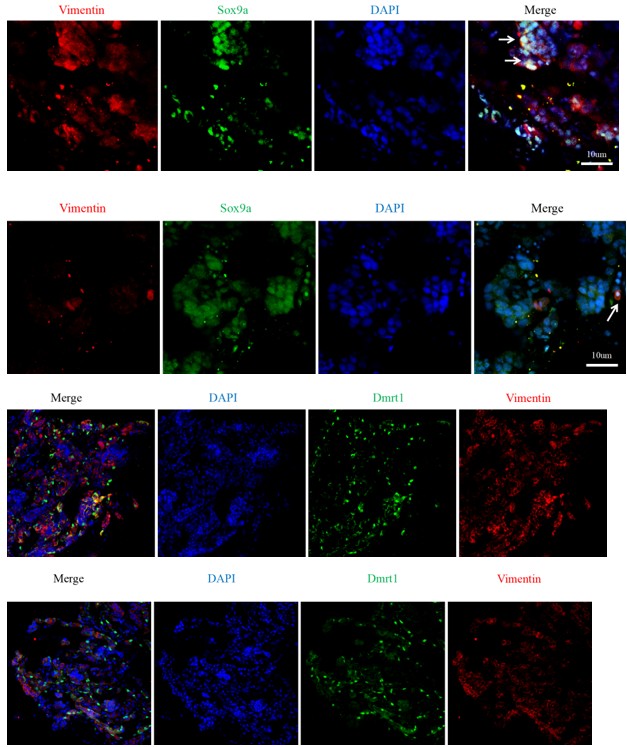
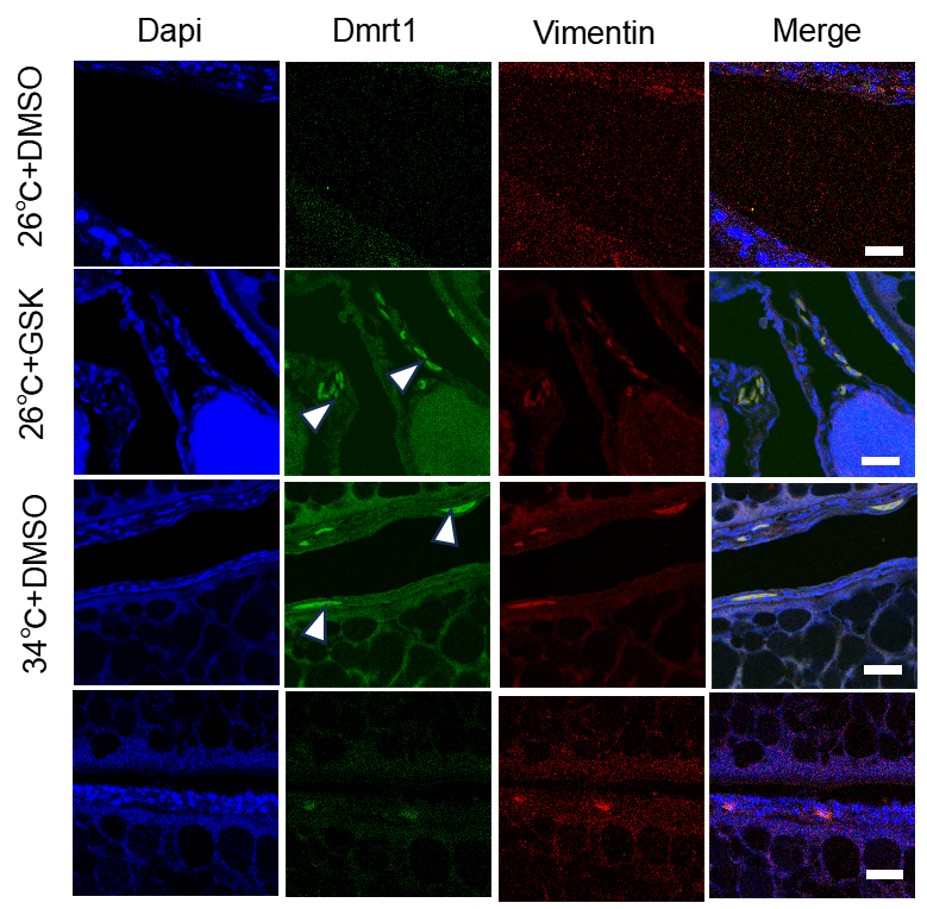
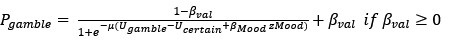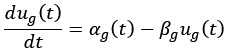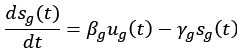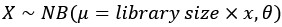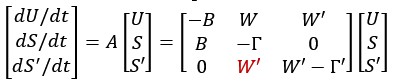Reviewer #1 (Evidence, reproducibility and clarity (Required)):
This study from the Niedergang lab establishes SNAT7 as a host-dependency factor in human macrophages that supports HIV-1 replication. They show a modest increase in SNAT7 levels HIV-1 infected macrophages and suggest that SNAT7 levels are transiently increased. Employing siRNA against SNAT7 they show reduction in HIV-1 protein levels and viral RNAs and claim that there is a block of reverse transcription in SNAT7 KD cells. Focusing on a known HIV-1 restriction factor in macrophages, SAMHD1, they interconnect the SNAT7 depletion with a reduction in phosphorylated, i.e. catalytical inactive SAMHD1 arguing that SNAT7 regulates the phosphorylation and thereby antiviral activity of SAMHD1. Since SNAT7 is a glutamine transporter that provides this AA from lysosomes, they lastly supplement glutamine and this somehow rescues the reduction of HIV-1 production in SNAT7 KD cells.
Major comments:
The strength of this manuscript is the clear focus on primary human macrophages that are HIV-1 infected and the interconnection of HIV-1 replication to the SNAT7 siRNA KD experiments in combination with SAMHD1 depletion and lastly glutamine supplementation. This establishes a stringent and coherent story line.
The effects reported are modest; high variability is not a problem since using primary hMDM this is expected and can be addressed by testing several donors and applying stringent statistics.
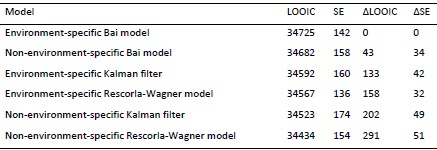
- Having said so, I realize that while they give information on the statistical test used, i.e. one-way ANOVA they miss to explain the post-test used to assess significance (i.e. Bonferroni, Fishers LSD, whatsoever). Please add this information.
We thank the reviewer for this comment. The figure legends have been updated to include more details of all the statistical tests used.
- Another issue that might underestimate the effects of HIV-1 infection on SNAT7 levels and vice versa of SNAT7 KD on HIV-1 replication is the non-single cell approach employed, i.e. WBlots. I assume that HIV-1 infection rates in macrophages are not super high, usually not exceeding 20-30%. So indeed the effects the authors observe could be much higher, when checking at the single cell level. I do not know about the SNAT7 ab, but all the other reagents should work via flow cytometry and could hence improve the readout a lot.
We agree with the reviewer and indeed, in previous studies on HIV-1 infection of human macrophages performed in the lab, we observed via immunofluorescence that the proportion of infected cells ranged from 20 to 40 %. At the time of submission, we did not have the possibility to label the native SNAT7 protein by immunofluorescence, as the commercial antibody used only works for western blotting.
In the meantime, we have been validating a new antibody (Proteintech) targeting SNAT7 for immunofluorescence. If this is confirmed, we will be able to detect and quantify HIV-1 p24 by immunofluorescence in SNAT7-depleted human macrophages and control cells, thus confirming our results in single-cell analysis.
Flow cytometry analyses are difficult to perform on primary human macrophages because these cells are highly adherent and must be detached first. The process induces significant cell death and damage. This is why we would prefer to carry out these analyses using immunofluorescence and microscopy on adhered cells. This option will be undoubtedly pursued.
- Furthermore the authors never commented about a dose-response effect in terms of HIV-1 infection levels. There is a MOI dependency described for Suppl.Fig.1 C-F, unfortunately the data is missing in the manuscript.
We apologize for this omission. The figures showing the increase in SNAT7 protein expression following HIV-1 infection at MOIs ranging from 0.05 to 0.5 were added to the new version of the manuscript (Supp. Fig. 1 C-F).
- Figure1: specify circulating T lymphocytes. I would expect to see levels of SNAT7 in PHA or CD3/CD28 activated lymphocytes versus resting T cells and a time course of SNAT7 levels upon activation. I think even though SNAT7 levels in T cells might be low, they could also be increased by HIV-1 infection and it is essential that the authors test for this. If not, the result is a valid negative control. For this they should employ HIV-1 primary strains with a tropism for T cells, or at least lab-adapted HIV-1 NL4-3
We thank the reviewer for this comment. Circulating T lymphocytes isolated from the blood of healthy donors are now referred to resting lymphocytes in the new version of the manuscript, as opposed to activated T lymphocytes stimulated with IL2 and PHA-P for several days (Fig. 1 A-C).
The expression levels of SNAT7, both at the gene and protein levels, are lower in resting or IL2/PHA-P-activated T cells than in macrophages from the same donors. As suggested, we will perform a kinetic of T-cell activation upon HIV-1 infection to investigate how SNAT7 expression varies in these conditions.
- Figure 2 again single cell measurements could reveal much more pronounced effects; it is a bit counterintuitive that siRNA #2 is more efficient in SNAT7 KD but has higher levels of HIV-1 replication in terms of Gag levels. I assume when looking at the stats it is always a comparison to the Ctl treated cells (C-G), but this is not entirely clear. Unify labeling as compared to the stats in Fig.2 I (this also applies for all the other figs).
We thank the reviewer for this comment. Fig. 2B indeed shows one of the different donors analyzed. However, protein quantification across six different donors shows that SNAT7 is more depleted with siRNA #2 (Fig. 2C), and that Gag Pr55 protein levels are consequently more reduced, than with siRNA #1 (Fig. 2D).
We use GraphPad Prism software to perform statistical analysis. Depending on the test used, the software automatically plots the comparison bar and displays the p-value above it. We changed the representation of statistics as suggested.
Figure 3: It is a bit odd that they finally conclude on RT as essential step that is reduced in the absence of SNAT7 and then they fail to provide statistical significance for this (Fig.3 panels F and G). One would expect that RT is much more affected given the huge effects on HIV-1 capsid and particle production shown in Fig.2 F, G and I.
The reviewer is right in pointing that we observed a stronger effect during the later stages of the viral cycle, from transcription of viral RNAs (Fig. 2I and Supp. Fig. 2G) to the production of viral particles in the supernatant (Fig. 2D-G), than during the earlier stage of reverse transcription (Fig. 3F, G). Also, it is also possible that we might have missed the peak in ERT/LRT production, which is transient.
It should be noted that SAMHD1 exhibits both dNTPase (Goldstone et al., 2011) and nuclease (Beloglazova et al., 2013) activities. The ability of SAMHD1 to restrict the virus, through dephosphorylation at T592, is mediated by its RNase activity (Ryoo et al., 2014), and not by the dNTPase activity (Welbourn et al., 2013; White et al., 2013).This could explain why SNAT7 exhibit a stronger impact on viral transcription than on reverse transcription.
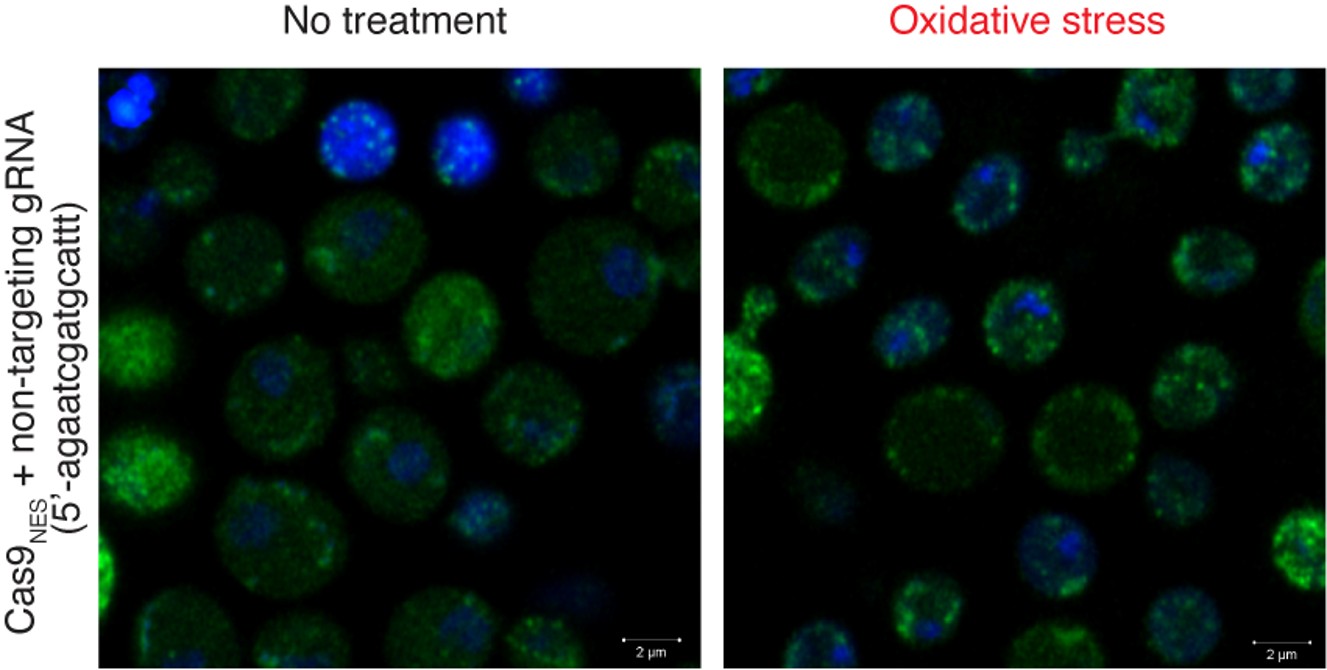
Figure 4; again single cell flow measurements of SAMHD1, pSAMHD1 and p24 /SNAT7 might help to more clearly discriminate effects that are specifically induced upon infection or happen in virally infected cells. Maybe alternatively IF?
We thank the reviewer for this suggestion. As mentioned under comment #2, flow cytometry analyses are difficult to perform on strongly adherent primary human macrophages.
With regard to immunofluorescence, there is a technical limitation based on the species in which the antibodies are produced. The antibody that targets the native SNAT7 protein, which is currently being validated in our laboratory, is produced in rabbits. An anti-CAp24 antibody produced in goats can be used. It will then be necessary to co-label the cells with anti SAMHD1 and phospho-SAMHD1produced in mouse. We will try to find options to co-label the cells.
The wblot shown in panel D does not really reflect the point the authors want to make by the quantification in panels G-I. Primary data (D) suggests that SNAT7 KD reduces HIV-1 production even in the absence of SAMHD1. The quantification rather indicates that SNAT7 KD does not affect HIV-1 production in the absence of SAMHD1. This needs clarification/corroboration by orthogonal approaches.
We respectfully disagree with the reviewer.
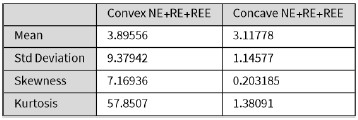
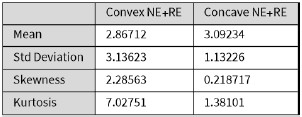
Figure 4D shows a representative blot of the six donors analysed. As mentioned, the depletion of SNAT7 in the absence of SAMHD1 reduces the production of the viral proteins GagPr55 and CAp24 (see Fig. 4D). This is illustrated by the quantifications (Fig. 4G–I). Following treatment with Vpx, GagPr55 protein expression in SNAT7 KD macrophages is reduced by a factor of 2.6 for siRNA #1 (mean = 1.48, light grey bar) and by a factor of 1.83 for siRNA #2 (mean = 2.13, orange bar), compared to the control (mean = 3.9, pink bar) (Fig. 4G). Similarly, CAp24 protein expression was reduced by a factor of 2.2 for siRNA #1 (mean = 2.05, light grey bar) and by a factor of 1.36 for siRNA #2 (mean = 3.34, orange bar), compared to the control (mean = 4.52, pink bar) (Fig. 4H).
These differences are therefore consistent between the Western blot and the quantifications. However, they are not significantly different to those observed in cells treated with Vpx and depleted with control siRNA, suggesting that the viral restriction observed in SNAT7 KD cells is primarily due to SAMHD1.
Figure 5: show SAMHD1 and pSAMHD1 levels upon glutamine supplementation.
We thank the reviewer for this comment, we will perform the suggested experiment.
- I think the discussion is very thin, mainly summarizing the results; but fails to give broader context or critically discuss the limitations and further directions.
We thank the reviewer for this comment. The discussion will be modified further accordingly.
