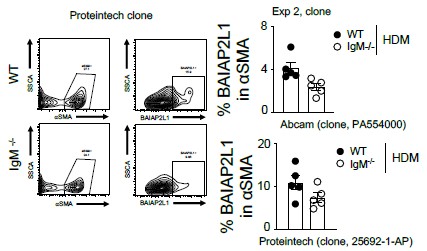
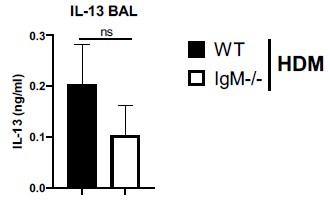
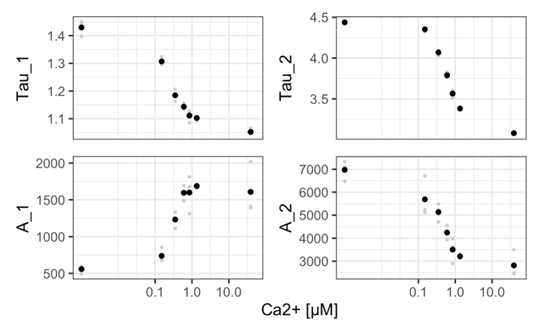
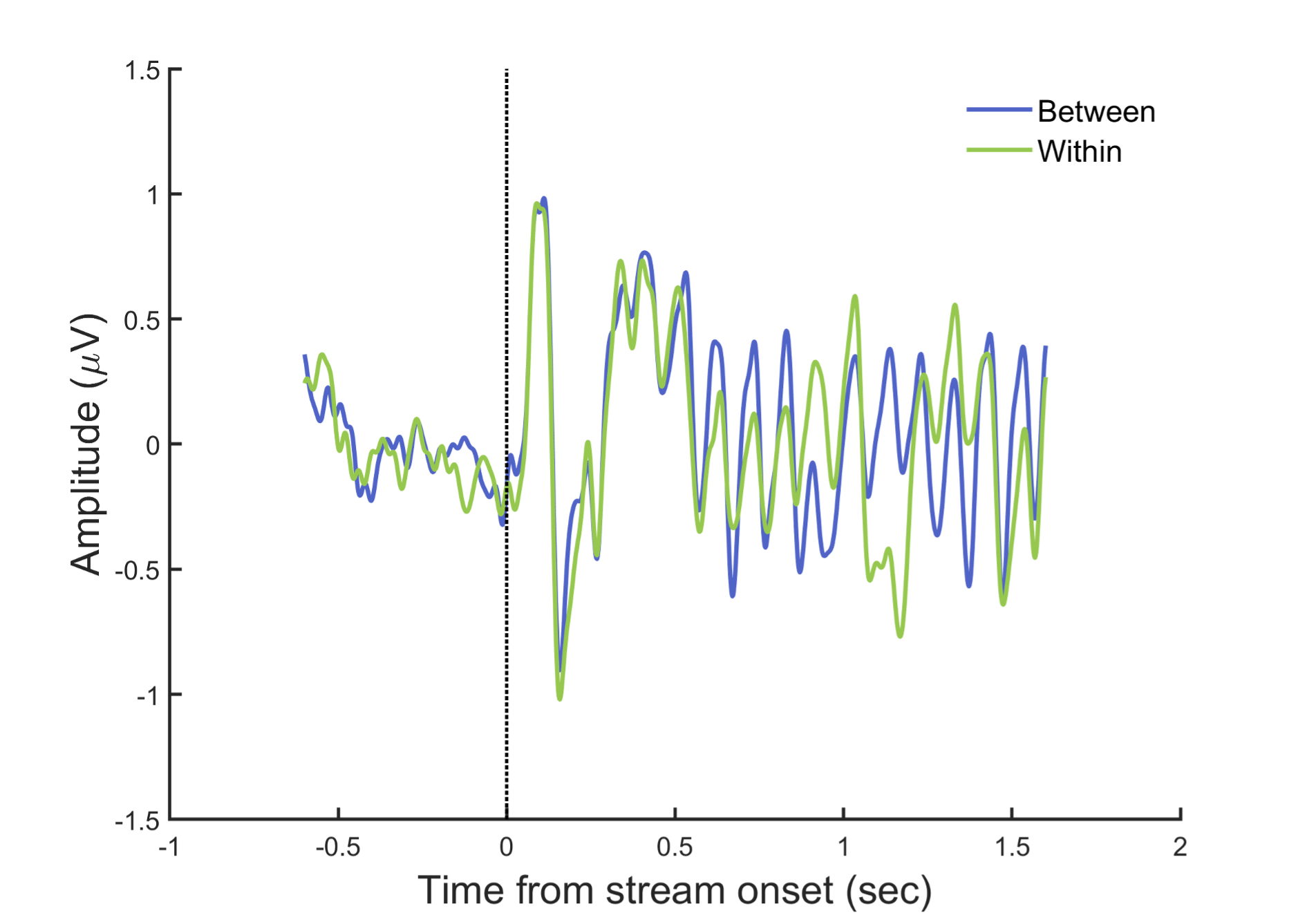
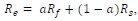Author response:
The following is the authors’ response to the original reviews
Public Reviews:
Reviewer #1 (Public review):
Domínguez-Rodrigo and colleagues make a moderately convincing case for habitual elephant butchery by Early Pleistocene hominins at Olduvai Gorge (Tanzania), ca. 1.8-1.7 million years ago. They present this at the site scale (the EAK locality, which they excavated), as well as across the penecontemporaneous landscape, analyzing a series of findspots that contain stone tools and large-mammal bones. The latter are primarily elephants, but giraffids and bovids were also butchered in a few localities. The authors claim that this is the earliest well-documented evidence for elephant butchery; doing so requires debunking other purported cases of elephant butchery in the literature, or in one case, reinterpreting elephant bone manipulation as being nutritional (fracturing to obtain marrow) rather than technological (to make bone tools). The authors' critical discussion of these cases may not be consensual, but it surely advances the scientific discourse. The authors conclude by suggesting that an evolutionary threshold was achieved at ca. 1.8 ma, whereby regular elephant consumption rich in fats and perhaps food surplus, more advanced extractive technology (the Acheulian toolkit), and larger human group size had coincided.
The fieldwork and spatial statistics methods are presented in detail and are solid and helpful, especially the excellent description (all too rare in zooarchaeology papers) of bone conservation and preservation procedures. However, the methods of the zooarchaeological and taphonomic analysis - the core of the study - are peculiarly missing. Some of these are explained along the manuscript, but not in a standard Methods paragraph with suitable references and an explicit account of how the authors recorded bone-surface modifications and the mode of bone fragmentation. This seems more of a technical omission that can be easily fixed than a true shortcoming of the study. The results are detailed and clearly presented.
By and large, the authors achieved their aims, showcasing recurring elephant butchery in 1.8-1.7 million-year-old archaeological contexts. Nevertheless, some ambiguity surrounds the evolutionary significance part. The authors emphasize the temporal and spatial correlation of (1) elephant butchery, (2) Acheulian toolkits, and (3) larger sites, but do not actually discuss how these elements may be causally related. Is it not possible that larger group size or the adoption of Acheulian technology have nothing to do with megafaunal exploitation? Alternative hypotheses exist, and at least, the authors should try to defend the causation, not just put forward the correlation. The only exception is briefly mentioning food surplus as a "significant advantage", but how exactly, in the absence of food-preservation technologies? Moreover, in a landscape full of aggressive scavengers, such excess carcass parts may become a death trap for hominins, not an advantage. I do think that demonstrating habitual butchery bears very significant implications for human evolution, but more effort should be invested in explaining how this might have worked.
