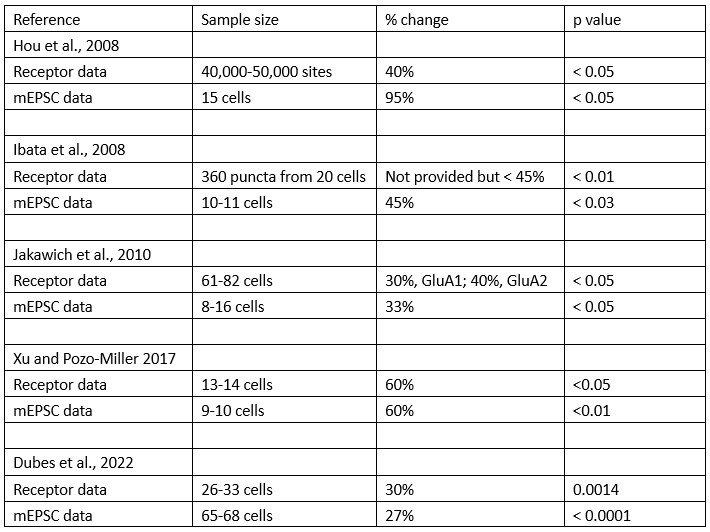
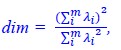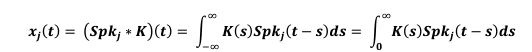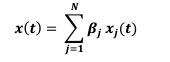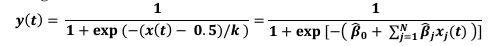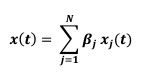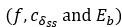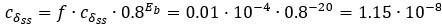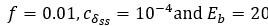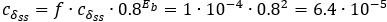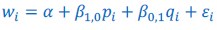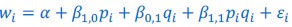Author response:
The following is the authors’ response to the current reviews.
eLife Assessment
This neuroimaging and electrophysiology study in a small cohort of congenital cataract patients with sight recovery aims to characterize the effects of early visual deprivation on excitatory and inhibitory balance in visual cortex. While contrasting sight-recovery with visually intact controls suggested the existence of persistent alterations in Glx/GABA ratio and aperiodic EEG signals, it provided only incomplete evidence supporting claims about the effects of early deprivation itself. The reported data were considered valuable, given the rare study population. However, the small sample sizes, lack of a specific control cohort and multiple methodological limitations will likely restrict usefulness to scientists working in this particular subfield.
We thank the reviewing editors for their consideration and updated assessment of our manuscript after its first revision.
In order to assess the effects of early deprivation, we included an age-matched, normally sighted control group recruited from the same community, measured in the same scanner and laboratory. This study design is analogous to numerous studies in permanently congenitally blind humans, which typically recruited sighted controls, but hardly ever individuals with a different, e.g. late blindness history. In order to improve the specificity of our conclusions, we used a frontal cortex voxel in addition to a visual cortex voxel (MRS). Analogously, we separately analyzed occipital and frontal electrodes (EEG).
Moreover, we relate our findings in congenital cataract reversal individuals to findings in the literature on permanent congenital blindness. Note, there are, to the best of our knowledge, neither MRS nor resting-state EEG studies in individuals with permanent late blindness.
Our participants necessarily have nystagmus and low visual acuity due to their congenital deprivation phase, and the existence of nystagmus is a recruitment criterion to diagnose congenital cataracts.
It might be interesting for future studies to investigate individuals with transient late blindness. However, such a study would be ill-motivated had we not found differences between the most “extreme” of congenital visual deprivation conditions and normally sighted individuals (analogous to why earlier research on permanent blindness investigated permanent congenitally blind humans first, rather than permanently late blind humans, or both in the same study). Any result of these future work would need the reference to our study, and neither results in these additional groups would invalidate our findings.
Since all our congenital cataract reversal individuals by definition had visual impairments, we included an eyes closed condition, both in the MRS and EEG assessment. Any group effect during the eyes closed condition cannot be due to visual acuity deficits changing the bottom-up driven visual activation.
As we detail in response to review 3, our EEG analyses followed the standards in the field.
Public Reviews:
Reviewer (1 (Public review):
Summary
In this human neuroimaging and electrophysiology study, the authors aimed to characterise effects of a period of visual deprivation in the sensitive period on excitatory and inhibitory balance in the visual cortex. They attempted to do so by comparing neurochemistry conditions ('eyes open', 'eyes closed') and resting state, and visually evoked EEG activity between ten congenital cataract patients with recovered sight (CC), and ten age-matched control participants (SC) with normal sight.
First, they used magnetic resonance spectroscopy to measure in vivo neurochemistry from two locations, the primary location of interest in the visual cortex, and a control location in the frontal cortex. Such voxels are used to provide a control for the spatial specificity of any effects, because the single-voxel MRS method provides a single sampling location. Using MR-visible proxies of excitatory and inhibitory neurotransmission, Glx and GABA+ respectively, the authors report no group effects in GABA+ or Glx, no difference in the functional conditions 'eyes closed' and 'eyes open'. They found an effect of group in the ratio of Glx/GABA+ and no similar effect in the control voxel location. They then perform multiple exploratory correlations between MRS measures and visual acuity, and report a weak positive correlation between the 'eyes open' condition and visual acuity in CC participants.