Looking at the data as a whole, I think the results support a modest functional importance of SNAT7 for HIV-1 production in macrophages. I acknowledge that the experiments in primary macrophages are prone to high variability in different donors and the authors transparently depicted their data. However clearly, I would advice the authors to tune down the extend in which they claim SNAT7-dependency given this huge variability and the sometimes-borderline statistics.
We respectfully disagree with the reviewer.
The cells used here imply greater variability than a cell line, but are also more relevant.
Indeed, the effects observed in the late stages of HIV-1 production are:
-
~80 % decrease in viral transcription compared to the control (Fig. 2I),
-
~85 % decrease in CAp24 protein expression compared to the control, as quantified by western blot (Fig. 2E), or ~90 % by ELISA measurement (Fig. 2F),
-
a reduction of more than 90 % in the release of infectious particles (Fig. 2G).
These results were all significant across donors, while SNAT7 depletion was always partial (Fig. 2C, between 31 to 62 % of depletion compared to the control in infected cells).
Therefore, the data were obtained from a mixture of depleted and non-depleted macrophages. This means that the results may be underestimated.
Together, our results show that SNAT7 is necessary for HIV-1 production.
However, reading the comments, we realized that our conclusions regarding reverse transcription were too strong. SNAT7 depletion does not affect viral fusion and reverse transcription. The manuscript was modified accordingly.
On top, there are a lot of optional experiments I am sure the authors are aware of that should be done at least in the future.
For instance, how does HIV-1 upregulate SNAT7, is a viral accessory protein involved?
What is the mechanism of SNAT7 dependent SAMHD1 phosphorylation? Does SNAT7 (or glutamine) regulate the activity of the SAMHD1 associated kinase / phosphatase) If so, does this impact on other targets of these enzymes?
We thank the reviewer for these questions.
To address the role of accessory viral proteins, we have already performed one experiment infecting hMDM with HIV-1 strains deleted for genes such as Nef, Vpr, Vpu and Vif, and have found no clear effect on SNAT7 protein expression compared to WT strains. As an alternative experiment, we could overexpress individual viral genes, such as Nef or Vpr, in HeLa cells and analyze their impact on SNAT7 expression by Western blot.
It is also possible that SNAT7 expression and recycling of lysosomal glutamine are modulated by the macrophage intrinsic immunity in response to HIV-1 infection.
The Thr592 motif of the SAMHD1 protein is phosphorylated by Cyclin A2/CDK1 and type 1 IFN in non-cycling cells, such as MDMs (Cribier et al., 2013). For now, the relationship between SNAT7 and SAMHD1 remains unclear. However, (Meng et al., 2022) demonstrated that SNAT7 positively regulates mTORC1 activity at the lysosomal membrane through release of lysosomal glutamine, and (Dias et al., 2024) showed that inhibiting mTORC1 activity decreases SAMHD1 Thr592 phosphorylation in hMDM. Therefore, we could speculate that the absence of SNAT7 down-regulates mTORC1 activity, which then leads to decreased SAMHD1 phosphorylation. This has been added to the discussion to explain the relationship between the 3 partners.
**Referees cross-commenting**
I think the comments from the other referees are reasonable and consistent with my assessment
Reviewer #1 (Significance (Required)):
Strength and limitations see above;
Significance: I think this work is of high interest for virologists working in the field of HIV-1 and infection of myeloid cells. In case SNAT7 (and hence glutamine) indeed regulates the phosphorylation of SAMHD1, there could potentially be broad relevance of this work. However unfortunately, this aspect remains underdeveloped and is also not discussed
Field of expertise: HIV-1, immunology, cell biology
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
In this report, Herit and colleagues describe the role of a HIV-1 dependency factor that promotes virus replication in macrophages. The authors suggest that the lysosomal membrane-associated SNAT7 glutamine transporter is a HIV dependency factor, that promotes virus replication by enhancing reverse transcription and Gag synthesis. The authors use transient knock-down approaches in primary macrophages to identify that SNAT7 depletion does not impact viral entry but inhibits early reverse transcription which was reversed by exogenous glutamine addition. While reverse transcription enhancement was likely due to selective increase in phosho-SAMHD1 expression, mechanisms by which SNAT7 enhanced viral gene expression were not clearly defined. These are well-controlled studies that pinpoint the role of SNAT7 in the early steps of viral life cycle and highlight the intricate interplay between macrophage metabolism and HIV-1 replication. While the question that is addressed is important, and the hypothesis overall sound, the data presented needs to be strengthened to support the conclusions. There are numerous weaknesses in data interpretation as well.
- Figure 1: SNAT7 expression was selectively enhanced upon differentiation of monocytes into macrophages but absent in CD4+ T cells. Though there is a claim of enhancement of SNAT7 expression upon HIV-1 infection of macrophages, RT-qPCR analysis shows the opposite trend (Fig 1E) and SNAT7 protein expression changes are modest. Statistical analysis in Fig. 1H needs to be revisited. The number of replicates vary for the lysates harvested at different day post infection, which might have an impact on the statistical test. To determine if SNAT7 expression enhancement is dependent on establishment of virus infection, as the authors imply, control lysates of virus infections in presence of replication inhibitors should be included.
We thank the reviewer for this comment. Indeed, there is a modest, but statistically significant increase in SNAT7 protein expression upon HIV-1 infection over time (Fig. 1G, H), without any modulation of SNAT7 gene expression (Fig. 1E). This indicates that the regulation of SNAT7 expression in this context is only at the translation level (i.e. increase of translation or stabilization of the SNAT7 protein).
As mentioned, Fig. 1H aggregates between 3 to 7 independent experiments on different donors depending on the infection time point. SNAT7 protein expression is increased already at 1 day post-infection and until 8 days. The statistical test used here, i.e. 2 way-ANOVA, compared Mock-infected and HIV-1-infected condition for each time point with the same number of donors. In this figure, the comparison is statistically different only at day 6 of the time course (7 donors). We agree that increasing the number of donors of the other time points could help to improve the statistical difference between control and infection condition.
We thank the reviewer for the suggestion mentioning the use of replication inhibitors in this experiment. We plan to use inhibitors of reverse transcription (Nevirapin) and integration (Dolutegravir).
The authors rely exclusively on western blot analysis for HIV-1 Gag expression in cell lysates as a measure of effects of SNAT7 on virus replication. Single cell analysis such as intracellular p24gag analysis by FACS should be included; this will provide a better measure of effects of SNAT7 onHIV-1 infection establishment.
We respectfully disagree with the reviewer for this question. Indeed, to evaluate the effects of SNAT7 on HIV-1 replication, we measured Gag Pr55 and Cap24 using a Western blot approach (Fig. 2B, D and E), but also assessed the quantity of Cap24 in the supernatants and lysates using an ELISA measurement, the quantity of infectious particles using TZM reporter cells, and total viral transcription or more specifically Gag Pr55 transcription using qPCR (Fig. 2F, G and I and Supp. Fig. 2G).
Regarding the quantification of CAp24 at the cell single level, please refer to comment #2 under Reviewer #1.
Knockdown of SNAT7 in MDMs was partial at best; only 30-50% decrease in expression (Fig 2C), but the effects on viral gene expression (Fig. 2I), p24 release and infectious particle production is dramatic (Fig. 2F and G). This discrepancy is not addressed. Does SNAT7 knock-down negatively impact virus particle release? Please note that the representative WB in Fig 2B does not correlate with the quantification in Fig. 2D. There are no p55gag or p24gag bands in SNAT7#1 siRNA condition (Fig. 2B)? Data could also be rearranged to follow the logical sequence of virus replication cycle (viral RNa expression followed by Gag expression, and then release).
We thank the reviewer for this comment. Our samples are indeed a mixture of SNAT7-depleted and non-depleted macrophages and RNA interference in these cells often leads to a decrease of 50 % of the protein expression.
To determine whether SNAT7 is involved in the release of particles, we quantified Cap24 in cell lysates and in the cell culture medium separately, and normalized the results to the total protein content. The absence of SNAT7 reduced the amount of Cap24 measured by ELISA in both samples to the same extent, showing that there is no storage of Cap24-positive viral particles inside the infected macrophages. These data were initially pooled in one graph (Fig. 2F), but separate graphs are now provided in new Supp. Fig. 2 E, F.
Regarding the western blot shown in Fig. 2B, please refer to comment #5 under Reviewer #1.
In the new version of the manuscript, we arranged the figures and placed the later stages of the viral cycle in Fig. 2 and the earlier stages, such as fusion, reverse transcription and transcription, in Fig. 3.
Data interpretation would be greatly improved by including infection controls (RT or integrase inhibitors) to confirm that measurements of viral RNA and Gag are indeed modulated by SNAT7 expression.
We thank the reviewer for this suggestion to include inhibitors of viral replication as controls. In our experiments, cells were Mock-infected in parallel as a negative control of viral detection. We provide the results in the new version of the manuscript to show that (i) there is no detection of viral or Gag RNA in the absence of the virus, (ii) the expression of viral genes measured in HIV-1-infected SNAT7-depleted cells is not different from Mock-infected cells, indicating almost complete inhibition of viral transcription (Fig. 3H and Supp. Fig. 3B), also confirmed at the protein level (Fig. 2B, D-F).
Figure 3: Decrease in SNAT7 expression in macrophages resulted in lower levels of early reverse transcripts. But surprisingly, LRT levels were not as affected by decreases in SNAT7 expression. The authors go on to suggest that decreases in early RT are due to loss of phospho-SAMHD1 and increases in catalytically active form of SAMHD1. Mechanistically this does not make sense: LRT should be similarly affected by increase in catalytically active SAMHD1. dNTP concentrations should be measured to determine if the rescue of RT is dependent on SAMHD1 dNTPase activity.
We thank the reviewer for this comment. LRT concentrations are very low in human macrophages and more challenging to detect than ERT concentrations. This might explain why the differences observed between the SNAT7-depleted and control conditions appear less pronounced for LRT than for ERT.
Furthermore, we cannot rule out the possibility that SNAT7 has a cumulative effect throughout the viral cycle. While reverse transcription remains statistically unaltered, and despite the reduced levels of ERT and LRT in SNAT7-depleted macrophages (Fig. 3 F, G), there is a significant impact on the transcription of viral RNAs (Fig. 2I) and Gag (Supp. Fig. 2G). This step may also be altered by the ribonuclease activity of SAMHD1 (Beloglazova et al., 2013; Ryoo et al., 2014).
Finally, with the help of Dr Baek Kim in Atlanta, we attempted to quantify dNTP concentrations in our human macrophages. Unfortunately, it was not possible to draw any conclusions, as the concentrations of dNTPs extracted from our cells were far too low.
Furthermore, it should be noted that SAMHD1 viral restriction through its phosphorylation at T592 is not correlated with its dNTPase activity (Welbourn et al., 2013; White et al., 2013), but with its ribonuclease activity (Beloglazova et al., 2013; Ryoo et al., 2014). This is supporting why SNAT7, by modulating the ribonuclease activity of SAMHD1, could have a greater effect on viral transcription than on reverse transcription.
There is lack of consistency in the data: p24 release upon SNAT7 depletion is highly variable. While there is a dramatic >90-95% decrease in p24 release (Fig. 2G), the effects are much more moderate in Fig. 4H (50-60% attenuation), even though siRNA-mediated depletion was similar across the data sets. The authors should comment on the variability in their findings.
We thank the reviewer for this comment, but believe that Figure 2E rather than Figure 2G is to be mentioned regarding the quantification of CAp24 by Western blot and to be compared with Figure 4H.
In Fig. 2E, we observed an average reduction of 85 % in CAp24 expression normalized to Clathrin HC expression across different donors for both siRNAs targeting SNAT7. For Fig. 4H, there was a 73 % reduction in CAp24 levels for siRNA #1 and a 56 % reduction for siRNA #2. In addition, it should be noted that the reduction in Gag levels is greater in Fig. 4G (between 77 % and 83 %) than in Fig. 2D (between 55 % and 72 %).
Therefore, there is some variation in the results obtained with the different donors, which could be explained by variations in Gag cleavage among donors, but this does not impact the conclusions for both figures.
SNAT7 is postulated to affect 2 steps in the virus life cycle: reverse transcription and viral transcription. But Vpx-mediated SAMHD1 degradation reversed both. Its not clear to me as to how SAMHD1 degradation impacts the role of SNAT7 in viral transcription. No explanation is provided.
We thank the reviewer for this comment. As suggested, we will perform experiments to assess the impact of Vpx-mediated SAMHD1 degradation on viral transcription.
Exogenous addition of glutamine only partially restored Gag synthesis and p24 release, which could be attributed to increased cytoplasmic levels and viral protein synthesis. What about effects on reverse transcription and viral gene expression?
We thank the reviewer for this comment. We will perform the suggested experiments to assess the impact of glutamine supplementation on viral transcription.
Reviewer #2 (Significance (Required)):
This is a novel finding, as there are limited number of studies on amino acid transporters and HIV-1 replication enhancement in macrophages. Most of the previous work has focused on CD4 T cells. These studies on SNAT7 and HIV-1 infection establishment in macrophages might better inform the influences of macrophage metabolism on HIV-1 persistence and inflammatory responses.
Reviewer #3 (Evidence, reproducibility and clarity (Required)):
This study investigates the role of the lysosomal glutamine transporter SLC38A7/SNAT7 in HIV‑1 replication in primary human macrophages. The authors demonstrate that SNAT7 is highly expressed in macrophages and upregulated upon HIV‑1 infection. They show that SNAT7 depletion inhibits HIV‑1 production at the reverse transcription step without affecting viral fusion or global cellular translation/transcription. Mechanistically, SNAT7 knockdown reduces the inhibitory phosphorylation of SAMHD1 at T592, and degradation of SAMHD1 by Vpx fully rescues viral replication. Extracellular glutamine supplementation partially restores HIV‑1 production in SNAT7‑deficient cells. Overall, the authors report interesting observations; however, the mechanistic investigation remains preliminary, raising concerns about whether the data fully support all the conclusions drawn.
Major Concerns:
1. The mechanistic depth is insufficient. The authors do not elucidate how glutamine regulates SAMHD1 T592 phosphorylation, whether through metabolite‑mediated control of kinases/phosphatases or via indirect effects.
We thank the reviewer for this comment. It is worth noting that (Meng et al., 2022) demonstrated that SNAT7 positively regulates mTORC1 activity at the lysosomal membrane through release of lysosomal glutamine, and (Dias et al., 2024) showed that inhibiting mTORC1 activity using drugs decreases SAMHD1 Thr592 phosphorylation in hMDM. Therefore, we could speculate that the absence of SNAT7 down-regulates mTORC1 activity, which then leads to decreased SAMHD1 phosphorylation. This is now further discussed in the discussion section of the manuscript.
The authors do not measure intracellular dNTP levels upon SNAT7 knockdown, which is the key functional substrate of SAMHD1. They also do not directly demonstrate that glutamine supplementation restores dNTP pools.
We thank the reviewer for this comment. Please, refer to comment #5 under Reviewer #2.
Extracellular glutamine only partially rescues viral production, implying the existence of transport‑independent functions of SNAT7 or additional pathways. This important observation is not discussed.
We thank the reviewer for this comment. The discussion has been modified accordingly.
It is suggested that the key findings be validated in immortalized THP‑1 cells differentiated into macrophage‑like cells by PMA.
We thank the reviewer for this suggestion but don’t really understand why this would strengthen our conclusions. Indeed, despite the known variability between donors and technical limitations to transduce cells, we chose human blood monocyte-derived macrophages as a relevant non-transformed model for HIV-1 infection of macrophages. They also represent to some extent the human diversity.
The Discussion section should be expanded to include the potential translational implications and limitations of the present study.
We thank the reviewer for this comment. The discussion points to some elements of potential translation and limitations of the study.
Reviewer #3 (Significance (Required)):
General assessment: This study identifies the lysosomal glutamine transporter SLC38A7/SNAT7 as a novel host dependency factor for HIV‑1 replication in primary human macrophages. The major strengths include the use of physiologically relevant primary macrophage models, a well-organized experimental pipeline from expression profiling to functional validation, and the establishment of a link between SNAT7, glutamine metabolism, and the HIV restriction factor SAMHD1.
Advance: It extends current understanding of HIV‑1 host dependency factors and immunometabolism by revealing a compartment‑specific metabolic pathway that supports viral reverse transcription.
Audience:This work will primarily interest specialized researchers in HIV‑1 biology, host-virus interactions, restriction factors, and antiviral innate immunity.