Overall, this is an interesting manuscript of broad interest that presents original data and interpretations from the Early Pleistocene archaeology of Olduvai Gorge. These observations and the authors' critical review of previously published evidence are an important contribution that will form the basis for building models of Early Pleistocene hominin adaptation.
This is a good example of the advantages of the eLife reviewing process. It has become much too common, among traditional peer-reviewing journals, to reject articles when there is no coincident agreement in the reviews, regardless of the heuristics (i.e., empirically-supported weight) of the arguments on both reviewers. Reviewers 1 and 2 provide contrasting evaluations, and the eLife dialogue between authors and reviewers enable us to address their comments differentially. Reviewer 1 (R1), whose evaluation is overall positive, remarks that the methods of the zooarchaeological and taphonomic analysis are missing. We have added them now in the revised version of our manuscript. R1 also remarks that our work highlights correlation of events, but not necessarily causation. We did not establish causation because such interpretations bear a considerable amount of speculation (and they might have fostered further criticism by R2); however, in the revised version, we expanded our discussion of these issues substantially. Establishing causation among the events described is impossible, but we certainly provide arguments to link them.
Reviewer #2 (Public review):
The authors argue that the Emiliano Aguirre Korongo (EAK) assemblage from the base of Bed II at Olduvai Gorge shows systematic exploitation of elephants by hominins about 1.78 million years ago. They describe it as the earliest clear case of proboscidean butchery at Olduvai and link it to a larger behavioral shift from the Oldowan to the Acheulean.
The paper includes detailed faunal and spatial data. The excavation and mapping methods appear to be careful, and the figures and tables effectively document the assemblage. The data presentation is strong, but the behavioral interpretation is not supported by the evidence.
The claim for butchery is based mainly on the presence of green-bone fractures and the proximity of bones and stone artifacts. These observations do not prove human activity. Fractures of this kind can form naturally when bones break while still fresh, and spatial overlap can result from post-depositional processes. The studies cited to support these points, including work by Haynes and colleagues, explain that such traces alone are not diagnostic of butchery, but this paper presents them as if they were.
The spatial analyses are technically correct, but their interpretation extends beyond what they can demonstrate. Clustering indicates proximity, not behavior. The claim that statistical results demonstrate a functional link between bones and artifacts is not justified. Other studies that use these methods combine them with direct modification evidence, which is lacking in this case.
The discussion treats different bodies of evidence unevenly. Well-documented cut-marked specimens from Nyayanga and other sites are described as uncertain, while less direct evidence at EAK is treated as decisive. This selective approach weakens the argument and creates inconsistency in how evidence is judged.
The broader evolutionary conclusions are not supported by the data. The paper presents EAK as marking the start of systematic megafaunal exploitation, but the evidence does not show this. The assemblage is described well, but the behavioral and evolutionary interpretations extend far beyond what can be demonstrated.
We disagree with the arguments provided by Reviewer 2 (R2). The arguments are based on two issues: bone breakage and spatial association. We will treat both separately here.
Bone breakage
R2 argues that:
“The claim for butchery is based mainly on the presence of green-bone fractures and the proximity of bones and stone artifacts. These observations do not prove human activity. Fractures of this kind can form naturally when bones break while still fresh, and spatial overlap can result from post-depositional processes. The studies cited to support these points, including work by Haynes and colleagues, explain that such traces alone are not diagnostic of butchery, but this paper presents them as if they were.”
In our manuscript, we argued that green-breakage provides an equally good (or even better) taphonomic evidence of butchery if documented following clear taphonomic indicators. Not all green breaks are equal and not all “cut marks” are unambiguously identifiable as such. First, “natural” elephant long limb breaks have been documented only in pre/peri-mortem stages when an elephant breaks a leg. As a matter of fact, they have only been reported in publication on femora, the thinnest long bone (Haynes et al., 2021). Unfortunately, they have been studied many months after the death of the individuals, and the published diagnosis is made under the assumption that no other process intervened in the modification of those bones during this vast time span. Most of the breaks resulting from pre-mortem fractures produce long smooth, oblique/helical outlines. Occasionally, some flake scarring may occur on the cortical surface. This has been documented as uneven, small-sized, spaced, and we are not sure if it resulted from rubbing of broken fragments while the animal was alive and attempting to walk or some may have resulted from dessication of the bone after one year. When looking at them in detail, such breaks contain sometimes step-microfractures and angular (butterfly-like) outlines. Sometimes, they may be accompanied by pseudo-notches, which are distinct and not comparable to the deep notches that hammerstone breaking generates on the same types of bones. Commonly, the edges of the breaks show some polishing, probably from separate break planes rubbing against each other. It should be emphasized that the experimental work on hammerstone breaking documented by Haynes et al. (2021) is based on bone fracture properties of bones that are no longer completely green. The cracking documented in their hammerstone experimentation, with very irregular outlines differs from the cracking that we are documented in butchery of recently dead elephants.
All this contrasts with the overlapping notches and flake scars (mostly occurring on the medullary side of the bone), both of them bigger in size, with clear smooth, spiral and longitudinal trajectories, with a more intensive modification on the medullary surface, and with sharp break edges resulting from hammerstone breaking of the green bone. No “natural” break has been documented replicating the same morphologies displayed in the Supplementary File to our paper. We display specimens with inflection points, hackle marks on the breaks, overlapping scarring on the medullary surface, with several specimens displaying percussion marks and pitting (also most likely percussion marks). Most importantly, we document this patterned modification on elements other than femora, for which no example has been documented of purported morphological equifinality caused by pre-mortem “natural” breaking. In contrast, such morphologies are documented in hammerstone-broken completely green bones (work in progress). We cited the works of Haynes to support this, because they do not show otherwise. As a matter of fact, Haynes himself had the courtesy of making a thorough reading of our manuscript and did not encounter any contradiction with his work.
Spatial association
R2 argues in this regard:
“The spatial analyses are technically correct, but their interpretation extends beyond what they can demonstrate. Clustering indicates proximity, not behavior. The claim that statistical results demonstrate a functional link between bones and artifacts is not justified. Other studies that use these methods combine them with direct modification evidence, which is lacking in this case.”
We should emphasize that there is some confusion in the use and interpretation of clustering by R2 when applied to EAK. R2 appears to interpret clustering as the typical naked-eye perception of the spatial association of different items. In contrast, we rely on the statistical concept of clustering, more specifically on spatial interdependence or covariance, which is different. Items may appear visually clustered but still be statistically independent. This could, for example, result from two independent depositional episodes that happen to overlap spatially. In such cases, the item-to-item relationship does not necessarily show any spatial interdependence between classes other than simple clustering (i.e., spatial coincidence in intensity).
Spatial statistical interdependence, on the other hand, reflects a spatial relationship or co-dependence between different items. This goes beyond the mere fact that classes appear clustered: items between classes may show specific spatial relationships — they may avoid each other or occupy distinct positions in space (regular co-dependence), or they may interact within the same spatial area (clustering co-dependence). Our tests indicate the latter for EAK.
Such patterns are difficult to explain when depositional events are unrelated, since the probability that two independent events would generate identical spatial patterns in the same loci is very low. They are also difficult to reconcile when post-depositional processes intervene and resediment part of the assemblage (Domínguez-Rodrigo et al. 2018).
