.