Reviewer #1 (Evidence, reproducibility and clarity (Required)):
This study from the Niedergang lab establishes SNAT7 as a host-dependency factor in human macrophages that supports HIV-1 replication. They show a modest increase in SNAT7 levels HIV-1 infected macrophages and suggest that SNAT7 levels are transiently increased. Employing siRNA against SNAT7 they show reduction in HIV-1 protein levels and viral RNAs and claim that there is a block of reverse transcription in SNAT7 KD cells. Focusing on a known HIV-1 restriction factor in macrophages, SAMHD1, they interconnect the SNAT7 depletion with a reduction in phosphorylated, i.e. catalytical inactive SAMHD1 arguing that SNAT7 regulates the phosphorylation and thereby antiviral activity of SAMHD1. Since SNAT7 is a glutamine transporter that provides this AA from lysosomes, they lastly supplement glutamine and this somehow rescues the reduction of HIV-1 production in SNAT7 KD cells.
Major comments:
The strength of this manuscript is the clear focus on primary human macrophages that are HIV-1 infected and the interconnection of HIV-1 replication to the SNAT7 siRNA KD experiments in combination with SAMHD1 depletion and lastly glutamine supplementation. This establishes a stringent and coherent story line.
The effects reported are modest; high variability is not a problem since using primary hMDM this is expected and can be addressed by testing several donors and applying stringent statistics.
- Having said so, I realize that while they give information on the statistical test used, i.e. one-way ANOVA they miss to explain the post-test used to assess significance (i.e. Bonferroni, Fishers LSD, whatsoever). Please add this information.
We thank the reviewer for this comment. The figure legends have been updated to include more details of all the statistical tests used.
- Another issue that might underestimate the effects of HIV-1 infection on SNAT7 levels and vice versa of SNAT7 KD on HIV-1 replication is the non-single cell approach employed, i.e. WBlots. I assume that HIV-1 infection rates in macrophages are not super high, usually not exceeding 20-30%. So indeed the effects the authors observe could be much higher, when checking at the single cell level. I do not know about the SNAT7 ab, but all the other reagents should work via flow cytometry and could hence improve the readout a lot.
We agree with the reviewer and indeed, in previous studies on HIV-1 infection of human macrophages performed in the lab, we observed via immunofluorescence that the proportion of infected cells ranged from 20 to 40 %. At the time of submission, we did not have the possibility to label the native SNAT7 protein by immunofluorescence, as the commercial antibody used only works for western blotting.
In the meantime, we have been validating a new antibody (Proteintech) targeting SNAT7 for immunofluorescence. If this is confirmed, we will be able to detect and quantify HIV-1 p24 by immunofluorescence in SNAT7-depleted human macrophages and control cells, thus confirming our results in single-cell analysis.
Flow cytometry analyses are difficult to perform on primary human macrophages because these cells are highly adherent and must be detached first. The process induces significant cell death and damage. This is why we would prefer to carry out these analyses using immunofluorescence and microscopy on adhered cells. This option will be undoubtedly pursued.
- Furthermore the authors never commented about a dose-response effect in terms of HIV-1 infection levels. There is a MOI dependency described for Suppl.Fig.1 C-F, unfortunately the data is missing in the manuscript.
We apologize for this omission. The figures showing the increase in SNAT7 protein expression following HIV-1 infection at MOIs ranging from 0.05 to 0.5 were added to the new version of the manuscript (Supp. Fig. 1 C-F).
- Figure1: specify circulating T lymphocytes. I would expect to see levels of SNAT7 in PHA or CD3/CD28 activated lymphocytes versus resting T cells and a time course of SNAT7 levels upon activation. I think even though SNAT7 levels in T cells might be low, they could also be increased by HIV-1 infection and it is essential that the authors test for this. If not, the result is a valid negative control. For this they should employ HIV-1 primary strains with a tropism for T cells, or at least lab-adapted HIV-1 NL4-3
We thank the reviewer for this comment. Circulating T lymphocytes isolated from the blood of healthy donors are now referred to resting lymphocytes in the new version of the manuscript, as opposed to activated T lymphocytes stimulated with IL2 and PHA-P for several days (Fig. 1 A-C).
The expression levels of SNAT7, both at the gene and protein levels, are lower in resting or IL2/PHA-P-activated T cells than in macrophages from the same donors. As suggested, we will perform a kinetic of T-cell activation upon HIV-1 infection to investigate how SNAT7 expression varies in these conditions.
- Figure 2 again single cell measurements could reveal much more pronounced effects; it is a bit counterintuitive that siRNA #2 is more efficient in SNAT7 KD but has higher levels of HIV-1 replication in terms of Gag levels. I assume when looking at the stats it is always a comparison to the Ctl treated cells (C-G), but this is not entirely clear. Unify labeling as compared to the stats in Fig.2 I (this also applies for all the other figs).
We thank the reviewer for this comment. Fig. 2B indeed shows one of the different donors analyzed. However, protein quantification across six different donors shows that SNAT7 is more depleted with siRNA #2 (Fig. 2C), and that Gag Pr55 protein levels are consequently more reduced, than with siRNA #1 (Fig. 2D).
We use GraphPad Prism software to perform statistical analysis. Depending on the test used, the software automatically plots the comparison bar and displays the p-value above it. We changed the representation of statistics as suggested.
Figure 3: It is a bit odd that they finally conclude on RT as essential step that is reduced in the absence of SNAT7 and then they fail to provide statistical significance for this (Fig.3 panels F and G). One would expect that RT is much more affected given the huge effects on HIV-1 capsid and particle production shown in Fig.2 F, G and I.
The reviewer is right in pointing that we observed a stronger effect during the later stages of the viral cycle, from transcription of viral RNAs (Fig. 2I and Supp. Fig. 2G) to the production of viral particles in the supernatant (Fig. 2D-G), than during the earlier stage of reverse transcription (Fig. 3F, G). Also, it is also possible that we might have missed the peak in ERT/LRT production, which is transient.
It should be noted that SAMHD1 exhibits both dNTPase (Goldstone et al., 2011) and nuclease (Beloglazova et al., 2013) activities. The ability of SAMHD1 to restrict the virus, through dephosphorylation at T592, is mediated by its RNase activity (Ryoo et al., 2014), and not by the dNTPase activity (Welbourn et al., 2013; White et al., 2013).This could explain why SNAT7 exhibit a stronger impact on viral transcription than on reverse transcription.
Figure 4; again single cell flow measurements of SAMHD1, pSAMHD1 and p24 /SNAT7 might help to more clearly discriminate effects that are specifically induced upon infection or happen in virally infected cells. Maybe alternatively IF?
We thank the reviewer for this suggestion. As mentioned under comment #2, flow cytometry analyses are difficult to perform on strongly adherent primary human macrophages.
With regard to immunofluorescence, there is a technical limitation based on the species in which the antibodies are produced. The antibody that targets the native SNAT7 protein, which is currently being validated in our laboratory, is produced in rabbits. An anti-CAp24 antibody produced in goats can be used. It will then be necessary to co-label the cells with anti SAMHD1 and phospho-SAMHD1produced in mouse. We will try to find options to co-label the cells.
The wblot shown in panel D does not really reflect the point the authors want to make by the quantification in panels G-I. Primary data (D) suggests that SNAT7 KD reduces HIV-1 production even in the absence of SAMHD1. The quantification rather indicates that SNAT7 KD does not affect HIV-1 production in the absence of SAMHD1. This needs clarification/corroboration by orthogonal approaches.
We respectfully disagree with the reviewer.
Figure 4D shows a representative blot of the six donors analysed. As mentioned, the depletion of SNAT7 in the absence of SAMHD1 reduces the production of the viral proteins GagPr55 and CAp24 (see Fig. 4D). This is illustrated by the quantifications (Fig. 4G–I). Following treatment with Vpx, GagPr55 protein expression in SNAT7 KD macrophages is reduced by a factor of 2.6 for siRNA #1 (mean = 1.48, light grey bar) and by a factor of 1.83 for siRNA #2 (mean = 2.13, orange bar), compared to the control (mean = 3.9, pink bar) (Fig. 4G). Similarly, CAp24 protein expression was reduced by a factor of 2.2 for siRNA #1 (mean = 2.05, light grey bar) and by a factor of 1.36 for siRNA #2 (mean = 3.34, orange bar), compared to the control (mean = 4.52, pink bar) (Fig. 4H).
These differences are therefore consistent between the Western blot and the quantifications. However, they are not significantly different to those observed in cells treated with Vpx and depleted with control siRNA, suggesting that the viral restriction observed in SNAT7 KD cells is primarily due to SAMHD1.
Figure 5: show SAMHD1 and pSAMHD1 levels upon glutamine supplementation.
We thank the reviewer for this comment, we will perform the suggested experiment.
- I think the discussion is very thin, mainly summarizing the results; but fails to give broader context or critically discuss the limitations and further directions.
We thank the reviewer for this comment. The discussion will be modified further accordingly.
Looking at the data as a whole, I think the results support a modest functional importance of SNAT7 for HIV-1 production in macrophages. I acknowledge that the experiments in primary macrophages are prone to high variability in different donors and the authors transparently depicted their data. However clearly, I would advice the authors to tune down the extend in which they claim SNAT7-dependency given this huge variability and the sometimes-borderline statistics.
We respectfully disagree with the reviewer.
The cells used here imply greater variability than a cell line, but are also more relevant.
Indeed, the effects observed in the late stages of HIV-1 production are:
-
~80 % decrease in viral transcription compared to the control (Fig. 2I),
-
~85 % decrease in CAp24 protein expression compared to the control, as quantified by western blot (Fig. 2E), or ~90 % by ELISA measurement (Fig. 2F),
-
a reduction of more than 90 % in the release of infectious particles (Fig. 2G).
These results were all significant across donors, while SNAT7 depletion was always partial (Fig. 2C, between 31 to 62 % of depletion compared to the control in infected cells).
Therefore, the data were obtained from a mixture of depleted and non-depleted macrophages. This means that the results may be underestimated.
Together, our results show that SNAT7 is necessary for HIV-1 production.
However, reading the comments, we realized that our conclusions regarding reverse transcription were too strong. SNAT7 depletion does not affect viral fusion and reverse transcription. The manuscript was modified accordingly.
On top, there are a lot of optional experiments I am sure the authors are aware of that should be done at least in the future.
For instance, how does HIV-1 upregulate SNAT7, is a viral accessory protein involved?
What is the mechanism of SNAT7 dependent SAMHD1 phosphorylation? Does SNAT7 (or glutamine) regulate the activity of the SAMHD1 associated kinase / phosphatase) If so, does this impact on other targets of these enzymes?
We thank the reviewer for these questions.
To address the role of accessory viral proteins, we have already performed one experiment infecting hMDM with HIV-1 strains deleted for genes such as Nef, Vpr, Vpu and Vif, and have found no clear effect on SNAT7 protein expression compared to WT strains. As an alternative experiment, we could overexpress individual viral genes, such as Nef or Vpr, in HeLa cells and analyze their impact on SNAT7 expression by Western blot.
It is also possible that SNAT7 expression and recycling of lysosomal glutamine are modulated by the macrophage intrinsic immunity in response to HIV-1 infection.
The Thr592 motif of the SAMHD1 protein is phosphorylated by Cyclin A2/CDK1 and type 1 IFN in non-cycling cells, such as MDMs (Cribier et al., 2013). For now, the relationship between SNAT7 and SAMHD1 remains unclear. However, (Meng et al., 2022) demonstrated that SNAT7 positively regulates mTORC1 activity at the lysosomal membrane through release of lysosomal glutamine, and (Dias et al., 2024) showed that inhibiting mTORC1 activity decreases SAMHD1 Thr592 phosphorylation in hMDM. Therefore, we could speculate that the absence of SNAT7 down-regulates mTORC1 activity, which then leads to decreased SAMHD1 phosphorylation. This has been added to the discussion to explain the relationship between the 3 partners.
**Referees cross-commenting**
I think the comments from the other referees are reasonable and consistent with my assessment
Reviewer #1 (Significance (Required)):
Strength and limitations see above;
Significance: I think this work is of high interest for virologists working in the field of HIV-1 and infection of myeloid cells. In case SNAT7 (and hence glutamine) indeed regulates the phosphorylation of SAMHD1, there could potentially be broad relevance of this work. However unfortunately, this aspect remains underdeveloped and is also not discussed
Field of expertise: HIV-1, immunology, cell biology
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
In this report, Herit and colleagues describe the role of a HIV-1 dependency factor that promotes virus replication in macrophages. The authors suggest that the lysosomal membrane-associated SNAT7 glutamine transporter is a HIV dependency factor, that promotes virus replication by enhancing reverse transcription and Gag synthesis. The authors use transient knock-down approaches in primary macrophages to identify that SNAT7 depletion does not impact viral entry but inhibits early reverse transcription which was reversed by exogenous glutamine addition. While reverse transcription enhancement was likely due to selective increase in phosho-SAMHD1 expression, mechanisms by which SNAT7 enhanced viral gene expression were not clearly defined. These are well-controlled studies that pinpoint the role of SNAT7 in the early steps of viral life cycle and highlight the intricate interplay between macrophage metabolism and HIV-1 replication. While the question that is addressed is important, and the hypothesis overall sound, the data presented needs to be strengthened to support the conclusions. There are numerous weaknesses in data interpretation as well.
- Figure 1: SNAT7 expression was selectively enhanced upon differentiation of monocytes into macrophages but absent in CD4+ T cells. Though there is a claim of enhancement of SNAT7 expression upon HIV-1 infection of macrophages, RT-qPCR analysis shows the opposite trend (Fig 1E) and SNAT7 protein expression changes are modest. Statistical analysis in Fig. 1H needs to be revisited. The number of replicates vary for the lysates harvested at different day post infection, which might have an impact on the statistical test. To determine if SNAT7 expression enhancement is dependent on establishment of virus infection, as the authors imply, control lysates of virus infections in presence of replication inhibitors should be included.
We thank the reviewer for this comment. Indeed, there is a modest, but statistically significant increase in SNAT7 protein expression upon HIV-1 infection over time (Fig. 1G, H), without any modulation of SNAT7 gene expression (Fig. 1E). This indicates that the regulation of SNAT7 expression in this context is only at the translation level (i.e. increase of translation or stabilization of the SNAT7 protein).
As mentioned, Fig. 1H aggregates between 3 to 7 independent experiments on different donors depending on the infection time point. SNAT7 protein expression is increased already at 1 day post-infection and until 8 days. The statistical test used here, i.e. 2 way-ANOVA, compared Mock-infected and HIV-1-infected condition for each time point with the same number of donors. In this figure, the comparison is statistically different only at day 6 of the time course (7 donors). We agree that increasing the number of donors of the other time points could help to improve the statistical difference between control and infection condition.
We thank the reviewer for the suggestion mentioning the use of replication inhibitors in this experiment. We plan to use inhibitors of reverse transcription (Nevirapin) and integration (Dolutegravir).
The authors rely exclusively on western blot analysis for HIV-1 Gag expression in cell lysates as a measure of effects of SNAT7 on virus replication. Single cell analysis such as intracellular p24gag analysis by FACS should be included; this will provide a better measure of effects of SNAT7 onHIV-1 infection establishment.
We respectfully disagree with the reviewer for this question. Indeed, to evaluate the effects of SNAT7 on HIV-1 replication, we measured Gag Pr55 and Cap24 using a Western blot approach (Fig. 2B, D and E), but also assessed the quantity of Cap24 in the supernatants and lysates using an ELISA measurement, the quantity of infectious particles using TZM reporter cells, and total viral transcription or more specifically Gag Pr55 transcription using qPCR (Fig. 2F, G and I and Supp. Fig. 2G).
Regarding the quantification of CAp24 at the cell single level, please refer to comment #2 under Reviewer #1.
Knockdown of SNAT7 in MDMs was partial at best; only 30-50% decrease in expression (Fig 2C), but the effects on viral gene expression (Fig. 2I), p24 release and infectious particle production is dramatic (Fig. 2F and G). This discrepancy is not addressed. Does SNAT7 knock-down negatively impact virus particle release? Please note that the representative WB in Fig 2B does not correlate with the quantification in Fig. 2D. There are no p55gag or p24gag bands in SNAT7#1 siRNA condition (Fig. 2B)? Data could also be rearranged to follow the logical sequence of virus replication cycle (viral RNa expression followed by Gag expression, and then release).
We thank the reviewer for this comment. Our samples are indeed a mixture of SNAT7-depleted and non-depleted macrophages and RNA interference in these cells often leads to a decrease of 50 % of the protein expression.
To determine whether SNAT7 is involved in the release of particles, we quantified Cap24 in cell lysates and in the cell culture medium separately, and normalized the results to the total protein content. The absence of SNAT7 reduced the amount of Cap24 measured by ELISA in both samples to the same extent, showing that there is no storage of Cap24-positive viral particles inside the infected macrophages. These data were initially pooled in one graph (Fig. 2F), but separate graphs are now provided in new Supp. Fig. 2 E, F.
Regarding the western blot shown in Fig. 2B, please refer to comment #5 under Reviewer #1.