Finally, R2 concludes:
“The discussion treats different bodies of evidence unevenly. Well-documented cut-marked specimens from Nyayanga and other sites are described as uncertain, while less direct evidence at EAK is treated as decisive. This selective approach weakens the argument and creates inconsistency in how evidence is judged.”
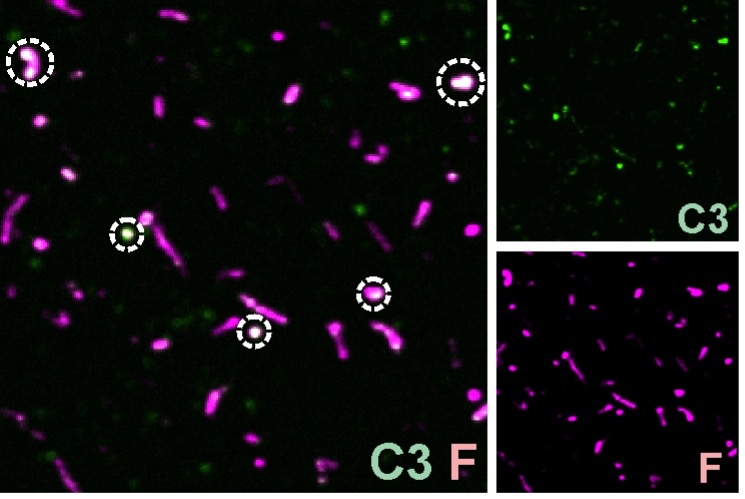
The Nyayanga hippo remains bearing modifications have not been well-documented cut marks. Neither R2 nor we can differentiate those marks from those inflicted by natural abrasive processes in coarse-grained sedimentary contexts, where the carcasses are found. The fact that the observable microscopic features (through low-quality photographs as appear in the original publication) differ between the cut marks documented on smaller animals and those inferred for the hippo remains makes them even more ambiguous. Nowhere in our manuscript do we treat the EAK evidence (or any other evidence) as decisive, but as the most likely given the methods used and the results reported.
References
Haynes G, Krasinski K, Wojtal P. 2021. A Study of Fractured Proboscidean Bones in Recent and Fossil Assemblages. Journal of Archaeological Method and Theory 28:956–1025.
Domínguez-Rodrigo, M., Cobo-Sánchez, L., yravedra, J., Uribelarrea, D., Arriaza, C., Organista, E., Baquedano, E. 2018. Fluvial spatial taphonomy: a new method for the study of post-depositional processes. Archaeological and Anthropological Sciences 10: 1769-1789.
Recommendations for authors:
Reviewer #1 (Recommendations for the authors):
I have several recommendations that, in my opinion, could enhance the communication of this study to the readers. The first point is the only crucial one.
(1) A detailed zooarchaeological methods section must be added, with explanations (or references to them) of precisely how the authors defined and recorded bone-surface modifications and mode of bone fragmentation.
This appears in the revised version of the manuscript in the form of a new sub-section within the Methods section.
(2) The title could be improved to better represent the contents of the paper. It contains two parts: the earliest evidence for elephant butchery (that's ok), and revealing the evolutionary impact of megafaunal exploitation. The latter point is not actually revealed in the manuscript, just alluded to here and there (see also below).
We have elaborated on this in the revised version, linking megafaunal exploitation and anatomical changes (which appear discussed in much more detail in the references indicated).
(3) The abstract does not make it clear whether the authors think that the megafaunal adaptation strongly correlates with the Acheulian technocomplex. It seems that they do, so please make this point apparent in the abstract.
From a functional point of view, we document the correlation, but do not believe in the causation, since most butchering tools around these megafaunal carcasses are typologically non Acheulian. We have indicated so in the abstract.
(4) Please define what you mean by "megafauna". How large should an animal be to be considered as megafauna in this particular context?
We have added this definition: we identify as “megafauna” those animals heavier than 800 kg.
(5) In the literature survey, consider also this Middle Pleistocene case-study of elephant butchery, including a probable bone tool: Rabinovich, R., Ackermann, O., Aladjem, E., Barkai, R., Biton, R., Milevski, I., Solodenko, N., and Marder, O., 2012. Elephants at the middle Pleistocene Acheulian open-air site of Revadim Quarry, Israel. Quaternary International, 276, pp.183-197.
Added to the revised version
(6) The paragraph in lines 123-160 is unclear. Do the authors argue that the lack of evidence for processing elephant carcasses for marrow and grease is universal? They bring forth a single example of a much later (MIS 5) site in Germany. Then, the authors state the huge importance of fats for foragers (when? Where? Surely not in all latitudes and ecosystems). This left me confused - what exactly are you trying to claim here?
We have explained this a little more in the revised text. What we pointed out was that most prehistoric (and modern) elephant butchery sites leave grease-containing long bones intact. Evidence of anthropogenic breakage of these elements is rather limited. The most probably reason is the overabundance of meat and fat from the rest of the carcass and the time-consuming effort needed to access the medullary cavity of elephant long bones.
(7) The paragraph in lines 174-187 disrupts the flow of the text, contains previously mentioned information, ends with an unclear sentence, and could be cut.
(8) Results: please provide the MNI for the EAK site (presumably 1, but this is never mentioned).
Done in the revised version.
(9) Lines 292 - 295: The authors found no traces of carnivoran activity (carnivoran remains, coprolites, or gnawing marks on the elephant bones), yet they attribute the absence of some non-dense skeletal elements to carnivore ravaging. I cannot understand this rationale, given that other density-mediated processes could have deleted the missing bones and epiphysis.
This interpretation stems from our observations of several elephant carcasses in the Okavango delta in Botswana. Those that were monitored showed deletion of remains (i.e., disappearance of certain bones, like feet) without necessarily imprinting damage on the rest of the carcass. Carnivore intervention in an elephant death site can result in deletion of a few remains without much damage (if any), or if hyena clans access the carcass, much more conspicuous damage can be documented. There is a whole range of carnivore signatures in between. We are currently working on our study of several elephant carcasses subjected to these highly variable degrees of carnivore impact.
(10) Lines 412 - 422: "The clustering of the elephant (and hippopotamus) carcasses in the areas containing the highest densities of landscape surface artifacts is suggestive of a hominin agency in at least part of their consumption and modification." - how so? It could equally suggest that both hominins and elephants were drawn to the same lush environments.
We agree. Both hominins and megafauna must have been drawn to the same ecological loci for interaction to emerge. However, the fact that the highest density clusters of artifacts coincide with the highest density of carcasses “showing evidence of having been broken”, is suggestive of hominin use and consumption.
(11) Discussion: I suggest starting the Discussion with a concise appraisal of the lines of evidence detailed in the Results and their interpretation, and only then, the critical reassessment of other studies. Similarly, a new topic starts in line 508, but without any subheading or an introductory sentence that could assist the readers.
We added the introductory lines of the former Conclusion section to the revised Discussion section, as suggested by R1.
(12) Line 607: Neumark-Nord are Late Pleistocene sites (MIS 5), not Middle Pleistocene.
Corrected.
(13) Regarding the ambiguity in how megafaunal exploitation may be causally related to the other features of the early Acheulian, the authors can develop the discussion. Alternatively, they should explicitly state that correlation is not causation, and that the present study adds the megafaunal exploitation element to be considered in future discussion of the shifts in lifestyles 1.8 million years ago.
We have done so.