The same participants then took part in an EEG experiment. The authors selected two electrodes placed in the visual cortex for analysis and report a group difference in an EEG index of neural activity, the aperiodic intercept, as well as the aperiodic slope, considered a proxy for cortical inhibition. Control electrodes in the frontal region did not present with the same pattern. They report an exploratory correlation between the aperiodic intercept and Glx in one out of three EEG conditions.
The authors report the difference in E/I ratio, and interpret the lower E/I ratio as representing an adaptation to visual deprivation, which would have initially caused a higher E/I ratio. Although intriguing, the strength of evidence in support of this view is not strong. Amongst the limitations are the low sample size, a critical control cohort that could provide evidence for higher E/I ratio in CC patients without recovered sight for example, and lower data quality in the control voxel. Nevertheless, the study provides a rare and valuable insight into experience-dependent plasticity in the human brain.
Strengths of study
How sensitive period experience shapes the developing brain is an enduring and important question in neuroscience. This question has been particularly difficult to investigate in humans. The authors recruited a small number of sight-recovered participants with bilateral congenital cataracts to investigate the effect of sensitive period deprivation on the balance of excitation and inhibition in the visual brain using measures of brain chemistry and brain electrophysiology. The research is novel, and the paper was interesting and well written.
Limitations
Low sample size. Ten for CC and ten for SC, and further two SC participants were rejected due to lack of frontal control voxel data. The sample size limits the statistical power of the dataset and increases the likelihood of effect inflation.
In the updated manuscript, the authors have provided justification for their sample size by pointing to prior studies and the inherent difficulties in recruiting individuals with bilateral congenital cataracts. Importantly, this highlights the value the study brings to the field while also acknowledging the need to replicate the effects in a larger cohort.
Lack of specific control cohort. The control cohort has normal vision. The control cohort is not specific enough to distinguish between people with sight loss due to different causes and patients with congenital cataracts with co-morbidities. Further data from a more specific populations, such as patients whose cataracts have not been removed, with developmental cataracts, or congenitally blind participants, would greatly improve the interpretability of the main finding. The lack of a more specific control cohort is a major caveat that limits a conclusive interpretation of the results.
In the updated version, the authors have indicated that future studies can pursue comparisons between congenital cataract participants and cohorts with later sight loss.
MRS data quality differences. Data quality in the control voxel appears worse than in the visual cortex voxel. The frontal cortex MRS spectrum shows far broader linewidth than the visual cortex (Supplementary Figures). Compared to the visual voxel, the frontal cortex voxel has less defined Glx and GABA+ peaks; lower GABA+ and Glx concentrations, lower NAA SNR values; lower NAA concentrations. If the data quality is a lot worse in the FC, then small effects may not be detectable.
In the updated version, the authors have added more information that informs the reader of the MRS quality differences between voxel locations. This increases the transparency of their reporting and enhances the assessment of the results.
Because of the direction of the difference in E/I, the authors interpret their findings as representing signatures of sight improvement after surgery without further evidence, either within the study or from the literature. However, the literature suggests that plasticity and visual deprivation drives the E/I index up rather than down. Decreasing GABA+ is thought to facilitate experience dependent remodelling. What evidence is there that cortical inhibition increases in response to a visual cortex that is over-sensitised to due congenital cataracts? Without further experimental or literature support this interpretation remains very speculative.
The updated manuscript contains key reference from non-human work to justify their interpretation.
Heterogeneity in patient group. Congenital cataract (CC) patients experienced a variety of duration of visual impairment and were of different ages. They presented with co-morbidities (absorbed lens, strabismus, nystagmus). Strabismus has been associated with abnormalities in GABAergic inhibition in the visual cortex. The possible interactions with residual vision and confounds of co-morbidities are not experimentally controlled for in the correlations, and not discussed.
The updated document has addressed this caveat.
Multiple exploratory correlations were performed to relate MRS measures to visual acuity (shown in Supplementary Materials), and only specific ones shown in the main document. The authors describe the analysis as exploratory in the 'Methods' section. Furthermore, the correlation between visual acuity and E/I metric is weak, not corrected for multiple comparisons. The results should be presented as preliminary, as no strong conclusions can be made from them. They can provide a hypothesis to test in a future study.
This has now been done throughout the document and increases the transparency of the reporting.
P.16 Given the correlation of the aperiodic intercept with age ("Age negatively correlated with the aperiodic intercept across CC and SC individuals, that is, a flattening of the intercept was observed with age"), age needs to be controlled for in the correlation between neurochemistry and the aperiodic intercept. Glx has also been shown to negatively correlates with age.
This caveat has been addressed in the revised manuscript.