In the new version of the manuscript, we arranged the figures and placed the later stages of the viral cycle in Fig. 2 and the earlier stages, such as fusion, reverse transcription and transcription, in Fig. 3.
Data interpretation would be greatly improved by including infection controls (RT or integrase inhibitors) to confirm that measurements of viral RNA and Gag are indeed modulated by SNAT7 expression.
We thank the reviewer for this suggestion to include inhibitors of viral replication as controls. In our experiments, cells were Mock-infected in parallel as a negative control of viral detection. We provide the results in the new version of the manuscript to show that (i) there is no detection of viral or Gag RNA in the absence of the virus, (ii) the expression of viral genes measured in HIV-1-infected SNAT7-depleted cells is not different from Mock-infected cells, indicating almost complete inhibition of viral transcription (Fig. 3H and Supp. Fig. 3B), also confirmed at the protein level (Fig. 2B, D-F).
Figure 3: Decrease in SNAT7 expression in macrophages resulted in lower levels of early reverse transcripts. But surprisingly, LRT levels were not as affected by decreases in SNAT7 expression. The authors go on to suggest that decreases in early RT are due to loss of phospho-SAMHD1 and increases in catalytically active form of SAMHD1. Mechanistically this does not make sense: LRT should be similarly affected by increase in catalytically active SAMHD1. dNTP concentrations should be measured to determine if the rescue of RT is dependent on SAMHD1 dNTPase activity.
We thank the reviewer for this comment. LRT concentrations are very low in human macrophages and more challenging to detect than ERT concentrations. This might explain why the differences observed between the SNAT7-depleted and control conditions appear less pronounced for LRT than for ERT.
Furthermore, we cannot rule out the possibility that SNAT7 has a cumulative effect throughout the viral cycle. While reverse transcription remains statistically unaltered, and despite the reduced levels of ERT and LRT in SNAT7-depleted macrophages (Fig. 3 F, G), there is a significant impact on the transcription of viral RNAs (Fig. 2I) and Gag (Supp. Fig. 2G). This step may also be altered by the ribonuclease activity of SAMHD1 (Beloglazova et al., 2013; Ryoo et al., 2014).
Finally, with the help of Dr Baek Kim in Atlanta, we attempted to quantify dNTP concentrations in our human macrophages. Unfortunately, it was not possible to draw any conclusions, as the concentrations of dNTPs extracted from our cells were far too low.
Furthermore, it should be noted that SAMHD1 viral restriction through its phosphorylation at T592 is not correlated with its dNTPase activity (Welbourn et al., 2013; White et al., 2013), but with its ribonuclease activity (Beloglazova et al., 2013; Ryoo et al., 2014). This is supporting why SNAT7, by modulating the ribonuclease activity of SAMHD1, could have a greater effect on viral transcription than on reverse transcription.
There is lack of consistency in the data: p24 release upon SNAT7 depletion is highly variable. While there is a dramatic >90-95% decrease in p24 release (Fig. 2G), the effects are much more moderate in Fig. 4H (50-60% attenuation), even though siRNA-mediated depletion was similar across the data sets. The authors should comment on the variability in their findings.
We thank the reviewer for this comment, but believe that Figure 2E rather than Figure 2G is to be mentioned regarding the quantification of CAp24 by Western blot and to be compared with Figure 4H.
In Fig. 2E, we observed an average reduction of 85 % in CAp24 expression normalized to Clathrin HC expression across different donors for both siRNAs targeting SNAT7. For Fig. 4H, there was a 73 % reduction in CAp24 levels for siRNA #1 and a 56 % reduction for siRNA #2. In addition, it should be noted that the reduction in Gag levels is greater in Fig. 4G (between 77 % and 83 %) than in Fig. 2D (between 55 % and 72 %).
Therefore, there is some variation in the results obtained with the different donors, which could be explained by variations in Gag cleavage among donors, but this does not impact the conclusions for both figures.
SNAT7 is postulated to affect 2 steps in the virus life cycle: reverse transcription and viral transcription. But Vpx-mediated SAMHD1 degradation reversed both. Its not clear to me as to how SAMHD1 degradation impacts the role of SNAT7 in viral transcription. No explanation is provided.
We thank the reviewer for this comment. As suggested, we will perform experiments to assess the impact of Vpx-mediated SAMHD1 degradation on viral transcription.
Exogenous addition of glutamine only partially restored Gag synthesis and p24 release, which could be attributed to increased cytoplasmic levels and viral protein synthesis. What about effects on reverse transcription and viral gene expression?
We thank the reviewer for this comment. We will perform the suggested experiments to assess the impact of glutamine supplementation on viral transcription.
Reviewer #2 (Significance (Required)):
This is a novel finding, as there are limited number of studies on amino acid transporters and HIV-1 replication enhancement in macrophages. Most of the previous work has focused on CD4 T cells. These studies on SNAT7 and HIV-1 infection establishment in macrophages might better inform the influences of macrophage metabolism on HIV-1 persistence and inflammatory responses.
Reviewer #3 (Evidence, reproducibility and clarity (Required)):
This study investigates the role of the lysosomal glutamine transporter SLC38A7/SNAT7 in HIV‑1 replication in primary human macrophages. The authors demonstrate that SNAT7 is highly expressed in macrophages and upregulated upon HIV‑1 infection. They show that SNAT7 depletion inhibits HIV‑1 production at the reverse transcription step without affecting viral fusion or global cellular translation/transcription. Mechanistically, SNAT7 knockdown reduces the inhibitory phosphorylation of SAMHD1 at T592, and degradation of SAMHD1 by Vpx fully rescues viral replication. Extracellular glutamine supplementation partially restores HIV‑1 production in SNAT7‑deficient cells. Overall, the authors report interesting observations; however, the mechanistic investigation remains preliminary, raising concerns about whether the data fully support all the conclusions drawn.
Major Concerns:
1. The mechanistic depth is insufficient. The authors do not elucidate how glutamine regulates SAMHD1 T592 phosphorylation, whether through metabolite‑mediated control of kinases/phosphatases or via indirect effects.
We thank the reviewer for this comment. It is worth noting that (Meng et al., 2022) demonstrated that SNAT7 positively regulates mTORC1 activity at the lysosomal membrane through release of lysosomal glutamine, and (Dias et al., 2024) showed that inhibiting mTORC1 activity using drugs decreases SAMHD1 Thr592 phosphorylation in hMDM. Therefore, we could speculate that the absence of SNAT7 down-regulates mTORC1 activity, which then leads to decreased SAMHD1 phosphorylation. This is now further discussed in the discussion section of the manuscript.
The authors do not measure intracellular dNTP levels upon SNAT7 knockdown, which is the key functional substrate of SAMHD1. They also do not directly demonstrate that glutamine supplementation restores dNTP pools.
We thank the reviewer for this comment. Please, refer to comment #5 under Reviewer #2.
Extracellular glutamine only partially rescues viral production, implying the existence of transport‑independent functions of SNAT7 or additional pathways. This important observation is not discussed.
We thank the reviewer for this comment. The discussion has been modified accordingly.
It is suggested that the key findings be validated in immortalized THP‑1 cells differentiated into macrophage‑like cells by PMA.
We thank the reviewer for this suggestion but don’t really understand why this would strengthen our conclusions. Indeed, despite the known variability between donors and technical limitations to transduce cells, we chose human blood monocyte-derived macrophages as a relevant non-transformed model for HIV-1 infection of macrophages. They also represent to some extent the human diversity.
The Discussion section should be expanded to include the potential translational implications and limitations of the present study.
We thank the reviewer for this comment. The discussion points to some elements of potential translation and limitations of the study.
Reviewer #3 (Significance (Required)):
General assessment: This study identifies the lysosomal glutamine transporter SLC38A7/SNAT7 as a novel host dependency factor for HIV‑1 replication in primary human macrophages. The major strengths include the use of physiologically relevant primary macrophage models, a well-organized experimental pipeline from expression profiling to functional validation, and the establishment of a link between SNAT7, glutamine metabolism, and the HIV restriction factor SAMHD1.
Advance: It extends current understanding of HIV‑1 host dependency factors and immunometabolism by revealing a compartment‑specific metabolic pathway that supports viral reverse transcription.
Audience:This work will primarily interest specialized researchers in HIV‑1 biology, host-virus interactions, restriction factors, and antiviral innate immunity.
Reviewer #1 (Evidence, reproducibility and clarity (Required)):
This study from the Niedergang lab establishes SNAT7 as a host-dependency factor in human macrophages that supports HIV-1 replication. They show a modest increase in SNAT7 levels HIV-1 infected macrophages and suggest that SNAT7 levels are transiently increased. Employing siRNA against SNAT7 they show reduction in HIV-1 protein levels and viral RNAs and claim that there is a block of reverse transcription in SNAT7 KD cells. Focusing on a known HIV-1 restriction factor in macrophages, SAMHD1, they interconnect the SNAT7 depletion with a reduction in phosphorylated, i.e. catalytical inactive SAMHD1 arguing that SNAT7 regulates the phosphorylation and thereby antiviral activity of SAMHD1. Since SNAT7 is a glutamine transporter that provides this AA from lysosomes, they lastly supplement glutamine and this somehow rescues the reduction of HIV-1 production in SNAT7 KD cells.
Major comments:
The strength of this manuscript is the clear focus on primary human macrophages that are HIV-1 infected and the interconnection of HIV-1 replication to the SNAT7 siRNA KD experiments in combination with SAMHD1 depletion and lastly glutamine supplementation. This establishes a stringent and coherent story line.
The effects reported are modest; high variability is not a problem since using primary hMDM this is expected and can be addressed by testing several donors and applying stringent statistics.
- Having said so, I realize that while they give information on the statistical test used, i.e. one-way ANOVA they miss to explain the post-test used to assess significance (i.e. Bonferroni, Fishers LSD, whatsoever). Please add this information.
We thank the reviewer for this comment. The figure legends have been updated to include more details of all the statistical tests used.
- Another issue that might underestimate the effects of HIV-1 infection on SNAT7 levels and vice versa of SNAT7 KD on HIV-1 replication is the non-single cell approach employed, i.e. WBlots. I assume that HIV-1 infection rates in macrophages are not super high, usually not exceeding 20-30%. So indeed the effects the authors observe could be much higher, when checking at the single cell level. I do not know about the SNAT7 ab, but all the other reagents should work via flow cytometry and could hence improve the readout a lot.
We agree with the reviewer and indeed, in previous studies on HIV-1 infection of human macrophages performed in the lab, we observed via immunofluorescence that the proportion of infected cells ranged from 20 to 40 %. At the time of submission, we did not have the possibility to label the native SNAT7 protein by immunofluorescence, as the commercial antibody used only works for western blotting.
In the meantime, we have been validating a new antibody (Proteintech) targeting SNAT7 for immunofluorescence. If this is confirmed, we will be able to detect and quantify HIV-1 p24 by immunofluorescence in SNAT7-depleted human macrophages and control cells, thus confirming our results in single-cell analysis.
Flow cytometry analyses are difficult to perform on primary human macrophages because these cells are highly adherent and must be detached first. The process induces significant cell death and damage. This is why we would prefer to carry out these analyses using immunofluorescence and microscopy on adhered cells. This option will be undoubtedly pursued.
- Furthermore the authors never commented about a dose-response effect in terms of HIV-1 infection levels. There is a MOI dependency described for Suppl.Fig.1 C-F, unfortunately the data is missing in the manuscript.
We apologize for this omission. The figures showing the increase in SNAT7 protein expression following HIV-1 infection at MOIs ranging from 0.05 to 0.5 were added to the new version of the manuscript (Supp. Fig. 1 C-F).
- Figure1: specify circulating T lymphocytes. I would expect to see levels of SNAT7 in PHA or CD3/CD28 activated lymphocytes versus resting T cells and a time course of SNAT7 levels upon activation. I think even though SNAT7 levels in T cells might be low, they could also be increased by HIV-1 infection and it is essential that the authors test for this. If not, the result is a valid negative control. For this they should employ HIV-1 primary strains with a tropism for T cells, or at least lab-adapted HIV-1 NL4-3
We thank the reviewer for this comment. Circulating T lymphocytes isolated from the blood of healthy donors are now referred to resting lymphocytes in the new version of the manuscript, as opposed to activated T lymphocytes stimulated with IL2 and PHA-P for several days (Fig. 1 A-C).
The expression levels of SNAT7, both at the gene and protein levels, are lower in resting or IL2/PHA-P-activated T cells than in macrophages from the same donors. As suggested, we will perform a kinetic of T-cell activation upon HIV-1 infection to investigate how SNAT7 expression varies in these conditions.
- Figure 2 again single cell measurements could reveal much more pronounced effects; it is a bit counterintuitive that siRNA #2 is more efficient in SNAT7 KD but has higher levels of HIV-1 replication in terms of Gag levels. I assume when looking at the stats it is always a comparison to the Ctl treated cells (C-G), but this is not entirely clear. Unify labeling as compared to the stats in Fig.2 I (this also applies for all the other figs).
We thank the reviewer for this comment. Fig. 2B indeed shows one of the different donors analyzed. However, protein quantification across six different donors shows that SNAT7 is more depleted with siRNA #2 (Fig. 2C), and that Gag Pr55 protein levels are consequently more reduced, than with siRNA #1 (Fig. 2D).
We use GraphPad Prism software to perform statistical analysis. Depending on the test used, the software automatically plots the comparison bar and displays the p-value above it. We changed the representation of statistics as suggested.
Figure 3: It is a bit odd that they finally conclude on RT as essential step that is reduced in the absence of SNAT7 and then they fail to provide statistical significance for this (Fig.3 panels F and G). One would expect that RT is much more affected given the huge effects on HIV-1 capsid and particle production shown in Fig.2 F, G and I.
The reviewer is right in pointing that we observed a stronger effect during the later stages of the viral cycle, from transcription of viral RNAs (Fig. 2I and Supp. Fig. 2G) to the production of viral particles in the supernatant (Fig. 2D-G), than during the earlier stage of reverse transcription (Fig. 3F, G). Also, it is also possible that we might have missed the peak in ERT/LRT production, which is transient.
It should be noted that SAMHD1 exhibits both dNTPase (Goldstone et al., 2011) and nuclease (Beloglazova et al., 2013) activities. The ability of SAMHD1 to restrict the virus, through dephosphorylation at T592, is mediated by its RNase activity (Ryoo et al., 2014), and not by the dNTPase activity (Welbourn et al., 2013; White et al., 2013).This could explain why SNAT7 exhibit a stronger impact on viral transcription than on reverse transcription.
Figure 4; again single cell flow measurements of SAMHD1, pSAMHD1 and p24 /SNAT7 might help to more clearly discriminate effects that are specifically induced upon infection or happen in virally infected cells. Maybe alternatively IF?
We thank the reviewer for this suggestion. As mentioned under comment #2, flow cytometry analyses are difficult to perform on strongly adherent primary human macrophages.
With regard to immunofluorescence, there is a technical limitation based on the species in which the antibodies are produced. The antibody that targets the native SNAT7 protein, which is currently being validated in our laboratory, is produced in rabbits. An anti-CAp24 antibody produced in goats can be used. It will then be necessary to co-label the cells with anti SAMHD1 and phospho-SAMHD1produced in mouse. We will try to find options to co-label the cells.
The wblot shown in panel D does not really reflect the point the authors want to make by the quantification in panels G-I. Primary data (D) suggests that SNAT7 KD reduces HIV-1 production even in the absence of SAMHD1. The quantification rather indicates that SNAT7 KD does not affect HIV-1 production in the absence of SAMHD1. This needs clarification/corroboration by orthogonal approaches.
We respectfully disagree with the reviewer.
Figure 4D shows a representative blot of the six donors analysed. As mentioned, the depletion of SNAT7 in the absence of SAMHD1 reduces the production of the viral proteins GagPr55 and CAp24 (see Fig. 4D). This is illustrated by the quantifications (Fig. 4G–I). Following treatment with Vpx, GagPr55 protein expression in SNAT7 KD macrophages is reduced by a factor of 2.6 for siRNA #1 (mean = 1.48, light grey bar) and by a factor of 1.83 for siRNA #2 (mean = 2.13, orange bar), compared to the control (mean = 3.9, pink bar) (Fig. 4G). Similarly, CAp24 protein expression was reduced by a factor of 2.2 for siRNA #1 (mean = 2.05, light grey bar) and by a factor of 1.36 for siRNA #2 (mean = 3.34, orange bar), compared to the control (mean = 4.52, pink bar) (Fig. 4H).
These differences are therefore consistent between the Western blot and the quantifications. However, they are not significantly different to those observed in cells treated with Vpx and depleted with control siRNA, suggesting that the viral restriction observed in SNAT7 KD cells is primarily due to SAMHD1.
Figure 5: show SAMHD1 and pSAMHD1 levels upon glutamine supplementation.
We thank the reviewer for this comment, we will perform the suggested experiment.