Reviewer #2 (Recommendations for the authors):
The following detailed comments are provided to help clarify arguments, ensure accurate representation of cited literature, and strengthen the logical and methodological framing of the paper. Line numbers refer to the version provided for review.
(1) Line 55: Such concurrency (sometimes in conjunction with other variables)
The term "other variables" is very vague. I would suggest expanding on this or taking it out altogether.
(2) Line 146: Megafaunal long bone green breakage (linked to continuous spiral fractures on thick cortical bone) is probably a less ambiguous trace of butchery than "cut marks", since many of the latter could be equifinal and harder to identify, especially in contexts of high abrasion and trampling (Haynes et al., 2021, 2020).
This reasoning is not supported by the evidence or the cited sources. Green-bone spiral fractures only show that a bone broke while it was fresh and do not reveal who or what caused it. Carnivore feeding, trampling, and natural sediment pressure can all create the same patterns, so these fractures are not clearer evidence of butchery than cut marks. Cut marks, when they are preserved and morphologically clear, remain the most reliable indicator of human activity. The Haynes papers actually show the opposite of what is claimed here. They warn that spiral fractures and surface marks can form naturally and that fracture patterns alone cannot be used to infer butchery. This section should be revised to reflect what those studies actually demonstrate.
The reasoning referred to in line 146 is further explained below in the original text as follows:
“Despite the occurrence of green fractures on naturally-broken bones, such as those trampled by elephants (Haynes et al., 2020), those occurring through traumatic fracturing or gnawed by carnivores (Haynes and Hutson, 2020), these fail to reproduce the elongated, extensive, or helicoidal spiral fractures (uninterrupted by stepped sections), accompanied by the overlapping conchoidal scars (both cortical and medullary), the reflected scarring, the inflection points, or the impact hackled break surfaces and flakes typical of dynamic percussive breakage. Evidence of this type of green breakage had not been documented earlier for the Early Pleistocene proboscidean or hippopotamid carcasses, beyond the documentation of flaked bone with the purpose of elaboration of bone tools (Backwell and d’Errico, 2004; Pante et al., 2020; Sano et al., 2020).”
The problem in the way that R2 uses Haynes et al.´s works is that R2 uses features separately. Natural breaks occurring while the bone is green can generate spiral smooth breaks, for example, but it is not the presence of a single feature that invalidates the diagnosis of agency or that is taphonomically relevant, but the concurrence of several of them. The best example of a naturally (pre-mortem) broken bone was published by Haynes et al.
The natural break shows helical fractures, subjugated to linear (angular) fracture outlines. Notice how the crack displays a zig-zag. The break is smooth but most damage occurs on the cortical surface, with flaking adjacent to the break and step micro-fracturing on the edges. The cortical scarring is discontinuous (almost marginal) and very small, almost limited to the very edge of the break. No modification occurs on the medullary surface. No extensive conchoidal fractures are documented, and certainly none inside the medullary surface of the break.
Compare with Figure S8, S10, S17 and S34 (all specimens are shown in their medullary surface):
In these examples, we see clearly modified medullary surfaces with multiple green breaks and large-sized step fractures, accompanied in some examples by hackle marks. Some show large overlapping scars (of substantially bigger size than those documented in the natural break image). Not a single example of naturally-broken bones has been documented displaying these morphologies simultaneously. It is the comprehensive analysis of the co-occurrence of these features and not their marginal and isolated occurrence in naturally-broken bones that make a difference in the attribution of agency. Likewise, no example of naturally-broken bone has been published that could mimic any of the two green-broken bones documented at EAK. In contrast, we do have bones from our on-going experimentation with green elephant carcasses that jointly reproduce these features. See also Figure 6 of the article to find another example without any modern referent in the naturally-broken bones documented.
We should emphasize that R2 is inaccurately portraying what Haynes et al.´s results really document. Contrary to R2´s assertion, trampling does not reproduce any of the examples shown above. Neither do carnivores. It should be stressed that Haynes & Harrod only document similar overlapping scarring on the medullary surface of bones, when using much smaller animals. In all the carnivore damage repertoire that they document for elephants, durophagous spotted hyenas can only inflict furrowing on the ends of the biggest long bones, especially if they are adults. Long bone midshafts remain inaccessible to them. The mid-shaft portions of bones that we document in our Supplementary File and at EAK cannot be the result of hyena (or carnivore damage) for this reason, and also because their intense gnawing on elephant bones leaves tooth marking on most of the elements that they modify, being absent in our sample.
(3) Line 176: other than hominins accessed them in different taphonomically-defined stages- stages - the "Stages" is repeated twice
Defined in the revised version
(4) Line 174: Regardless of the type of butchery evidence - and with the taphonomic caveat that no unambiguous evidence exists to confirm that megafaunal carcasses were hunted or scavenged other than hominins accessed them in different taphonomically-defined stages- stages - the principal reasons for exploring megafaunal consumption in early human evolution is its origin, its episodic or temporally-patterned occurrence, its impact on hominin adaptation to certain landscapes, and its reflection on hominin group size and site functionality.
This sentence is confusing and needs to be rewritten for clarity. It tries to combine too many ideas at once, and the phrasing makes it hard to tell what the main point is. The taphonomic caveat in the middle interrupts the sentence and obscures the argument. It should be broken into separate, clearer statements that distinguish what evidence exists, what remains uncertain, and what the broader goals of the discussion are.
We believe the ideas are displayed clearly
(5) Line 179: landscapes, and its reflection on hominin group size and site functionality. If hominins actively sought the exploitation of megafauna, especially if targeting early stages of carcass consumption, the recovery of an apparent surplus of resources reflects a substantially different behavior from the small-group/small-site pattern documented at several earlier Oldowan anthropogenic sites (Domínguez-Rodrigo et al., 2019) -or some modern foragers, like the Hadza, who only exploit megafaunal carcasses very sporadically, mostly upon opportunistic encounters (Marlowe, 2010; O'Connell et al., 1992; Wood, 2010; Wood and Marlowe, 2013).
This sentence makes a reasonable point, but is written in a confusing way. The idea that early, deliberate access to megafauna would represent a different behavioral pattern from smaller Oldowan or modern foraging contexts is valid, but the sentence is awkward and hard to follow. It should be rephrased to make the logic clearer and more direct.
We believe the ideas are displayed clearly
(6) Line 186: When the process started of becoming megafaunal commensal started has major implications for human evolution.
This sentence is awkward and needs to be rewritten for clarity. The phrasing "when the process started of becoming megafaunal commensal started" is confusing and grammatically incorrect. It could be revised to something like "Determining when hominins first began to interact regularly with megafauna has major implications for human evolution," or another version that clearly identifies the process being discussed.
Modified in the revised version
(7) Line189: The multiple taphonomic biases intervening in the palimpsestic nature of most of these butchery sites often prevent the detection of the causal traces linking megafaunal carcasses and hominins. Functional links have commonly been assumed through the spatial concurrence of tools and carcass remains; however, this perception may be utterly unjustified as we argued above. Functional association of both archaeological elements can more securely be detected through objective spatial statistical methods. This has been argued to be foundational for heuristic interpretations of proboscidean butchery sites (Giusti, 2021). Such an approach removes ambiguity and solidifies spatial functional association, as demonstrated at sites like Marathousa 1 (Konidaris et al., 2018) or TK Sivatherium (Panera et al., 2019). This method will play a major role in the present study.
