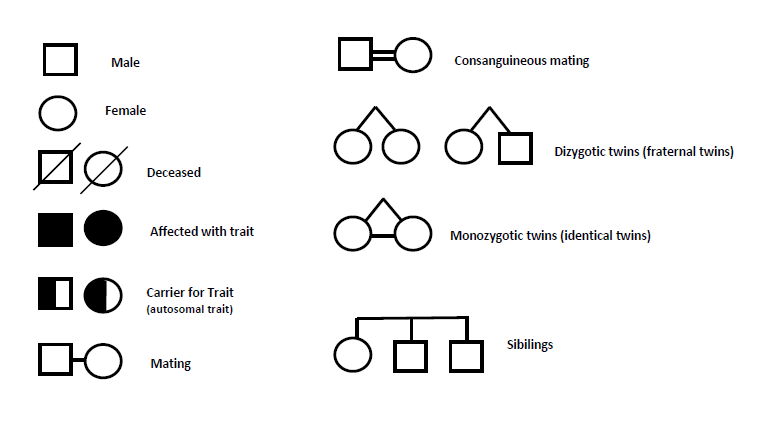
The Homozygosity Mapping Collaborative for Autism (HMCA) (21) has recruited 104 families (79 simplex and 25 multiplex) from the Arabic Middle East, Turkey, and Pakistan (table S1 and fig. S1), of which 88 pedigrees (69 simplex and 19 multiplex) have cousin marriages (i.e., parental consanguinity)
The authors selected families with one or more individuals affected by autism.
Among those families, about half have cousin marriages, which are referred to as consanguineous pedigrees.
A simplex family is one in which there is only one person with autism. A multiplex family has multiple people with autism.
 It has been shown that CNVs at specific locations increase the risk to develop autism.
It has been shown that CNVs at specific locations increase the risk to develop autism.