- I think the discussion is very thin, mainly summarizing the results; but fails to give broader context or critically discuss the limitations and further directions.
We thank the reviewer for this comment. The discussion will be modified further accordingly.
Looking at the data as a whole, I think the results support a modest functional importance of SNAT7 for HIV-1 production in macrophages. I acknowledge that the experiments in primary macrophages are prone to high variability in different donors and the authors transparently depicted their data. However clearly, I would advice the authors to tune down the extend in which they claim SNAT7-dependency given this huge variability and the sometimes-borderline statistics.
We respectfully disagree with the reviewer.
The cells used here imply greater variability than a cell line, but are also more relevant.
Indeed, the effects observed in the late stages of HIV-1 production are:
-
~80 % decrease in viral transcription compared to the control (Fig. 2I),
-
~85 % decrease in CAp24 protein expression compared to the control, as quantified by western blot (Fig. 2E), or ~90 % by ELISA measurement (Fig. 2F),
-
a reduction of more than 90 % in the release of infectious particles (Fig. 2G).
These results were all significant across donors, while SNAT7 depletion was always partial (Fig. 2C, between 31 to 62 % of depletion compared to the control in infected cells).
Therefore, the data were obtained from a mixture of depleted and non-depleted macrophages. This means that the results may be underestimated.
Together, our results show that SNAT7 is necessary for HIV-1 production.
However, reading the comments, we realized that our conclusions regarding reverse transcription were too strong. SNAT7 depletion does not affect viral fusion and reverse transcription. The manuscript was modified accordingly.
On top, there are a lot of optional experiments I am sure the authors are aware of that should be done at least in the future.
For instance, how does HIV-1 upregulate SNAT7, is a viral accessory protein involved?
What is the mechanism of SNAT7 dependent SAMHD1 phosphorylation? Does SNAT7 (or glutamine) regulate the activity of the SAMHD1 associated kinase / phosphatase) If so, does this impact on other targets of these enzymes?
We thank the reviewer for these questions.
To address the role of accessory viral proteins, we have already performed one experiment infecting hMDM with HIV-1 strains deleted for genes such as Nef, Vpr, Vpu and Vif, and have found no clear effect on SNAT7 protein expression compared to WT strains. As an alternative experiment, we could overexpress individual viral genes, such as Nef or Vpr, in HeLa cells and analyze their impact on SNAT7 expression by Western blot.
It is also possible that SNAT7 expression and recycling of lysosomal glutamine are modulated by the macrophage intrinsic immunity in response to HIV-1 infection.
The Thr592 motif of the SAMHD1 protein is phosphorylated by Cyclin A2/CDK1 and type 1 IFN in non-cycling cells, such as MDMs (Cribier et al., 2013). For now, the relationship between SNAT7 and SAMHD1 remains unclear. However, (Meng et al., 2022) demonstrated that SNAT7 positively regulates mTORC1 activity at the lysosomal membrane through release of lysosomal glutamine, and (Dias et al., 2024) showed that inhibiting mTORC1 activity decreases SAMHD1 Thr592 phosphorylation in hMDM. Therefore, we could speculate that the absence of SNAT7 down-regulates mTORC1 activity, which then leads to decreased SAMHD1 phosphorylation. This has been added to the discussion to explain the relationship between the 3 partners.
**Referees cross-commenting**
I think the comments from the other referees are reasonable and consistent with my assessment
Reviewer #1 (Significance (Required)):
Strength and limitations see above;
Significance: I think this work is of high interest for virologists working in the field of HIV-1 and infection of myeloid cells. In case SNAT7 (and hence glutamine) indeed regulates the phosphorylation of SAMHD1, there could potentially be broad relevance of this work. However unfortunately, this aspect remains underdeveloped and is also not discussed
Field of expertise: HIV-1, immunology, cell biology
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
In this report, Herit and colleagues describe the role of a HIV-1 dependency factor that promotes virus replication in macrophages. The authors suggest that the lysosomal membrane-associated SNAT7 glutamine transporter is a HIV dependency factor, that promotes virus replication by enhancing reverse transcription and Gag synthesis. The authors use transient knock-down approaches in primary macrophages to identify that SNAT7 depletion does not impact viral entry but inhibits early reverse transcription which was reversed by exogenous glutamine addition. While reverse transcription enhancement was likely due to selective increase in phosho-SAMHD1 expression, mechanisms by which SNAT7 enhanced viral gene expression were not clearly defined. These are well-controlled studies that pinpoint the role of SNAT7 in the early steps of viral life cycle and highlight the intricate interplay between macrophage metabolism and HIV-1 replication. While the question that is addressed is important, and the hypothesis overall sound, the data presented needs to be strengthened to support the conclusions. There are numerous weaknesses in data interpretation as well.
- Figure 1: SNAT7 expression was selectively enhanced upon differentiation of monocytes into macrophages but absent in CD4+ T cells. Though there is a claim of enhancement of SNAT7 expression upon HIV-1 infection of macrophages, RT-qPCR analysis shows the opposite trend (Fig 1E) and SNAT7 protein expression changes are modest. Statistical analysis in Fig. 1H needs to be revisited. The number of replicates vary for the lysates harvested at different day post infection, which might have an impact on the statistical test. To determine if SNAT7 expression enhancement is dependent on establishment of virus infection, as the authors imply, control lysates of virus infections in presence of replication inhibitors should be included.
We thank the reviewer for this comment. Indeed, there is a modest, but statistically significant increase in SNAT7 protein expression upon HIV-1 infection over time (Fig. 1G, H), without any modulation of SNAT7 gene expression (Fig. 1E). This indicates that the regulation of SNAT7 expression in this context is only at the translation level (i.e. increase of translation or stabilization of the SNAT7 protein).
As mentioned, Fig. 1H aggregates between 3 to 7 independent experiments on different donors depending on the infection time point. SNAT7 protein expression is increased already at 1 day post-infection and until 8 days. The statistical test used here, i.e. 2 way-ANOVA, compared Mock-infected and HIV-1-infected condition for each time point with the same number of donors. In this figure, the comparison is statistically different only at day 6 of the time course (7 donors). We agree that increasing the number of donors of the other time points could help to improve the statistical difference between control and infection condition.
We thank the reviewer for the suggestion mentioning the use of replication inhibitors in this experiment. We plan to use inhibitors of reverse transcription (Nevirapin) and integration (Dolutegravir).
The authors rely exclusively on western blot analysis for HIV-1 Gag expression in cell lysates as a measure of effects of SNAT7 on virus replication. Single cell analysis such as intracellular p24gag analysis by FACS should be included; this will provide a better measure of effects of SNAT7 onHIV-1 infection establishment.
We respectfully disagree with the reviewer for this question. Indeed, to evaluate the effects of SNAT7 on HIV-1 replication, we measured Gag Pr55 and Cap24 using a Western blot approach (Fig. 2B, D and E), but also assessed the quantity of Cap24 in the supernatants and lysates using an ELISA measurement, the quantity of infectious particles using TZM reporter cells, and total viral transcription or more specifically Gag Pr55 transcription using qPCR (Fig. 2F, G and I and Supp. Fig. 2G).
Regarding the quantification of CAp24 at the cell single level, please refer to comment #2 under Reviewer #1.
Knockdown of SNAT7 in MDMs was partial at best; only 30-50% decrease in expression (Fig 2C), but the effects on viral gene expression (Fig. 2I), p24 release and infectious particle production is dramatic (Fig. 2F and G). This discrepancy is not addressed. Does SNAT7 knock-down negatively impact virus particle release? Please note that the representative WB in Fig 2B does not correlate with the quantification in Fig. 2D. There are no p55gag or p24gag bands in SNAT7#1 siRNA condition (Fig. 2B)? Data could also be rearranged to follow the logical sequence of virus replication cycle (viral RNa expression followed by Gag expression, and then release).
We thank the reviewer for this comment. Our samples are indeed a mixture of SNAT7-depleted and non-depleted macrophages and RNA interference in these cells often leads to a decrease of 50 % of the protein expression.
To determine whether SNAT7 is involved in the release of particles, we quantified Cap24 in cell lysates and in the cell culture medium separately, and normalized the results to the total protein content. The absence of SNAT7 reduced the amount of Cap24 measured by ELISA in both samples to the same extent, showing that there is no storage of Cap24-positive viral particles inside the infected macrophages. These data were initially pooled in one graph (Fig. 2F), but separate graphs are now provided in new Supp. Fig. 2 E, F.
Regarding the western blot shown in Fig. 2B, please refer to comment #5 under Reviewer #1.
In the new version of the manuscript, we arranged the figures and placed the later stages of the viral cycle in Fig. 2 and the earlier stages, such as fusion, reverse transcription and transcription, in Fig. 3.
Data interpretation would be greatly improved by including infection controls (RT or integrase inhibitors) to confirm that measurements of viral RNA and Gag are indeed modulated by SNAT7 expression.
We thank the reviewer for this suggestion to include inhibitors of viral replication as controls. In our experiments, cells were Mock-infected in parallel as a negative control of viral detection. We provide the results in the new version of the manuscript to show that (i) there is no detection of viral or Gag RNA in the absence of the virus, (ii) the expression of viral genes measured in HIV-1-infected SNAT7-depleted cells is not different from Mock-infected cells, indicating almost complete inhibition of viral transcription (Fig. 3H and Supp. Fig. 3B), also confirmed at the protein level (Fig. 2B, D-F).
Figure 3: Decrease in SNAT7 expression in macrophages resulted in lower levels of early reverse transcripts. But surprisingly, LRT levels were not as affected by decreases in SNAT7 expression. The authors go on to suggest that decreases in early RT are due to loss of phospho-SAMHD1 and increases in catalytically active form of SAMHD1. Mechanistically this does not make sense: LRT should be similarly affected by increase in catalytically active SAMHD1. dNTP concentrations should be measured to determine if the rescue of RT is dependent on SAMHD1 dNTPase activity.
We thank the reviewer for this comment. LRT concentrations are very low in human macrophages and more challenging to detect than ERT concentrations. This might explain why the differences observed between the SNAT7-depleted and control conditions appear less pronounced for LRT than for ERT.
Furthermore, we cannot rule out the possibility that SNAT7 has a cumulative effect throughout the viral cycle. While reverse transcription remains statistically unaltered, and despite the reduced levels of ERT and LRT in SNAT7-depleted macrophages (Fig. 3 F, G), there is a significant impact on the transcription of viral RNAs (Fig. 2I) and Gag (Supp. Fig. 2G). This step may also be altered by the ribonuclease activity of SAMHD1 (Beloglazova et al., 2013; Ryoo et al., 2014).
Finally, with the help of Dr Baek Kim in Atlanta, we attempted to quantify dNTP concentrations in our human macrophages. Unfortunately, it was not possible to draw any conclusions, as the concentrations of dNTPs extracted from our cells were far too low.
Furthermore, it should be noted that SAMHD1 viral restriction through its phosphorylation at T592 is not correlated with its dNTPase activity (Welbourn et al., 2013; White et al., 2013), but with its ribonuclease activity (Beloglazova et al., 2013; Ryoo et al., 2014). This is supporting why SNAT7, by modulating the ribonuclease activity of SAMHD1, could have a greater effect on viral transcription than on reverse transcription.
There is lack of consistency in the data: p24 release upon SNAT7 depletion is highly variable. While there is a dramatic >90-95% decrease in p24 release (Fig. 2G), the effects are much more moderate in Fig. 4H (50-60% attenuation), even though siRNA-mediated depletion was similar across the data sets. The authors should comment on the variability in their findings.
We thank the reviewer for this comment, but believe that Figure 2E rather than Figure 2G is to be mentioned regarding the quantification of CAp24 by Western blot and to be compared with Figure 4H.
In Fig. 2E, we observed an average reduction of 85 % in CAp24 expression normalized to Clathrin HC expression across different donors for both siRNAs targeting SNAT7. For Fig. 4H, there was a 73 % reduction in CAp24 levels for siRNA #1 and a 56 % reduction for siRNA #2. In addition, it should be noted that the reduction in Gag levels is greater in Fig. 4G (between 77 % and 83 %) than in Fig. 2D (between 55 % and 72 %).
Therefore, there is some variation in the results obtained with the different donors, which could be explained by variations in Gag cleavage among donors, but this does not impact the conclusions for both figures.
SNAT7 is postulated to affect 2 steps in the virus life cycle: reverse transcription and viral transcription. But Vpx-mediated SAMHD1 degradation reversed both. Its not clear to me as to how SAMHD1 degradation impacts the role of SNAT7 in viral transcription. No explanation is provided.
We thank the reviewer for this comment. As suggested, we will perform experiments to assess the impact of Vpx-mediated SAMHD1 degradation on viral transcription.
Exogenous addition of glutamine only partially restored Gag synthesis and p24 release, which could be attributed to increased cytoplasmic levels and viral protein synthesis. What about effects on reverse transcription and viral gene expression?
We thank the reviewer for this comment. We will perform the suggested experiments to assess the impact of glutamine supplementation on viral transcription.
Reviewer #2 (Significance (Required)):
This is a novel finding, as there are limited number of studies on amino acid transporters and HIV-1 replication enhancement in macrophages. Most of the previous work has focused on CD4 T cells. These studies on SNAT7 and HIV-1 infection establishment in macrophages might better inform the influences of macrophage metabolism on HIV-1 persistence and inflammatory responses.
Reviewer #3 (Evidence, reproducibility and clarity (Required)):
This study investigates the role of the lysosomal glutamine transporter SLC38A7/SNAT7 in HIV‑1 replication in primary human macrophages. The authors demonstrate that SNAT7 is highly expressed in macrophages and upregulated upon HIV‑1 infection. They show that SNAT7 depletion inhibits HIV‑1 production at the reverse transcription step without affecting viral fusion or global cellular translation/transcription. Mechanistically, SNAT7 knockdown reduces the inhibitory phosphorylation of SAMHD1 at T592, and degradation of SAMHD1 by Vpx fully rescues viral replication. Extracellular glutamine supplementation partially restores HIV‑1 production in SNAT7‑deficient cells. Overall, the authors report interesting observations; however, the mechanistic investigation remains preliminary, raising concerns about whether the data fully support all the conclusions drawn.
Major Concerns:
1. The mechanistic depth is insufficient. The authors do not elucidate how glutamine regulates SAMHD1 T592 phosphorylation, whether through metabolite‑mediated control of kinases/phosphatases or via indirect effects.
We thank the reviewer for this comment. It is worth noting that (Meng et al., 2022) demonstrated that SNAT7 positively regulates mTORC1 activity at the lysosomal membrane through release of lysosomal glutamine, and (Dias et al., 2024) showed that inhibiting mTORC1 activity using drugs decreases SAMHD1 Thr592 phosphorylation in hMDM. Therefore, we could speculate that the absence of SNAT7 down-regulates mTORC1 activity, which then leads to decreased SAMHD1 phosphorylation. This is now further discussed in the discussion section of the manuscript.
The authors do not measure intracellular dNTP levels upon SNAT7 knockdown, which is the key functional substrate of SAMHD1. They also do not directly demonstrate that glutamine supplementation restores dNTP pools.
We thank the reviewer for this comment. Please, refer to comment #5 under Reviewer #2.
Extracellular glutamine only partially rescues viral production, implying the existence of transport‑independent functions of SNAT7 or additional pathways. This important observation is not discussed.
We thank the reviewer for this comment. The discussion has been modified accordingly.
It is suggested that the key findings be validated in immortalized THP‑1 cells differentiated into macrophage‑like cells by PMA.
We thank the reviewer for this suggestion but don’t really understand why this would strengthen our conclusions. Indeed, despite the known variability between donors and technical limitations to transduce cells, we chose human blood monocyte-derived macrophages as a relevant non-transformed model for HIV-1 infection of macrophages. They also represent to some extent the human diversity.
The Discussion section should be expanded to include the potential translational implications and limitations of the present study.
We thank the reviewer for this comment. The discussion points to some elements of potential translation and limitations of the study.
Reviewer #3 (Significance (Required)):
General assessment: This study identifies the lysosomal glutamine transporter SLC38A7/SNAT7 as a novel host dependency factor for HIV‑1 replication in primary human macrophages. The major strengths include the use of physiologically relevant primary macrophage models, a well-organized experimental pipeline from expression profiling to functional validation, and the establishment of a link between SNAT7, glutamine metabolism, and the HIV restriction factor SAMHD1.
Advance: It extends current understanding of HIV‑1 host dependency factors and immunometabolism by revealing a compartment‑specific metabolic pathway that supports viral reverse transcription.
Audience:This work will primarily interest specialized researchers in HIV‑1 biology, host-virus interactions, restriction factors, and antiviral innate immunity.