This section overstates what spatial analysis can demonstrate and misrepresents the cited studies. The works by Giusti (2021), Konidaris et al. (2018), and Panera et al. (2019) do use spatial statistics to examine relationships between artifacts and faunal remains, but they explicitly caution that spatial overlap alone does not prove functional or behavioral association. These studies argue that clustering can support such interpretations only when combined with detailed taphonomic and stratigraphic evidence. None of them claims that spatial analysis "removes ambiguity" or "solidifies" functional links. The text should be revised to reflect the more qualified conclusions of those papers and to avoid implying that spatial statistics can establish behavioral causation on their own.
We disagree. Both works (Giusti and Panera) use spatial statistical tools to create an inferential basis reinforcing a functional association of lithics and bones. In both cases, the anthropogenic agency inferred is based on that. We should stress that this only provides a basis for argumentation, not a definitive causation. Again, those analyses show much more than just apparent visual clustering.
(8) Line 200: Here, we present the discovery of a new elephant butchery site (Emiliano Aguirre Korongo, EAK), dated to 1.78 Ma, from the base of Bed II at Olduvai Gorge. It is the oldest unambiguous proboscidean butchery site at Olduvai.
It is fine to state the main finding in the introduction, but the phrasing here is too strong. Calling EAK "the oldest unambiguous proboscidean butchery site" asserts certainty before the evidence is presented. The claim should be stated more cautiously, for example, "a new site that provides early evidence for proboscidean butchery," so that the language reflects the strength of the data rather than pre-judging it.
We understand the caution by R2, but in this case, EAK is the oldest taphonomically-supported evidence of elephant butchery at Olduvai (see discussion about FLK North in the text). Whether this is declared at the beginning or the end of the text is irrelevant.
(9) Line 224: The drying that characterizes Bed II had not yet taken place during this moment.
This sentence reads like a literal translation. It should be rewritten for clarity.
Modified in the revised version
(10) Line 233: During the recent Holocene, the EAK site was affected by a small landslide which displaced the...
This section contains far more geological detail than is needed for the argument. The reader only needs to know that the site block was displaced by a small Holocene landslide but retains its stratigraphic integrity. The extended discussion of regional faults, seismicity, and slope processes goes well beyond what is necessary for context and distracts from the main focus of the paper.
We disagree. The geological information is what is most commonly missing from most archaeological reports. Here, it is relevant because of the atypical process and because it has been documented only twice with elephant butchery sites. Explaining the dynamic geological process that shaped the site helps to understand its spatial properties.
(11) Line 264: In June 2022, a partial elephant carcass was found at EAK on a fragmented stratigraphic block...
This section reads like field notes rather than a formal site description. Most of the details about the discovery sequence, trench setup, and excavation process are unnecessary for the main text. Only the basic contextual information about the find location, stratigraphic position, and anatomical composition is needed. The rest could be condensed or moved to the methods or supplementary material.
We disagree. See reply above.
(12) Line 291: hominins or other carnivores. Ongoing restoration work will provide an accurate estimate of well-preserved and modified fractions of the assemblage.
This sentence is unclear and needs to specify what kind of restoration work is being done and what is meant by well-preserved and modified fractions. It is not clear whether modified refers to surface marks, diagenetic alteration, or something else. If the bones are still being cleaned or prepared, the analysis is incomplete, and the counts cannot be considered final. If restoration only means conservation or stabilization, that should be stated clearly so the reader understands that it does not affect the results. As written, it is not clear whether the data presented here are preliminary or complete.
We added: For this reason, until restoration is concluded, we cannot produce any asssertion about the presence or absence of bone surface modifications.
(13) Line 294: The tibiae were well preserved, but the epiphyseal portions of the femora were missing, probably removed by carnivores, which would also explain why a large portion of the rib cage and almost all vertebrae are missing.
This explanation is not well supported. The missing elements could be the result of other forms of density-mediated destruction, such as sediment compaction or post-depositional fragmentation, especially since no tooth marks were found. Given the low density of ribs, vertebrae, and femoral epiphyses, these processes are more likely explanations than carnivore removal. The text should acknowledge these alternatives rather than attributing the pattern to carnivore activity without direct evidence.
Sediment compaction and post-depositional can break bones but cannot make them disappear. Our excavation process was careful enough to detect bone if present. Their absence indicates two possibilities: erosion through the years at the front of the excavation or carnivore intervention. Carnivores can take elephant bones without impacting the remaining assemblage (see our reply above to a similar comment).
(14) Line 304: The fact that the carcass was moved while encased in its sedimentary context, along with the close association of stone tools with the elephant bones, is in agreement with the inference that the animal was butchered by hominins. A more objective way to assess this association is through spatial statistical analysis.
The authors state that "the carcass was moved while encased in its sedimentary context, along with the close association of stone tools with the elephant bones, is in agreement with the inference that the animal was butchered by hominins." This does not logically follow. Movement of the block explains why the bones and tools remain together, not how that association was created. The preserved association alone does not demonstrate butchery, especially in the absence of cut marks or other direct evidence of hominin activity.
Again, we are sorry that R2 is completely overlooking the strong signal detected by the spatial statistical analysis. The way that the block moved, it preserved the original association of bones and tools. This statement is meant to clarify that despite the allochthonous nature of the block, the original autochthonous depositional process of both types of archaeological materials has been preserved. The spatial association, as statistically demonstrated, indicates that the functional link is more likely than any other alternative process. The additional fact that nowhere else in that portion of the outcrop do we identify scatters of tools (all appear clustered at a landscape scale with the elephant) adds more support to this interpretation. This would have been further supported by the presence of cut marks, no doubt, but their absence does not indicate lack of functional association, since as Haynes´ works have clearly shown, most bulk defleshing of modern elephant leaves no traces on most bones.
(15) Line 370: This also shows that the functional connection between the elephant bones and the tools has been maintained despite the block post-sedimentary movement.
The spatial analyses appear to have been carried out appropriately, and the interpretations of clustering and segregation are consistent with the reported results. However, the conclusion that the "functional connection" between bones and tools has been maintained goes beyond what spatial correlation alone can demonstrate. These analyses show spatial proximity and scale-dependent clustering but cannot, by themselves, confirm a behavioral or functional link.
R2 is making this comment repeatedly and we have addressed it more than once above. We disagree and we refer to our replies above to sustain it.
(16) Line 412: The clustering of the elephant (and hippopotamus) carcasses in the areas containing the highest densities of landscape surface artifacts is suggestive of a hominin agency in at least part of their consumption and modification. The presence of green broken elephant long bone elements in the area surveyed is only documented within such clusters, both for lower and upper Bed II. This constitutes inverse negative evidence for natural breaks occurring on those carcasses through natural (i.e., non-hominin) pre- and peri-mortem limb breaking (Haynes et al., 2021, 2020; Haynes and Hutson, 2020). In this latter case, it would be expected for green-broken bones to show a more random landscape distribution, and occur in similar frequencies in areas with intense hominin landscape use (as documented in high density artifact deposition) and those with marginal or non-hominin intervention (mostly devoid of anthropogenic lithic remains).
The clustering of green-bone fractures with stone tools is intriguing but should be interpreted cautiously. The Haynes references are misrepresented here. Those studies address both cut marks and green-bone (spiral) fractures, emphasizing that each can arise through non-hominin processes such as trampling, carcass collapse, and sediment loading. They do not treat green fractures as clearer evidence of butchery; in fact, they caution that such breakage patterns can occur naturally and even form clustered distributions in areas of repeated animal activity. The claim that these studies support spiral fractures as unambiguous indicators of hominin activity, or that natural breaks would be randomly distributed, is not accurate.