Reviewer #1 (Evidence, reproducibility and clarity (Required)):
This study from the Niedergang lab establishes SNAT7 as a host-dependency factor in human macrophages that supports HIV-1 replication. They show a modest increase in SNAT7 levels HIV-1 infected macrophages and suggest that SNAT7 levels are transiently increased. Employing siRNA against SNAT7 they show reduction in HIV-1 protein levels and viral RNAs and claim that there is a block of reverse transcription in SNAT7 KD cells. Focusing on a known HIV-1 restriction factor in macrophages, SAMHD1, they interconnect the SNAT7 depletion with a reduction in phosphorylated, i.e. catalytical inactive SAMHD1 arguing that SNAT7 regulates the phosphorylation and thereby antiviral activity of SAMHD1. Since SNAT7 is a glutamine transporter that provides this AA from lysosomes, they lastly supplement glutamine and this somehow rescues the reduction of HIV-1 production in SNAT7 KD cells.
Major comments:
The strength of this manuscript is the clear focus on primary human macrophages that are HIV-1 infected and the interconnection of HIV-1 replication to the SNAT7 siRNA KD experiments in combination with SAMHD1 depletion and lastly glutamine supplementation. This establishes a stringent and coherent story line.
The effects reported are modest; high variability is not a problem since using primary hMDM this is expected and can be addressed by testing several donors and applying stringent statistics.
- Having said so, I realize that while they give information on the statistical test used, i.e. one-way ANOVA they miss to explain the post-test used to assess significance (i.e. Bonferroni, Fishers LSD, whatsoever). Please add this information.
We thank the reviewer for this comment. The figure legends have been updated to include more details of all the statistical tests used.
- Another issue that might underestimate the effects of HIV-1 infection on SNAT7 levels and vice versa of SNAT7 KD on HIV-1 replication is the non-single cell approach employed, i.e. WBlots. I assume that HIV-1 infection rates in macrophages are not super high, usually not exceeding 20-30%. So indeed the effects the authors observe could be much higher, when checking at the single cell level. I do not know about the SNAT7 ab, but all the other reagents should work via flow cytometry and could hence improve the readout a lot.
We agree with the reviewer and indeed, in previous studies on HIV-1 infection of human macrophages performed in the lab, we observed via immunofluorescence that the proportion of infected cells ranged from 20 to 40 %. At the time of submission, we did not have the possibility to label the native SNAT7 protein by immunofluorescence, as the commercial antibody used only works for western blotting.
In the meantime, we have been validating a new antibody (Proteintech) targeting SNAT7 for immunofluorescence. If this is confirmed, we will be able to detect and quantify HIV-1 p24 by immunofluorescence in SNAT7-depleted human macrophages and control cells, thus confirming our results in single-cell analysis.
Flow cytometry analyses are difficult to perform on primary human macrophages because these cells are highly adherent and must be detached first. The process induces significant cell death and damage. This is why we would prefer to carry out these analyses using immunofluorescence and microscopy on adhered cells. This option will be undoubtedly pursued.
- Furthermore the authors never commented about a dose-response effect in terms of HIV-1 infection levels. There is a MOI dependency described for Suppl.Fig.1 C-F, unfortunately the data is missing in the manuscript.
We apologize for this omission. The figures showing the increase in SNAT7 protein expression following HIV-1 infection at MOIs ranging from 0.05 to 0.5 were added to the new version of the manuscript (Supp. Fig. 1 C-F).
- Figure1: specify circulating T lymphocytes. I would expect to see levels of SNAT7 in PHA or CD3/CD28 activated lymphocytes versus resting T cells and a time course of SNAT7 levels upon activation. I think even though SNAT7 levels in T cells might be low, they could also be increased by HIV-1 infection and it is essential that the authors test for this. If not, the result is a valid negative control. For this they should employ HIV-1 primary strains with a tropism for T cells, or at least lab-adapted HIV-1 NL4-3
We thank the reviewer for this comment. Circulating T lymphocytes isolated from the blood of healthy donors are now referred to resting lymphocytes in the new version of the manuscript, as opposed to activated T lymphocytes stimulated with IL2 and PHA-P for several days (Fig. 1 A-C).
The expression levels of SNAT7, both at the gene and protein levels, are lower in resting or IL2/PHA-P-activated T cells than in macrophages from the same donors. As suggested, we will perform a kinetic of T-cell activation upon HIV-1 infection to investigate how SNAT7 expression varies in these conditions.
- Figure 2 again single cell measurements could reveal much more pronounced effects; it is a bit counterintuitive that siRNA #2 is more efficient in SNAT7 KD but has higher levels of HIV-1 replication in terms of Gag levels. I assume when looking at the stats it is always a comparison to the Ctl treated cells (C-G), but this is not entirely clear. Unify labeling as compared to the stats in Fig.2 I (this also applies for all the other figs).
We thank the reviewer for this comment. Fig. 2B indeed shows one of the different donors analyzed. However, protein quantification across six different donors shows that SNAT7 is more depleted with siRNA #2 (Fig. 2C), and that Gag Pr55 protein levels are consequently more reduced, than with siRNA #1 (Fig. 2D).
We use GraphPad Prism software to perform statistical analysis. Depending on the test used, the software automatically plots the comparison bar and displays the p-value above it. We changed the representation of statistics as suggested.
Figure 3: It is a bit odd that they finally conclude on RT as essential step that is reduced in the absence of SNAT7 and then they fail to provide statistical significance for this (Fig.3 panels F and G). One would expect that RT is much more affected given the huge effects on HIV-1 capsid and particle production shown in Fig.2 F, G and I.
The reviewer is right in pointing that we observed a stronger effect during the later stages of the viral cycle, from transcription of viral RNAs (Fig. 2I and Supp. Fig. 2G) to the production of viral particles in the supernatant (Fig. 2D-G), than during the earlier stage of reverse transcription (Fig. 3F, G). Also, it is also possible that we might have missed the peak in ERT/LRT production, which is transient.
It should be noted that SAMHD1 exhibits both dNTPase (Goldstone et al., 2011) and nuclease (Beloglazova et al., 2013) activities. The ability of SAMHD1 to restrict the virus, through dephosphorylation at T592, is mediated by its RNase activity (Ryoo et al., 2014), and not by the dNTPase activity (Welbourn et al., 2013; White et al., 2013).This could explain why SNAT7 exhibit a stronger impact on viral transcription than on reverse transcription.
Figure 4; again single cell flow measurements of SAMHD1, pSAMHD1 and p24 /SNAT7 might help to more clearly discriminate effects that are specifically induced upon infection or happen in virally infected cells. Maybe alternatively IF?
We thank the reviewer for this suggestion. As mentioned under comment #2, flow cytometry analyses are difficult to perform on strongly adherent primary human macrophages.
With regard to immunofluorescence, there is a technical limitation based on the species in which the antibodies are produced. The antibody that targets the native SNAT7 protein, which is currently being validated in our laboratory, is produced in rabbits. An anti-CAp24 antibody produced in goats can be used. It will then be necessary to co-label the cells with anti SAMHD1 and phospho-SAMHD1produced in mouse. We will try to find options to co-label the cells.
The wblot shown in panel D does not really reflect the point the authors want to make by the quantification in panels G-I. Primary data (D) suggests that SNAT7 KD reduces HIV-1 production even in the absence of SAMHD1. The quantification rather indicates that SNAT7 KD does not affect HIV-1 production in the absence of SAMHD1. This needs clarification/corroboration by orthogonal approaches.
We respectfully disagree with the reviewer.
Figure 4D shows a representative blot of the six donors analysed. As mentioned, the depletion of SNAT7 in the absence of SAMHD1 reduces the production of the viral proteins GagPr55 and CAp24 (see Fig. 4D). This is illustrated by the quantifications (Fig. 4G–I). Following treatment with Vpx, GagPr55 protein expression in SNAT7 KD macrophages is reduced by a factor of 2.6 for siRNA #1 (mean = 1.48, light grey bar) and by a factor of 1.83 for siRNA #2 (mean = 2.13, orange bar), compared to the control (mean = 3.9, pink bar) (Fig. 4G). Similarly, CAp24 protein expression was reduced by a factor of 2.2 for siRNA #1 (mean = 2.05, light grey bar) and by a factor of 1.36 for siRNA #2 (mean = 3.34, orange bar), compared to the control (mean = 4.52, pink bar) (Fig. 4H).
These differences are therefore consistent between the Western blot and the quantifications. However, they are not significantly different to those observed in cells treated with Vpx and depleted with control siRNA, suggesting that the viral restriction observed in SNAT7 KD cells is primarily due to SAMHD1.
Figure 5: show SAMHD1 and pSAMHD1 levels upon glutamine supplementation.
We thank the reviewer for this comment, we will perform the suggested experiment.
- I think the discussion is very thin, mainly summarizing the results; but fails to give broader context or critically discuss the limitations and further directions.
We thank the reviewer for this comment. The discussion will be modified further accordingly.
Looking at the data as a whole, I think the results support a modest functional importance of SNAT7 for HIV-1 production in macrophages. I acknowledge that the experiments in primary macrophages are prone to high variability in different donors and the authors transparently depicted their data. However clearly, I would advice the authors to tune down the extend in which they claim SNAT7-dependency given this huge variability and the sometimes-borderline statistics.
We respectfully disagree with the reviewer.
The cells used here imply greater variability than a cell line, but are also more relevant.
Indeed, the effects observed in the late stages of HIV-1 production are:
-
~80 % decrease in viral transcription compared to the control (Fig. 2I),
-
~85 % decrease in CAp24 protein expression compared to the control, as quantified by western blot (Fig. 2E), or ~90 % by ELISA measurement (Fig. 2F),
-
a reduction of more than 90 % in the release of infectious particles (Fig. 2G).
These results were all significant across donors, while SNAT7 depletion was always partial (Fig. 2C, between 31 to 62 % of depletion compared to the control in infected cells).
Therefore, the data were obtained from a mixture of depleted and non-depleted macrophages. This means that the results may be underestimated.
Together, our results show that SNAT7 is necessary for HIV-1 production.
However, reading the comments, we realized that our conclusions regarding reverse transcription were too strong. SNAT7 depletion does not affect viral fusion and reverse transcription. The manuscript was modified accordingly.
On top, there are a lot of optional experiments I am sure the authors are aware of that should be done at least in the future.
For instance, how does HIV-1 upregulate SNAT7, is a viral accessory protein involved?
What is the mechanism of SNAT7 dependent SAMHD1 phosphorylation? Does SNAT7 (or glutamine) regulate the activity of the SAMHD1 associated kinase / phosphatase) If so, does this impact on other targets of these enzymes?
We thank the reviewer for these questions.
To address the role of accessory viral proteins, we have already performed one experiment infecting hMDM with HIV-1 strains deleted for genes such as Nef, Vpr, Vpu and Vif, and have found no clear effect on SNAT7 protein expression compared to WT strains. As an alternative experiment, we could overexpress individual viral genes, such as Nef or Vpr, in HeLa cells and analyze their impact on SNAT7 expression by Western blot.
It is also possible that SNAT7 expression and recycling of lysosomal glutamine are modulated by the macrophage intrinsic immunity in response to HIV-1 infection.
The Thr592 motif of the SAMHD1 protein is phosphorylated by Cyclin A2/CDK1 and type 1 IFN in non-cycling cells, such as MDMs (Cribier et al., 2013). For now, the relationship between SNAT7 and SAMHD1 remains unclear. However, (Meng et al., 2022) demonstrated that SNAT7 positively regulates mTORC1 activity at the lysosomal membrane through release of lysosomal glutamine, and (Dias et al., 2024) showed that inhibiting mTORC1 activity decreases SAMHD1 Thr592 phosphorylation in hMDM. Therefore, we could speculate that the absence of SNAT7 down-regulates mTORC1 activity, which then leads to decreased SAMHD1 phosphorylation. This has been added to the discussion to explain the relationship between the 3 partners.
**Referees cross-commenting**
I think the comments from the other referees are reasonable and consistent with my assessment
Reviewer #1 (Significance (Required)):
Strength and limitations see above;
Significance: I think this work is of high interest for virologists working in the field of HIV-1 and infection of myeloid cells. In case SNAT7 (and hence glutamine) indeed regulates the phosphorylation of SAMHD1, there could potentially be broad relevance of this work. However unfortunately, this aspect remains underdeveloped and is also not discussed
Field of expertise: HIV-1, immunology, cell biology
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
In this report, Herit and colleagues describe the role of a HIV-1 dependency factor that promotes virus replication in macrophages. The authors suggest that the lysosomal membrane-associated SNAT7 glutamine transporter is a HIV dependency factor, that promotes virus replication by enhancing reverse transcription and Gag synthesis. The authors use transient knock-down approaches in primary macrophages to identify that SNAT7 depletion does not impact viral entry but inhibits early reverse transcription which was reversed by exogenous glutamine addition. While reverse transcription enhancement was likely due to selective increase in phosho-SAMHD1 expression, mechanisms by which SNAT7 enhanced viral gene expression were not clearly defined. These are well-controlled studies that pinpoint the role of SNAT7 in the early steps of viral life cycle and highlight the intricate interplay between macrophage metabolism and HIV-1 replication. While the question that is addressed is important, and the hypothesis overall sound, the data presented needs to be strengthened to support the conclusions. There are numerous weaknesses in data interpretation as well.
- Figure 1: SNAT7 expression was selectively enhanced upon differentiation of monocytes into macrophages but absent in CD4+ T cells. Though there is a claim of enhancement of SNAT7 expression upon HIV-1 infection of macrophages, RT-qPCR analysis shows the opposite trend (Fig 1E) and SNAT7 protein expression changes are modest. Statistical analysis in Fig. 1H needs to be revisited. The number of replicates vary for the lysates harvested at different day post infection, which might have an impact on the statistical test. To determine if SNAT7 expression enhancement is dependent on establishment of virus infection, as the authors imply, control lysates of virus infections in presence of replication inhibitors should be included.
We thank the reviewer for this comment. Indeed, there is a modest, but statistically significant increase in SNAT7 protein expression upon HIV-1 infection over time (Fig. 1G, H), without any modulation of SNAT7 gene expression (Fig. 1E). This indicates that the regulation of SNAT7 expression in this context is only at the translation level (i.e. increase of translation or stabilization of the SNAT7 protein).
As mentioned, Fig. 1H aggregates between 3 to 7 independent experiments on different donors depending on the infection time point. SNAT7 protein expression is increased already at 1 day post-infection and until 8 days. The statistical test used here, i.e. 2 way-ANOVA, compared Mock-infected and HIV-1-infected condition for each time point with the same number of donors. In this figure, the comparison is statistically different only at day 6 of the time course (7 donors). We agree that increasing the number of donors of the other time points could help to improve the statistical difference between control and infection condition.
We thank the reviewer for the suggestion mentioning the use of replication inhibitors in this experiment. We plan to use inhibitors of reverse transcription (Nevirapin) and integration (Dolutegravir).
The authors rely exclusively on western blot analysis for HIV-1 Gag expression in cell lysates as a measure of effects of SNAT7 on virus replication. Single cell analysis such as intracellular p24gag analysis by FACS should be included; this will provide a better measure of effects of SNAT7 onHIV-1 infection establishment.
We respectfully disagree with the reviewer for this question. Indeed, to evaluate the effects of SNAT7 on HIV-1 replication, we measured Gag Pr55 and Cap24 using a Western blot approach (Fig. 2B, D and E), but also assessed the quantity of Cap24 in the supernatants and lysates using an ELISA measurement, the quantity of infectious particles using TZM reporter cells, and total viral transcription or more specifically Gag Pr55 transcription using qPCR (Fig. 2F, G and I and Supp. Fig. 2G).
Regarding the quantification of CAp24 at the cell single level, please refer to comment #2 under Reviewer #1.
Knockdown of SNAT7 in MDMs was partial at best; only 30-50% decrease in expression (Fig 2C), but the effects on viral gene expression (Fig. 2I), p24 release and infectious particle production is dramatic (Fig. 2F and G). This discrepancy is not addressed. Does SNAT7 knock-down negatively impact virus particle release? Please note that the representative WB in Fig 2B does not correlate with the quantification in Fig. 2D. There are no p55gag or p24gag bands in SNAT7#1 siRNA condition (Fig. 2B)? Data could also be rearranged to follow the logical sequence of virus replication cycle (viral RNa expression followed by Gag expression, and then release).
We thank the reviewer for this comment. Our samples are indeed a mixture of SNAT7-depleted and non-depleted macrophages and RNA interference in these cells often leads to a decrease of 50 % of the protein expression.
To determine whether SNAT7 is involved in the release of particles, we quantified Cap24 in cell lysates and in the cell culture medium separately, and normalized the results to the total protein content. The absence of SNAT7 reduced the amount of Cap24 measured by ELISA in both samples to the same extent, showing that there is no storage of Cap24-positive viral particles inside the infected macrophages. These data were initially pooled in one graph (Fig. 2F), but separate graphs are now provided in new Supp. Fig. 2 E, F.
Regarding the western blot shown in Fig. 2B, please refer to comment #5 under Reviewer #1.
In the new version of the manuscript, we arranged the figures and placed the later stages of the viral cycle in Fig. 2 and the earlier stages, such as fusion, reverse transcription and transcription, in Fig. 3.
Data interpretation would be greatly improved by including infection controls (RT or integrase inhibitors) to confirm that measurements of viral RNA and Gag are indeed modulated by SNAT7 expression.
We thank the reviewer for this suggestion to include inhibitors of viral replication as controls. In our experiments, cells were Mock-infected in parallel as a negative control of viral detection. We provide the results in the new version of the manuscript to show that (i) there is no detection of viral or Gag RNA in the absence of the virus, (ii) the expression of viral genes measured in HIV-1-infected SNAT7-depleted cells is not different from Mock-infected cells, indicating almost complete inhibition of viral transcription (Fig. 3H and Supp. Fig. 3B), also confirmed at the protein level (Fig. 2B, D-F).
Figure 3: Decrease in SNAT7 expression in macrophages resulted in lower levels of early reverse transcripts. But surprisingly, LRT levels were not as affected by decreases in SNAT7 expression. The authors go on to suggest that decreases in early RT are due to loss of phospho-SAMHD1 and increases in catalytically active form of SAMHD1. Mechanistically this does not make sense: LRT should be similarly affected by increase in catalytically active SAMHD1. dNTP concentrations should be measured to determine if the rescue of RT is dependent on SAMHD1 dNTPase activity.
We thank the reviewer for this comment. LRT concentrations are very low in human macrophages and more challenging to detect than ERT concentrations. This might explain why the differences observed between the SNAT7-depleted and control conditions appear less pronounced for LRT than for ERT.
Furthermore, we cannot rule out the possibility that SNAT7 has a cumulative effect throughout the viral cycle. While reverse transcription remains statistically unaltered, and despite the reduced levels of ERT and LRT in SNAT7-depleted macrophages (Fig. 3 F, G), there is a significant impact on the transcription of viral RNAs (Fig. 2I) and Gag (Supp. Fig. 2G). This step may also be altered by the ribonuclease activity of SAMHD1 (Beloglazova et al., 2013; Ryoo et al., 2014).
Finally, with the help of Dr Baek Kim in Atlanta, we attempted to quantify dNTP concentrations in our human macrophages. Unfortunately, it was not possible to draw any conclusions, as the concentrations of dNTPs extracted from our cells were far too low.
Furthermore, it should be noted that SAMHD1 viral restriction through its phosphorylation at T592 is not correlated with its dNTPase activity (Welbourn et al., 2013; White et al., 2013), but with its ribonuclease activity (Beloglazova et al., 2013; Ryoo et al., 2014). This is supporting why SNAT7, by modulating the ribonuclease activity of SAMHD1, could have a greater effect on viral transcription than on reverse transcription.
There is lack of consistency in the data: p24 release upon SNAT7 depletion is highly variable. While there is a dramatic >90-95% decrease in p24 release (Fig. 2G), the effects are much more moderate in Fig. 4H (50-60% attenuation), even though siRNA-mediated depletion was similar across the data sets. The authors should comment on the variability in their findings.
We thank the reviewer for this comment, but believe that Figure 2E rather than Figure 2G is to be mentioned regarding the quantification of CAp24 by Western blot and to be compared with Figure 4H.
In Fig. 2E, we observed an average reduction of 85 % in CAp24 expression normalized to Clathrin HC expression across different donors for both siRNAs targeting SNAT7. For Fig. 4H, there was a 73 % reduction in CAp24 levels for siRNA #1 and a 56 % reduction for siRNA #2. In addition, it should be noted that the reduction in Gag levels is greater in Fig. 4G (between 77 % and 83 %) than in Fig. 2D (between 55 % and 72 %).
Therefore, there is some variation in the results obtained with the different donors, which could be explained by variations in Gag cleavage among donors, but this does not impact the conclusions for both figures.
SNAT7 is postulated to affect 2 steps in the virus life cycle: reverse transcription and viral transcription. But Vpx-mediated SAMHD1 degradation reversed both. Its not clear to me as to how SAMHD1 degradation impacts the role of SNAT7 in viral transcription. No explanation is provided.
We thank the reviewer for this comment. As suggested, we will perform experiments to assess the impact of Vpx-mediated SAMHD1 degradation on viral transcription.
Exogenous addition of glutamine only partially restored Gag synthesis and p24 release, which could be attributed to increased cytoplasmic levels and viral protein synthesis. What about effects on reverse transcription and viral gene expression?
We thank the reviewer for this comment. We will perform the suggested experiments to assess the impact of glutamine supplementation on viral transcription.
Reviewer #2 (Significance (Required)):
This is a novel finding, as there are limited number of studies on amino acid transporters and HIV-1 replication enhancement in macrophages. Most of the previous work has focused on CD4 T cells. These studies on SNAT7 and HIV-1 infection establishment in macrophages might better inform the influences of macrophage metabolism on HIV-1 persistence and inflammatory responses.
Reviewer #3 (Evidence, reproducibility and clarity (Required)):
This study investigates the role of the lysosomal glutamine transporter SLC38A7/SNAT7 in HIV‑1 replication in primary human macrophages. The authors demonstrate that SNAT7 is highly expressed in macrophages and upregulated upon HIV‑1 infection. They show that SNAT7 depletion inhibits HIV‑1 production at the reverse transcription step without affecting viral fusion or global cellular translation/transcription. Mechanistically, SNAT7 knockdown reduces the inhibitory phosphorylation of SAMHD1 at T592, and degradation of SAMHD1 by Vpx fully rescues viral replication. Extracellular glutamine supplementation partially restores HIV‑1 production in SNAT7‑deficient cells. Overall, the authors report interesting observations; however, the mechanistic investigation remains preliminary, raising concerns about whether the data fully support all the conclusions drawn.
Major Concerns:
1. The mechanistic depth is insufficient. The authors do not elucidate how glutamine regulates SAMHD1 T592 phosphorylation, whether through metabolite‑mediated control of kinases/phosphatases or via indirect effects.
We thank the reviewer for this comment. It is worth noting that (Meng et al., 2022) demonstrated that SNAT7 positively regulates mTORC1 activity at the lysosomal membrane through release of lysosomal glutamine, and (Dias et al., 2024) showed that inhibiting mTORC1 activity using drugs decreases SAMHD1 Thr592 phosphorylation in hMDM. Therefore, we could speculate that the absence of SNAT7 down-regulates mTORC1 activity, which then leads to decreased SAMHD1 phosphorylation. This is now further discussed in the discussion section of the manuscript.
The authors do not measure intracellular dNTP levels upon SNAT7 knockdown, which is the key functional substrate of SAMHD1. They also do not directly demonstrate that glutamine supplementation restores dNTP pools.
We thank the reviewer for this comment. Please, refer to comment #5 under Reviewer #2.
Extracellular glutamine only partially rescues viral production, implying the existence of transport‑independent functions of SNAT7 or additional pathways. This important observation is not discussed.
We thank the reviewer for this comment. The discussion has been modified accordingly.
It is suggested that the key findings be validated in immortalized THP‑1 cells differentiated into macrophage‑like cells by PMA.
We thank the reviewer for this suggestion but don’t really understand why this would strengthen our conclusions. Indeed, despite the known variability between donors and technical limitations to transduce cells, we chose human blood monocyte-derived macrophages as a relevant non-transformed model for HIV-1 infection of macrophages. They also represent to some extent the human diversity.
The Discussion section should be expanded to include the potential translational implications and limitations of the present study.
We thank the reviewer for this comment. The discussion points to some elements of potential translation and limitations of the study.
Reviewer #3 (Significance (Required)):
General assessment: This study identifies the lysosomal glutamine transporter SLC38A7/SNAT7 as a novel host dependency factor for HIV‑1 replication in primary human macrophages. The major strengths include the use of physiologically relevant primary macrophage models, a well-organized experimental pipeline from expression profiling to functional validation, and the establishment of a link between SNAT7, glutamine metabolism, and the HIV restriction factor SAMHD1.
Advance: It extends current understanding of HIV‑1 host dependency factors and immunometabolism by revealing a compartment‑specific metabolic pathway that supports viral reverse transcription.
Audience:This work will primarily interest specialized researchers in HIV‑1 biology, host-virus interactions, restriction factors, and antiviral innate immunity.
2.15.1.0
Reviewer #1 (Evidence, reproducibility and clarity (Required)):
This study from the Niedergang lab establishes SNAT7 as a host-dependency factor in human macrophages that supports HIV-1 replication. They show a modest increase in SNAT7 levels HIV-1 infected macrophages and suggest that SNAT7 levels are transiently increased. Employing siRNA against SNAT7 they show reduction in HIV-1 protein levels and viral RNAs and claim that there is a block of reverse transcription in SNAT7 KD cells. Focusing on a known HIV-1 restriction factor in macrophages, SAMHD1, they interconnect the SNAT7 depletion with a reduction in phosphorylated, i.e. catalytical inactive SAMHD1 arguing that SNAT7 regulates the phosphorylation and thereby antiviral activity of SAMHD1. Since SNAT7 is a glutamine transporter that provides this AA from lysosomes, they lastly supplement glutamine and this somehow rescues the reduction of HIV-1 production in SNAT7 KD cells.
Major comments:
The strength of this manuscript is the clear focus on primary human macrophages that are HIV-1 infected and the interconnection of HIV-1 replication to the SNAT7 siRNA KD experiments in combination with SAMHD1 depletion and lastly glutamine supplementation. This establishes a stringent and coherent story line.
The effects reported are modest; high variability is not a problem since using primary hMDM this is expected and can be addressed by testing several donors and applying stringent statistics.
- Having said so, I realize that while they give information on the statistical test used, i.e. one-way ANOVA they miss to explain the post-test used to assess significance (i.e. Bonferroni, Fishers LSD, whatsoever). Please add this information.
We thank the reviewer for this comment. The figure legends have been updated to include more details of all the statistical tests used.
- Another issue that might underestimate the effects of HIV-1 infection on SNAT7 levels and vice versa of SNAT7 KD on HIV-1 replication is the non-single cell approach employed, i.e. WBlots. I assume that HIV-1 infection rates in macrophages are not super high, usually not exceeding 20-30%. So indeed the effects the authors observe could be much higher, when checking at the single cell level. I do not know about the SNAT7 ab, but all the other reagents should work via flow cytometry and could hence improve the readout a lot.
We agree with the reviewer and indeed, in previous studies on HIV-1 infection of human macrophages performed in the lab, we observed via immunofluorescence that the proportion of infected cells ranged from 20 to 40 %. At the time of submission, we did not have the possibility to label the native SNAT7 protein by immunofluorescence, as the commercial antibody used only works for western blotting.
In the meantime, we have been validating a new antibody (Proteintech) targeting SNAT7 for immunofluorescence. If this is confirmed, we will be able to detect and quantify HIV-1 p24 by immunofluorescence in SNAT7-depleted human macrophages and control cells, thus confirming our results in single-cell analysis.
Flow cytometry analyses are difficult to perform on primary human macrophages because these cells are highly adherent and must be detached first. The process induces significant cell death and damage. This is why we would prefer to carry out these analyses using immunofluorescence and microscopy on adhered cells. This option will be undoubtedly pursued.
- Furthermore the authors never commented about a dose-response effect in terms of HIV-1 infection levels. There is a MOI dependency described for Suppl.Fig.1 C-F, unfortunately the data is missing in the manuscript.
We apologize for this omission. The figures showing the increase in SNAT7 protein expression following HIV-1 infection at MOIs ranging from 0.05 to 0.5 were added to the new version of the manuscript (Supp. Fig. 1 C-F).
- Figure1: specify circulating T lymphocytes. I would expect to see levels of SNAT7 in PHA or CD3/CD28 activated lymphocytes versus resting T cells and a time course of SNAT7 levels upon activation. I think even though SNAT7 levels in T cells might be low, they could also be increased by HIV-1 infection and it is essential that the authors test for this. If not, the result is a valid negative control. For this they should employ HIV-1 primary strains with a tropism for T cells, or at least lab-adapted HIV-1 NL4-3
We thank the reviewer for this comment. Circulating T lymphocytes isolated from the blood of healthy donors are now referred to resting lymphocytes in the new version of the manuscript, as opposed to activated T lymphocytes stimulated with IL2 and PHA-P for several days (Fig. 1 A-C).
The expression levels of SNAT7, both at the gene and protein levels, are lower in resting or IL2/PHA-P-activated T cells than in macrophages from the same donors. As suggested, we will perform a kinetic of T-cell activation upon HIV-1 infection to investigate how SNAT7 expression varies in these conditions.
- Figure 2 again single cell measurements could reveal much more pronounced effects; it is a bit counterintuitive that siRNA #2 is more efficient in SNAT7 KD but has higher levels of HIV-1 replication in terms of Gag levels. I assume when looking at the stats it is always a comparison to the Ctl treated cells (C-G), but this is not entirely clear. Unify labeling as compared to the stats in Fig.2 I (this also applies for all the other figs).
We thank the reviewer for this comment. Fig. 2B indeed shows one of the different donors analyzed. However, protein quantification across six different donors shows that SNAT7 is more depleted with siRNA #2 (Fig. 2C), and that Gag Pr55 protein levels are consequently more reduced, than with siRNA #1 (Fig. 2D).
We use GraphPad Prism software to perform statistical analysis. Depending on the test used, the software automatically plots the comparison bar and displays the p-value above it. We changed the representation of statistics as suggested.
Figure 3: It is a bit odd that they finally conclude on RT as essential step that is reduced in the absence of SNAT7 and then they fail to provide statistical significance for this (Fig.3 panels F and G). One would expect that RT is much more affected given the huge effects on HIV-1 capsid and particle production shown in Fig.2 F, G and I.
The reviewer is right in pointing that we observed a stronger effect during the later stages of the viral cycle, from transcription of viral RNAs (Fig. 2I and Supp. Fig. 2G) to the production of viral particles in the supernatant (Fig. 2D-G), than during the earlier stage of reverse transcription (Fig. 3F, G). Also, it is also possible that we might have missed the peak in ERT/LRT production, which is transient.
It should be noted that SAMHD1 exhibits both dNTPase (Goldstone et al., 2011) and nuclease (Beloglazova et al., 2013) activities. The ability of SAMHD1 to restrict the virus, through dephosphorylation at T592, is mediated by its RNase activity (Ryoo et al., 2014), and not by the dNTPase activity (Welbourn et al., 2013; White et al., 2013).This could explain why SNAT7 exhibit a stronger impact on viral transcription than on reverse transcription.
Figure 4; again single cell flow measurements of SAMHD1, pSAMHD1 and p24 /SNAT7 might help to more clearly discriminate effects that are specifically induced upon infection or happen in virally infected cells. Maybe alternatively IF?
We thank the reviewer for this suggestion. As mentioned under comment #2, flow cytometry analyses are difficult to perform on strongly adherent primary human macrophages.
With regard to immunofluorescence, there is a technical limitation based on the species in which the antibodies are produced. The antibody that targets the native SNAT7 protein, which is currently being validated in our laboratory, is produced in rabbits. An anti-CAp24 antibody produced in goats can be used. It will then be necessary to co-label the cells with anti SAMHD1 and phospho-SAMHD1produced in mouse. We will try to find options to co-label the cells.
The wblot shown in panel D does not really reflect the point the authors want to make by the quantification in panels G-I. Primary data (D) suggests that SNAT7 KD reduces HIV-1 production even in the absence of SAMHD1. The quantification rather indicates that SNAT7 KD does not affect HIV-1 production in the absence of SAMHD1. This needs clarification/corroboration by orthogonal approaches.
We respectfully disagree with the reviewer.
Figure 4D shows a representative blot of the six donors analysed. As mentioned, the depletion of SNAT7 in the absence of SAMHD1 reduces the production of the viral proteins GagPr55 and CAp24 (see Fig. 4D). This is illustrated by the quantifications (Fig. 4G–I). Following treatment with Vpx, GagPr55 protein expression in SNAT7 KD macrophages is reduced by a factor of 2.6 for siRNA #1 (mean = 1.48, light grey bar) and by a factor of 1.83 for siRNA #2 (mean = 2.13, orange bar), compared to the control (mean = 3.9, pink bar) (Fig. 4G). Similarly, CAp24 protein expression was reduced by a factor of 2.2 for siRNA #1 (mean = 2.05, light grey bar) and by a factor of 1.36 for siRNA #2 (mean = 3.34, orange bar), compared to the control (mean = 4.52, pink bar) (Fig. 4H).
These differences are therefore consistent between the Western blot and the quantifications. However, they are not significantly different to those observed in cells treated with Vpx and depleted with control siRNA, suggesting that the viral restriction observed in SNAT7 KD cells is primarily due to SAMHD1.