We would like to emphasize again that the Haynes´references are not misrepresented here. See our extensive reply above. If R2 can provide evidence of natural breakage patterns resulting from pre-mortem limb breaking or post-mortem trampling resulting in all limb bones being affected by these processes and resulting in smooth spiral breaks, accompanied with extensive and overlapping scarring on the medullary surface, in conjunction with the other features described in our replies above, then we would be willing to reconsider. With the evidence reported until now, that does not occur simultaneously on specimens resulting from studies on modern elephant bones.
R2 seems to contradict him(her)self here by saying that Haynes studies show that cut marks are not reliable because they can also be reproduced via trampling. Until this point, R2 had been saying that only cut marks could demonstrate a functional link and support butchery. Haynes´ studies do not deal experimentally with sediment loading.
(17) Line 424: This indicates that from lower Bed II (1.78 Ma) onwards, there is ample documented evidence of anthropogenic agency in the modification of proboscidean bones across the Olduvai paleolandscapes. The discovery of EAK constitutes, in this respect, the oldest evidence thereof at the gorge. The taphonomic evidence of dynamic proboscidean bone breaking across time and space supports, therefore, the inferences made by the spatial statistical analyses of bones and lithics at the site.
This conclusion is overstated. The claim of "ample documented evidence of anthropogenic agency" is too strong, given that the main support comes from indirect indicators like green-bone fractures and spatial clustering rather than clear butchery marks. It would be more accurate to say that the evidence suggests or is consistent with possible hominin involvement. The final sentence also conflates association with causation; spatial and taphonomic data can indicate a relationship, but do not confirm that the carcasses were butchered by hominins.
The evidence is based on spatially clustering (at a landscape scale) of tools and elephant (and other megafaunal taxa) bones, in conjunction with a large amount of green-broken elements. This interpretation, if we compare it against modern referents is supported even stronger. In the past few years, we have been conducting work on modern naturally dead elephant carcasses in Botswana and Zambia, and of the several carcasses that we have seen, we have not identified a single case of long bone shaft breaks like those described by Haynes as natural or like those we describe here as anthropogenic. This probably means that they are highly unlikely or marginal occurrences at a landscape scale. This seems to be supported by Haynes´ work too. Out of the hundreds of elephant carcasses that he has monitored and studied over the years for different works, we have managed to identify only two instances where he described natural pre-mortem breaks. This certainly qualifies as extremely marginal.
Most of the Results section is clearly descriptive, but beginning with "The clustering of the elephant (and hippopotamus) carcasses..." the text shifts from reporting observations to drawing behavioral conclusions. From this point on, it interprets the data as evidence of hominin activity rather than simply describing the patterns. This part would be more appropriate for the Discussion, or should be rewritten in a neutral, descriptive way if it is meant to stay in the Results.
This appears extensively discussed in the Discussion section, but the data presented in the results is also interpreted in that section, following a clear argumental chain.
(18) Line 433: A recent discovery of a couple of hippopotamus partial carcasses at the 3.0-2.6 Ma site of Nyayanga (Kenya), spatially concurrent with stone artifacts, has been argued to be causally linked by the presence of cut marks on some bones (Plummer et al., 2023). The only evidence published thereof is a series of bone surface modifications on a hippo rib and a tibial crest, which we suggest may be the result of byproduct of abiotic abrasive processes; the marks contrast noticeably with the well-defined cut marks found on smaller mammal bones (Plummer et al. ́s 2023: Figure 3C, D) associated with the hippo remains (Plummer et al., 2023).
The authors suggest that the Nyayanga marks could result from abiotic abrasion, but this claim does not engage with the detailed evidence presented by Plummer et al. (2023). Plummer and colleagues documented well-defined, morphologically consistent cut marks and considered the sedimentary context in their interpretation. Raising abrasion as a general possibility without addressing that analysis gives the impression of selective skepticism rather than an evaluation grounded in the published data.
We disagree again on this matter. R2 does not clarify what he/she means by well-defined or morphologically consistent. We provide an alternative interpretation of those marks that fit their morphology and features and that Plummer at al did not successfully exclude. We also emphasize that the interpretation of the Nyayanga marks was made descriptively, without any analytical approach and with a high degree of subjectivity by the researcher. All of this disqualifies the approach as well defined and keeps casting an old look at modern taphonomy. Descriptive taphonomy is a thing of the 1980´s. Today there are a plethora of analytical methods, from multivariate statistics, to geometric morphometrics to AI computer vision (so far the most reliable) which represent how taphonomy (and more specifically, analysis of bone surface modifications) should be conducted in the XXI century. This approaches would reinforce interpretations as preliminarily published by Plummer et al, provided they reject alternative explanations like those that we have provided.
(19) Line 459: It would have been essential to document that the FLK N6 tools associated with the elephant were either on the same depositional surface as the elephant bones and/or on the same vertical position. The ambiguity about the FLK N6 elephant renders EAK the oldest secure proboscidean butchery evidence at Olduvai, and also probably one of the oldest in the early Pleistocene elsewhere in Africa.
The concern about vertical mixing is fair, but the tone makes it sound like the association is definitely not real. It would be more accurate to say that the evidence is ambiguous, not that it should be dismissed altogether.
We have precisely done so. We do not dismiss it, but we cannot take it for anything solid since we excavated the site and show how easily one could make functional associations if forgetting about the third dimension. It is not a secure butchery site. This is what we said and we stick to this statement.
(20) Line 479: In all cases, these wet environments must have been preferred places for water-dependent megafauna, like elephants and hippos, and their overlapping ecological niches are reflected in the spatial co-occurrence of their carcasses. Both types of megafauna show traces of hominin use through either cutmarked or percussed bones, green-broken bones, or both (Supplementary Information).
The environmental part is good, but the behavioral interpretation is too strong. Saying elephants and hippos "must have been" drawn to these areas is too certain, and claiming that both "show traces of hominin use" makes it sound like every carcass was modified. It should be clearer that only some have possible evidence of this.
The sentence only refers to both types of fauna taxonomically. No inference can be drawn therefor that all carcasses are modified.
(21) Line 496: In most green-broken limb bones, we document the presence of a medullary cavity, despite the continuous presence of trabecular bone tissue on its walls.
This sentence is confusing and doesn't seem to add anything meaningful. All limb bones naturally have a medullary cavity lined with trabecular bone, so it's unclear why this is noted as significant. The authors should clarify what they mean here or remove it if it's simply describing normal bone structure.
No. Modern elephant long bones do not have a hollow medullary cavity. All the medullary volume is composed of trabecular tissue. Some elephants in the past had hollow medullary cavities, which probably contained larger amounts of marrow and fat.
(22) Line 518: We are not confident that the artefacts reported by de la Torre et al are indeed tools.
While I generally agree with this statement, the paragraph reads as defensive rather than comparative. It would help if they briefly summarized what de la Torre et al. actually argued before explaining why they disagree.
We devote two full pages of the Discussion section to do so precisely.
(23) Lines 518-574: They are similar to the green-broken specimens that we have reported here...
This part is very detailed but inconsistent. They argue that the T69 marks could come from natural processes, but they use similar evidence (green fractures, overlapping scars) to argue for human activity at EAK. If equifinality applies to one, it applies to both.