- Figure 5: show SAMHD1 and pSAMHD1 levels upon glutamine supplementation.
We thank the reviewer for this comment, we will perform the suggested experiment.
- I think the discussion is very thin, mainly summarizing the results; but fails to give broader context or critically discuss the limitations and further directions.
We thank the reviewer for this comment. The discussion will be modified further accordingly.
Looking at the data as a whole, I think the results support a modest functional importance of SNAT7 for HIV-1 production in macrophages. I acknowledge that the experiments in primary macrophages are prone to high variability in different donors and the authors transparently depicted their data. However clearly, I would advice the authors to tune down the extend in which they claim SNAT7-dependency given this huge variability and the sometimes-borderline statistics.
We respectfully disagree with the reviewer.
The cells used here imply greater variability than a cell line, but are also more relevant.
Indeed, the effects observed in the late stages of HIV-1 production are:
-
~80 % decrease in viral transcription compared to the control (Fig. 2I),
-
~85 % decrease in CAp24 protein expression compared to the control, as quantified by western blot (Fig. 2E), or ~90 % by ELISA measurement (Fig. 2F),
-
a reduction of more than 90 % in the release of infectious particles (Fig. 2G).
These results were all significant across donors, while SNAT7 depletion was always partial (Fig. 2C, between 31 to 62 % of depletion compared to the control in infected cells).
Therefore, the data were obtained from a mixture of depleted and non-depleted macrophages. This means that the results may be underestimated.
Together, our results show that SNAT7 is necessary for HIV-1 production.
However, reading the comments, we realized that our conclusions regarding reverse transcription were too strong. SNAT7 depletion does not affect viral fusion and reverse transcription. The manuscript was modified accordingly.
On top, there are a lot of optional experiments I am sure the authors are aware of that should be done at least in the future.
For instance, how does HIV-1 upregulate SNAT7, is a viral accessory protein involved?
What is the mechanism of SNAT7 dependent SAMHD1 phosphorylation? Does SNAT7 (or glutamine) regulate the activity of the SAMHD1 associated kinase / phosphatase) If so, does this impact on other targets of these enzymes?
We thank the reviewer for these questions.
To address the role of accessory viral proteins, we have already performed one experiment infecting hMDM with HIV-1 strains deleted for genes such as Nef, Vpr, Vpu and Vif, and have found no clear effect on SNAT7 protein expression compared to WT strains. As an alternative experiment, we could overexpress individual viral genes, such as Nef or Vpr, in HeLa cells and analyze their impact on SNAT7 expression by Western blot.
It is also possible that SNAT7 expression and recycling of lysosomal glutamine are modulated by the macrophage intrinsic immunity in response to HIV-1 infection.
The Thr592 motif of the SAMHD1 protein is phosphorylated by Cyclin A2/CDK1 and type 1 IFN in non-cycling cells, such as MDMs (Cribier et al., 2013). For now, the relationship between SNAT7 and SAMHD1 remains unclear. However, (Meng et al., 2022) demonstrated that SNAT7 positively regulates mTORC1 activity at the lysosomal membrane through release of lysosomal glutamine, and (Dias et al., 2024) showed that inhibiting mTORC1 activity decreases SAMHD1 Thr592 phosphorylation in hMDM. Therefore, we could speculate that the absence of SNAT7 down-regulates mTORC1 activity, which then leads to decreased SAMHD1 phosphorylation. This has been added to the discussion to explain the relationship between the 3 partners.
**Referees cross-commenting**
I think the comments from the other referees are reasonable and consistent with my assessment
Reviewer #1 (Significance (Required)):
Strength and limitations see above;
Significance: I think this work is of high interest for virologists working in the field of HIV-1 and infection of myeloid cells. In case SNAT7 (and hence glutamine) indeed regulates the phosphorylation of SAMHD1, there could potentially be broad relevance of this work. However unfortunately, this aspect remains underdeveloped and is also not discussed
Field of expertise: HIV-1, immunology, cell biology
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
In this report, Herit and colleagues describe the role of a HIV-1 dependency factor that promotes virus replication in macrophages. The authors suggest that the lysosomal membrane-associated SNAT7 glutamine transporter is a HIV dependency factor, that promotes virus replication by enhancing reverse transcription and Gag synthesis. The authors use transient knock-down approaches in primary macrophages to identify that SNAT7 depletion does not impact viral entry but inhibits early reverse transcription which was reversed by exogenous glutamine addition. While reverse transcription enhancement was likely due to selective increase in phosho-SAMHD1 expression, mechanisms by which SNAT7 enhanced viral gene expression were not clearly defined. These are well-controlled studies that pinpoint the role of SNAT7 in the early steps of viral life cycle and highlight the intricate interplay between macrophage metabolism and HIV-1 replication. While the question that is addressed is important, and the hypothesis overall sound, the data presented needs to be strengthened to support the conclusions. There are numerous weaknesses in data interpretation as well.
- Figure 1: SNAT7 expression was selectively enhanced upon differentiation of monocytes into macrophages but absent in CD4+ T cells. Though there is a claim of enhancement of SNAT7 expression upon HIV-1 infection of macrophages, RT-qPCR analysis shows the opposite trend (Fig 1E) and SNAT7 protein expression changes are modest. Statistical analysis in Fig. 1H needs to be revisited. The number of replicates vary for the lysates harvested at different day post infection, which might have an impact on the statistical test. To determine if SNAT7 expression enhancement is dependent on establishment of virus infection, as the authors imply, control lysates of virus infections in presence of replication inhibitors should be included.
We thank the reviewer for this comment. Indeed, there is a modest, but statistically significant increase in SNAT7 protein expression upon HIV-1 infection over time (Fig. 1G, H), without any modulation of SNAT7 gene expression (Fig. 1E). This indicates that the regulation of SNAT7 expression in this context is only at the translation level (i.e. increase of translation or stabilization of the SNAT7 protein).
As mentioned, Fig. 1H aggregates between 3 to 7 independent experiments on different donors depending on the infection time point. SNAT7 protein expression is increased already at 1 day post-infection and until 8 days. The statistical test used here, i.e. 2 way-ANOVA, compared Mock-infected and HIV-1-infected condition for each time point with the same number of donors. In this figure, the comparison is statistically different only at day 6 of the time course (7 donors). We agree that increasing the number of donors of the other time points could help to improve the statistical difference between control and infection condition.
We thank the reviewer for the suggestion mentioning the use of replication inhibitors in this experiment. We plan to use inhibitors of reverse transcription (Nevirapin) and integration (Dolutegravir).
The authors rely exclusively on western blot analysis for HIV-1 Gag expression in cell lysates as a measure of effects of SNAT7 on virus replication. Single cell analysis such as intracellular p24gag analysis by FACS should be included; this will provide a better measure of effects of SNAT7 onHIV-1 infection establishment.
We respectfully disagree with the reviewer for this question. Indeed, to evaluate the effects of SNAT7 on HIV-1 replication, we measured Gag Pr55 and Cap24 using a Western blot approach (Fig. 2B, D and E), but also assessed the quantity of Cap24 in the supernatants and lysates using an ELISA measurement, the quantity of infectious particles using TZM reporter cells, and total viral transcription or more specifically Gag Pr55 transcription using qPCR (Fig. 2F, G and I and Supp. Fig. 2G).
Regarding the quantification of CAp24 at the cell single level, please refer to comment #2 under Reviewer #1.
Knockdown of SNAT7 in MDMs was partial at best; only 30-50% decrease in expression (Fig 2C), but the effects on viral gene expression (Fig. 2I), p24 release and infectious particle production is dramatic (Fig. 2F and G). This discrepancy is not addressed. Does SNAT7 knock-down negatively impact virus particle release? Please note that the representative WB in Fig 2B does not correlate with the quantification in Fig. 2D. There are no p55gag or p24gag bands in SNAT7#1 siRNA condition (Fig. 2B)? Data could also be rearranged to follow the logical sequence of virus replication cycle (viral RNa expression followed by Gag expression, and then release).
We thank the reviewer for this comment. Our samples are indeed a mixture of SNAT7-depleted and non-depleted macrophages and RNA interference in these cells often leads to a decrease of 50 % of the protein expression.
To determine whether SNAT7 is involved in the release of particles, we quantified Cap24 in cell lysates and in the cell culture medium separately, and normalized the results to the total protein content. The absence of SNAT7 reduced the amount of Cap24 measured by ELISA in both samples to the same extent, showing that there is no storage of Cap24-positive viral particles inside the infected macrophages. These data were initially pooled in one graph (Fig. 2F), but separate graphs are now provided in new Supp. Fig. 2 E, F.
Regarding the western blot shown in Fig. 2B, please refer to comment #5 under Reviewer #1.
In the new version of the manuscript, we arranged the figures and placed the later stages of the viral cycle in Fig. 2 and the earlier stages, such as fusion, reverse transcription and transcription, in Fig. 3.
Data interpretation would be greatly improved by including infection controls (RT or integrase inhibitors) to confirm that measurements of viral RNA and Gag are indeed modulated by SNAT7 expression.
We thank the reviewer for this suggestion to include inhibitors of viral replication as controls. In our experiments, cells were Mock-infected in parallel as a negative control of viral detection. We provide the results in the new version of the manuscript to show that (i) there is no detection of viral or Gag RNA in the absence of the virus, (ii) the expression of viral genes measured in HIV-1-infected SNAT7-depleted cells is not different from Mock-infected cells, indicating almost complete inhibition of viral transcription (Fig. 3H and Supp. Fig. 3B), also confirmed at the protein level (Fig. 2B, D-F).
Figure 3: Decrease in SNAT7 expression in macrophages resulted in lower levels of early reverse transcripts. But surprisingly, LRT levels were not as affected by decreases in SNAT7 expression. The authors go on to suggest that decreases in early RT are due to loss of phospho-SAMHD1 and increases in catalytically active form of SAMHD1. Mechanistically this does not make sense: LRT should be similarly affected by increase in catalytically active SAMHD1. dNTP concentrations should be measured to determine if the rescue of RT is dependent on SAMHD1 dNTPase activity.
We thank the reviewer for this comment. LRT concentrations are very low in human macrophages and more challenging to detect than ERT concentrations. This might explain why the differences observed between the SNAT7-depleted and control conditions appear less pronounced for LRT than for ERT.
Furthermore, we cannot rule out the possibility that SNAT7 has a cumulative effect throughout the viral cycle. While reverse transcription remains statistically unaltered, and despite the reduced levels of ERT and LRT in SNAT7-depleted macrophages (Fig. 3 F, G), there is a significant impact on the transcription of viral RNAs (Fig. 2I) and Gag (Supp. Fig. 2G). This step may also be altered by the ribonuclease activity of SAMHD1 (Beloglazova et al., 2013; Ryoo et al., 2014).
Finally, with the help of Dr Baek Kim in Atlanta, we attempted to quantify dNTP concentrations in our human macrophages. Unfortunately, it was not possible to draw any conclusions, as the concentrations of dNTPs extracted from our cells were far too low.
Furthermore, it should be noted that SAMHD1 viral restriction through its phosphorylation at T592 is not correlated with its dNTPase activity (Welbourn et al., 2013; White et al., 2013), but with its ribonuclease activity (Beloglazova et al., 2013; Ryoo et al., 2014). This is supporting why SNAT7, by modulating the ribonuclease activity of SAMHD1, could have a greater effect on viral transcription than on reverse transcription.
There is lack of consistency in the data: p24 release upon SNAT7 depletion is highly variable. While there is a dramatic >90-95% decrease in p24 release (Fig. 2G), the effects are much more moderate in Fig. 4H (50-60% attenuation), even though siRNA-mediated depletion was similar across the data sets. The authors should comment on the variability in their findings.
We thank the reviewer for this comment, but believe that Figure 2E rather than Figure 2G is to be mentioned regarding the quantification of CAp24 by Western blot and to be compared with Figure 4H.
In Fig. 2E, we observed an average reduction of 85 % in CAp24 expression normalized to Clathrin HC expression across different donors for both siRNAs targeting SNAT7. For Fig. 4H, there was a 73 % reduction in CAp24 levels for siRNA #1 and a 56 % reduction for siRNA #2. In addition, it should be noted that the reduction in Gag levels is greater in Fig. 4G (between 77 % and 83 %) than in Fig. 2D (between 55 % and 72 %).
Therefore, there is some variation in the results obtained with the different donors, which could be explained by variations in Gag cleavage among donors, but this does not impact the conclusions for both figures.
SNAT7 is postulated to affect 2 steps in the virus life cycle: reverse transcription and viral transcription. But Vpx-mediated SAMHD1 degradation reversed both. Its not clear to me as to how SAMHD1 degradation impacts the role of SNAT7 in viral transcription. No explanation is provided.
We thank the reviewer for this comment. As suggested, we will perform experiments to assess the impact of Vpx-mediated SAMHD1 degradation on viral transcription.
Exogenous addition of glutamine only partially restored Gag synthesis and p24 release, which could be attributed to increased cytoplasmic levels and viral protein synthesis. What about effects on reverse transcription and viral gene expression?
We thank the reviewer for this comment. We will perform the suggested experiments to assess the impact of glutamine supplementation on viral transcription.
Reviewer #2 (Significance (Required)):
This is a novel finding, as there are limited number of studies on amino acid transporters and HIV-1 replication enhancement in macrophages. Most of the previous work has focused on CD4 T cells. These studies on SNAT7 and HIV-1 infection establishment in macrophages might better inform the influences of macrophage metabolism on HIV-1 persistence and inflammatory responses.
Reviewer #3 (Evidence, reproducibility and clarity (Required)):
This study investigates the role of the lysosomal glutamine transporter SLC38A7/SNAT7 in HIV‑1 replication in primary human macrophages. The authors demonstrate that SNAT7 is highly expressed in macrophages and upregulated upon HIV‑1 infection. They show that SNAT7 depletion inhibits HIV‑1 production at the reverse transcription step without affecting viral fusion or global cellular translation/transcription. Mechanistically, SNAT7 knockdown reduces the inhibitory phosphorylation of SAMHD1 at T592, and degradation of SAMHD1 by Vpx fully rescues viral replication. Extracellular glutamine supplementation partially restores HIV‑1 production in SNAT7‑deficient cells. Overall, the authors report interesting observations; however, the mechanistic investigation remains preliminary, raising concerns about whether the data fully support all the conclusions drawn.
Major Concerns:
1. The mechanistic depth is insufficient. The authors do not elucidate how glutamine regulates SAMHD1 T592 phosphorylation, whether through metabolite‑mediated control of kinases/phosphatases or via indirect effects.
We thank the reviewer for this comment. It is worth noting that (Meng et al., 2022) demonstrated that SNAT7 positively regulates mTORC1 activity at the lysosomal membrane through release of lysosomal glutamine, and (Dias et al., 2024) showed that inhibiting mTORC1 activity using drugs decreases SAMHD1 Thr592 phosphorylation in hMDM. Therefore, we could speculate that the absence of SNAT7 down-regulates mTORC1 activity, which then leads to decreased SAMHD1 phosphorylation. This is now further discussed in the discussion section of the manuscript.
The authors do not measure intracellular dNTP levels upon SNAT7 knockdown, which is the key functional substrate of SAMHD1. They also do not directly demonstrate that glutamine supplementation restores dNTP pools.
We thank the reviewer for this comment. Please, refer to comment #5 under Reviewer #2.
Extracellular glutamine only partially rescues viral production, implying the existence of transport‑independent functions of SNAT7 or additional pathways. This important observation is not discussed.
We thank the reviewer for this comment. The discussion has been modified accordingly.
It is suggested that the key findings be validated in immortalized THP‑1 cells differentiated into macrophage‑like cells by PMA.
We thank the reviewer for this suggestion but don’t really understand why this would strengthen our conclusions. Indeed, despite the known variability between donors and technical limitations to transduce cells, we chose human blood monocyte-derived macrophages as a relevant non-transformed model for HIV-1 infection of macrophages. They also represent to some extent the human diversity.
The Discussion section should be expanded to include the potential translational implications and limitations of the present study.
We thank the reviewer for this comment. The discussion points to some elements of potential translation and limitations of the study.
Reviewer #3 (Significance (Required)):
General assessment: This study identifies the lysosomal glutamine transporter SLC38A7/SNAT7 as a novel host dependency factor for HIV‑1 replication in primary human macrophages. The major strengths include the use of physiologically relevant primary macrophage models, a well-organized experimental pipeline from expression profiling to functional validation, and the establishment of a link between SNAT7, glutamine metabolism, and the HIV restriction factor SAMHD1.
Advance: It extends current understanding of HIV‑1 host dependency factors and immunometabolism by revealing a compartment‑specific metabolic pathway that supports viral reverse transcription.
Audience:This work will primarily interest specialized researchers in HIV‑1 biology, host-virus interactions, restriction factors, and antiviral innate immunity.