We are confused by this misinterpretation. Features like green fractures and overlapping scars (among others) can be used to detect anthropogenic agency in elephant bone breaking; that is, any given specimen can be determined to have been an “artifact” (in the sense of human-created item), but going from there to interpreting an artifact as a tool, there is a large distance. Whereas an artifact (something made by a human) can be created indirectly through several processes (for example, demarrowing a bone resulting in long bone fragments), a tool suggest either intentional manufacture and use or both. That is the difference between de la Torre et al.´s interpretation and ours. We believe that they are showing anthropogenically-made items, but they have provided no proof that they were tools.
(24) Line 576: A final argument used by the authors to justify the intentional artifactual nature of their bone implements is that the bone tools were found in situ within a single stratigraphic horizon securely dated to 1.5 million years ago, indicating systematic production rather than episodic use. This is taphonomically unjustified.
The reasoning here feels uneven in how clustering evidence is used. At EAK, clustering of bones and artifacts is taken as meaningful evidence of hominin activity, but here the same pattern at T69 is treated as a natural by-product of butchery or carnivore activity. If clustering alone cannot distinguish between intentional and incidental association, the authors should clarify why it is interpreted as diagnostic in one case but not in the other.
Again, we are confused by this misinterpretation. It applies to two different scenarios/questions:
a) is there a functional link between tools and bones at EAK and T69? We have statistically demonstrated that at EAK and we think de la Torre et al. is trying to do the same for T69, although using a different method.
b) Are the purported tools at T69 tools? Are those that we report here tools? In this regard there is no evidence for either case and given that several bones from T69 come from animals smaller than elephants, we do not discard that carnivores might have been responsible for those, whereas hominin butchery might have been responsible for the intense long limb breaking at that site. It remains to be seen how many (if any) of those specimens were tools.
(25) Line 600: If such a bone implement was a tool, it would be the oldest bone tool documented to date (>1.7 Ma).
The comparison to prior studies is useful, and the point about missing use-wear traces is well taken. However, the last lines feel speculative. If no clear use evidence has been found, it's premature to suggest that one specimen "would be the oldest bone tool." That claim should be either removed or clearly stated as hypothetical.
It clearly reads as hypothetical.
(26) Line 606: Evidence documents that the oldest systematic anthropogenic exploitation of proboscidean carcasses are documented (at several paleolandscape scales) in the Middle Pleistocene sites of Neumark-Nord (Germany)(Gaudzinski-Windheuser et al., 2023a, 2023b).
This is the first and only mention of Neumark-Nord in the paper, and it appears without any prior discussion or connection to the rest of the study. If this site is being used for comparison or as part of a broader temporal framework, it needs to be introduced and contextualized earlier. As written, it feels out of place and disconnected from the rest of the argument.
This is a Late Pleistocene site and we do not see the need to present it earlier, given that the scope of this work is Early Pleistocene.
(27) Line 608: Evidence of at least episodic access to proboscidean remains goes back in time (see review in Agam and Barkai, 2018; Ben-Dor et al., 2011; Haynes, 2022).
The distinction between "systematic" and "episodic" exploitation is useful, but the authors should clarify what criteria define each. The phrase "episodic access...goes back in time" is vague and could be replaced with a clearer statement summarizing the nature of the earlier evidence.
It is self-explanatory
(28) Line 610: Redundant megafaunal exploitation is well documented at some early Pleistocene sites from Olduvai Gorge (Domínguez-Rodrigo et al., 2014a, 2014b; Organista et al., 2019, 2017, 2016).
The phrase "redundant megafaunal exploitation" needs clarification. "Redundant" is not standard terminology in this context. Does this mean repeated, consistent, or overlapping behaviors? Also, while these same Olduvai sites are mentioned earlier, this phrasing also introduces new interpretive language not used before and implies a broader behavioral generalization than what the data actually show.
Webster: Redundant means repetitive, occurring multiple times.
(29) Line 612: At the very same sites, the stone artifactual assemblages, as well as the site dimensions, are substantially larger than those documented in the Bed I Oldowan sites (Diez-Martín et al., 2024, 2017, 2014, 2009).
The placement and logic of this comparison are unclear. The discussion moves from Middle Pleistocene Neumark-Nord to early Pleistocene Olduvai sites, then to Bed I Oldowan contexts without clearly signaling the temporal or geographic transitions. If the intent is to contrast Acheulean vs. Oldowan site scale or organization, that connection needs to be made explicit. As written, it reads as a disjointed shift rather than a continuation of the argument.
We disagree. Here, we finalize by bringing in some more recent assemblages where hominin agency is not in question.
(30) Line 616: Here, we have reported a significant change in hominin foraging behaviors during Bed I and Bed II times, roughly coinciding with the replacement of Oldowan industries by Acheulian tool kits -although during Bed II, both industries co-existed for a substantial amount of time (Domínguez-Rodrigo et al., 2023; Uribelarrea et al., 2019, 2017).
This section should be restructured for flow. The reference to behavioral change during Bed I-II and the overlap of Oldowan and Acheulean industries is important, but feels buried after a long detour. Consider moving this earlier or rephrasing so the main conclusion (behavioral change across Beds I-II) is clearly stated first, followed by supporting examples.
It is not within the scope of this work and is properly described in the references mentioned.
(31) Line 620: The evidence presented here, together with that documented by de la Torre et al. (2025), represents the most geographically extensive documentation of repeated access to proboscidean and other megafaunal remains at a single fossil locality.
The phrase "most geographically extensive documentation of repeated access" overstates what has been demonstrated. The evidence presented is site-specific and does not justify such a broad superlative. This should be toned down or supported with comparative quantitative data.
We disagree. There is no other example where such an abundant record of green-broken elements from megafauna is documented. Neumark-Nord is more similar because it shows extensive evidence of butchery, but not so much about degreasing.
(32) Line 623: The transition from Oldowan sites, where lithic and archaeofaunal assemblages are typically concentrated within 30-40 m2 clusters, to Acheulean sites that span hundreds or even over 1000 m2 (as in BK), with distinct internal spatial organization and redundancy in space use across multiple archaeological layers spanning meters of stratigraphic sequence (Domínguez-Rodrigo et al., 2014a, 2009b; Organista et al., 2017), reflects significant behavioral and technological shifts.
This sentence about site size and spatial organization repeats earlier claims without adding new insight. If it's meant as a synthesis, it should explicitly say how the spatial expansion relates to changes in behavior or mobility, not just describe the difference.
In the Conclusion section these correlations have been explained in more detail to add some causation.
(33) Line 628: This pattern likely signifies critical innovations in human evolution, coinciding with major anatomical and physiological transformations in early hominins (Dembitzer et al., 2022; Domínguez-Rodrigo et al., 2021, 2012).
The conclusion that this "signifies critical innovations in human evolution" is too sweeping, given the data presented. It introduces physiological and anatomical transformation without connecting it to any evidence in this paper. Either cite the relevant findings or limit the claim to behavioral implications.
The references cited elaboration in extension this. The revised version of the Conclusion section also elaborates on this.
Overall, the conclusions section reads as a loosely connected set of assertions rather than a focused synthesis. It introduces new interpretations and terminology not supported or developed earlier in the paper, and the argument jumps across temporal and geographic scales without clear transitions. The discussion should be restructured to summarize key results, clarify the scope of interpretation, and avoid speculative or overstated claims about evolutionary significance.
We have done so, supported by the references used in addition to extending some of the arguments
(34) Line 639: The systematic excavation of the stratigraphic layers involved a small crew.
This sentence is not necessary.
No comment
(35) Line 643: The orientation and inclination of the artifacts were recorded using a compass and an inclinometer, respectively.
What were these measurements used for (e.g., post-depositional movement analysis, spatial patterning)? A short note on the purpose would make this more meaningful.
Fabric analysis has been added to the revised version.
(36) Line 659: Restoration of the EAK elephant bones
This section could be streamlined and clarified. It includes procedural detail that doesn't contribute to scientific replicability (e.g., the texture of gauze, number of consolidant applications), while omitting some key information (such as how restoration may have affected analytical results). It also contains interpretive comments ("most of the assemblage has been successfully studied") that don't belong in Methods.
No comment
(37) Line 689: In the field laboratory, cleaning of the bone remains was carried out, along with adhesion of fragments and their consolidation when necessary.
Clarify whether cleaning or adhesion treatments might obscure or alter bone surface modifications, as this has analytical implications.
These protocols do not impact bone like that anymore.
(38) Line 711: (b) Percussion Tools - Includes hammerstones or cobbles exhibiting diagnostic battering, pitting, and/or impact scars consistent with percussive activities.
Define how diagnostic features (battering, pitting) were identified - visual inspection, magnification, or quantitative criteria?
Both macro and microscopically
(39) Line 734: We conducted the analysis in three different ways after selecting the spatial window, i.e., the analysed excavated area (52.56 m2).
Clarify why the 52.56 m<sup>2</sup> spatial window was chosen. Was this the total excavated area or a selected portion?
It was what was left of the elephant accumulation after erosion.
(40) Line 728: The spatial statistical analyses of EAK.
Adding one or two sentences at the start explaining the analytical objective, such as testing spatial association between faunal and lithic materials, would help readers understand how each analysis relates to the broader research questions.
This is well explained in the main text
(41) Line 782: An intensive survey seeking stratigraphically-associated megafaunal bones was carried out in the months of June 2023 and 2024.
It would help to specify whether the same areas were resurveyed in both field seasons or if different zones were covered each year. This information is important for understanding sampling consistency and potential spatial bias.
Both areas were surveyed in both field seasons. We were very consistent.
(42) Line 787: We focused on proboscidean bones and used hippopotamus bones, some of the most abundant in the megafaunal fossils, as a spatial control.
Clarify how the hippopotamus remains functional as a "spatial control." Are they used as a proxy for water-associated taxa to test habitat patterning, or as a baseline for comparing carcass distribution? The meaning of "control" in this context is ambiguous.
As a proxy for megafaunal distribution given their greater abundance over any other megafaunal taxa.
(43) Line 789: Stratigraphic association was carried out by direct observation of the geological context and with the presence of a Quaternary geologist during the whole survey.
This is good methodological practice, but it would be helpful to describe how stratigraphic boundaries were identified in the field (for example, by reference to tuffs or marker beds). That information would make the geological framework more replicable.
This is basic geological work. Of course, both tuffs and marker beds were followed.
(44) Line 791: When fossils found were ambiguously associated with specific strata, these were excluded from the present analysis.
You might specify what proportion of the total finds were excluded due to uncertain stratigraphic association. Reporting this would indicate the strength of the stratigraphic control.
This was not quantified but it was a very small amount compared to those whose stratigraphic provenience was certain.
(45) Line 799: The goals of this survey were: a) collect a spatial sample of proboscidean and megafaunal bones enabling us to understand if carcasses on the Olduvai paleolandscapes were randomly deposited or associated to specific habitats.
You might clarify how randomness or habitat association was tested.
Randomness was tested spatially and comparing density according to ecotone. Same for habitat association.
(46) The Methods section provides detailed information about excavation, restoration, and spatial analyses but omits critical details about the zooarchaeological and taphonomic procedures. There is no explanation of how faunal remains were analyzed once recovered, including how cut marks, percussion marks, or green bone fractures were identified or what magnification or diagnostic criteria were used. The authors also do not specify the analytical unit used for faunal quantification (e.g., NISP, MNI, MNE, or other), making it unclear how specimen counts were generated for spatial or taphonomic analyses. Even if these details are provided in the Supplementary Information, the main text should include at least a concise summary describing the analytical framework, the criteria for identifying surface modifications and fracture morphology, and the quantification system employed. This information is essential for transparency, replicability, and proper evaluation of the behavioral interpretations.
See reply above. There is a new subsection on taphonomic methods now.
Supplementary information:
(47) The Supplementary Information includes a large number of green-broken proboscidean specimens from other Olduvai localities (BK, LAS, SC, FLK West), but it is never explained why these are shown or how they relate to the EAK study. The main analysis focuses entirely on the EAK elephant, including so much unrelated material without any stated purpose, which makes the supplement confusing. If these examples are meant only to illustrate the appearance of green fractures, that should be stated. Otherwise, the extensive inclusion of non-EAK material gives the impression that they were part of the analyzed assemblage when they were not.
This is stated in the opening paragraph to the section.
(48) Line 96: A small collection of green-broken elephant bones was retrieved from the lower and upper Bed II units.
It would help to clarify whether these specimens are part of the EAK assemblage or derive from other Bed II localities. As written, it is not clear whether this description refers to material analyzed in the main text or to comparative examples shown only in the Supplementary Information.
No, EAK only occupies the lower Bed II section. They belong in the Bed II paleolandscape units.
(49) Line 97: One of them, a proximal femoral shaft found within the LAS unit, has all the traces of having been used as a tool (Figure 6).
This says the bone tool in Figure 6 is from LAS, but the main text caption identifies it as from EAK. If I am not mistaken, EAK is a site at the base of Bed II, and LAS is a separate stratigraphic unit higher in the sequence, so the authors should clarify which is correct.
Our mistake. It provenience is from LAS in the vicinity of EAK.
(50) Line 186: Figure S20. Example of other megafaunal long bone shafts showing green breaks.
Not cited in text or SI narrative. No indication where these bones come from or why they are relevant.
It appears justified in the revised version.
(51) Line 474: Figure S28-S30. Hyena-ravaged giraffe bones from Chobe (Botswana).
These figures are not discussed in the text or SI, and their relevance to the study is unclear. The authors should explain why these modern comparative examples were included and how they inform interpretations of the Olduvai assemblages.
It appears justified in the revised version.
(52) Line 498: Figure S31. Bos/Bison bone from Bois Roche (France).
This figure is not mentioned in the text or Supplementary Information. The authors should specify why this specimen is shown and how it contributes to the study's taphonomic or behavioral comparisons.
It appears justified in the revised version.
(53) Line 504: Figure S32. Miocene Gomphotherium femur from Spain.
This figure is never referenced in the paper. The authors should clarify the purpose of including a Miocene specimen from outside Africa and explain what it adds to the interpretation of Bed II material.
It appears justified in the revised version.
(54) Line 508: Figure S33. Elephant femoral shaft from BK (Olduvai).
This figure appears to show comparative material but is not cited or discussed in the text. The authors should explain why the BK material is presented here and how it relates to EAK or the broader analysis.
There are two figures labeled S33.
It appears justified in the revised version.
(55) Line 515: Figure S33. Tibia fragment from a large medium-sized bovid displaying multiple overlapping scars on both breakage planes inflicted by carnivore damage.
Because this figure repeats the S33 label and is not cited or explained in the text, it is unclear why this specimen is included or how it contributes to the study. The authors should correct the duplicate numbering and clarify the purpose of this figure.
It appears justified in the revised version.
(56) Line 522: Same specimen as shown in Figure S30, viewed on its medial side.
This is not the same bone as S30. This figure is not discussed in the text or Supplementary Information. The authors should clarify why it is included and how it relates to the rest of the analysis.
It appears justified in the revised version.