Multiple exploratory correlations were performed to relate MRS to EEG measures (shown in Supplementary Materials), and only specific ones shown in the main document. Given the multiple measures from the MRS, the correlations with the EEG measures were exploratory, as stated in the text, p.16, and in Fig.4. yet the introduction said that there was a prior hypothesis "We further hypothesized that neurotransmitter changes would relate to changes in the slope and intercept of the EEG aperiodic activity in the same subjects." It would be great if the text could be revised for consistency and the analysis described as exploratory.
This has been done throughout the document and increases the transparency of the reporting.
The analysis for the EEG needs to take more advantage of the available data. As far as I understand, only two electrodes were used, yet far more were available as seen in their previous study (Ossandon et al., 2023). The spatial specificity is not established. The authors could use the frontal cortex electrode (FP1, FP2) signals as a control for spatial specificity in the group effects, or even better, all available electrodes and correct for multiple comparisons. Furthermore, they could use the aperiodic intercept vs Glx in SC to evaluate the specificity of the correlation to CC.
This caveat has been addressed. The authors have added frontal electrodes to their analysis, providing an essential regional control for the visual cortex location.
Comments on the latest version:
The authors have made reasonable adjustments to their manuscript that addressed most of my comments by adding further justification for their methodology, essential literature support, pointing out exploratory analyses, limitations and adding key control analyses. Their revised manuscript has overall improved, providing valuable information, though the evidence that supports their claims is still incomplete.
We thank the reviewer for suggesting ways to improve our manuscript and carefully reassessing our revised manuscript.
Reviewer 2 (Public review):
Summary:
The study examined 10 congenitally blind patients who recovered vision through the surgical removal of bilateral dense cataracts, measuring neural activity and neuro chemical profiles from the visual cortex. The declared aim is to test whether restoring visual function after years of complete blindness impacts excitation/inhibition balance in the visual cortex.
Strengths:
The findings are undoubtedly useful for the community, as they contribute towards characterising the many ways in which this special population differs from normally sighted individuals. The combination of MRS and EEG measures is a promising strategy to estimate a fundamental physiological parameter - the balance between excitation and inhibition in the visual cortex, which animal studies show to be heavily dependent upon early visual experience. Thus, the reported results pave the way for further studies, which may use a similar approach to evaluate more patients and control groups.
Weaknesses:
The main methodological limitation is the lack of an appropriate comparison group or condition to delineate the effect of sight recovery (as opposed to the effect of congenital blindness). Few previous studies suggested that Excitation/Inhibition ratio in the visual cortex is increased in congenitally blind patients; the present study reports that E/I ratio decreases instead. The authors claim that this implies a change of E/I ratio following sight recovery. However, supporting this claim would require showing a shift of E/I after vs. before the sight-recovery surgery, or at least it would require comparing patients who did and did not undergo the sight-recovery surgery (as common in the field).
We thank the reviewer for suggesting ways to improve our manuscript and carefully reassessing our revised manuscript.
Since we have not been able to acquire longitudinal data with the experimental design of the present study in congenital cataract reversal individuals, we compared the MRS and EEG results of congenital cataract reversal individuals to published work in congenitally permanent blind individuals. We consider this as a resource saving approach. We think that the results of our cross-sectional study now justify the costs and enormous efforts (and time for the patients who often have to travel long distances) associated with longitudinal studies in this rare population.
There are also more technical limitations related to the correlation analyses, which are partly acknowledged in the manuscript. A bland correlation between GLX/GABA and the visual impairment is reported, but this is specific to the patients group (N=10) and would not hold across groups (the correlation is positive, predicting the lowest GLX/GABA ratio values for the sighted controls - opposite of what is found). There is also a strong correlation between GLX concentrations and the EEG power at the lowest temporal frequencies. Although this relation is intriguing, it only holds for a very specific combination of parameters (of the many tested): only with eyes open, only in the patients group.
Given the exploratory nature of the correlations, we do not base the majority of our conclusions on this analysis. There are no doubts that the reported correlations need replication; however, replication is only possible after a first report. Thus, we hope to motivate corresponding analyses in further studies.
It has to be noted that in the present study significance testing for correlations were corrected for multiple comparisons, and that some findings replicate earlier reports (e.g. effects on EEG aperiodic slope, alpha power, and correlations with chronological age).
Conclusions:
The main claim of the study is that sight recovery impacts the excitation/inhibition balance in the visual cortex, estimated with MRS or through indirect EEG indices. However, due to the weaknesses outlined above, the study cannot distinguish the effects of sight recovery from those of visual deprivation. Moreover, many aspects of the results are interesting but their validation and interpretation require additional experimental work.
We interpret the group differences between individuals tested years after congenital visual deprivation and normally sighted individuals as supportive of the E/I ratio being impacted by congenital visual deprivation. In the absence of a sensitive period for the development of an E/I ratio, individuals with a transient phase of congenital blindness might have developed a visual system indistinguishable from normally sighted individuals. As we demonstrate, this is not so. Comparing the results of congenitally blind humans with those of congenitally permanently blind humans (from previous studies) allowed us to identify changes of E/I ratio, which add to those found for congenital blindness.
We thank the reviewer for the helpful comments and suggestions related to the first submission and first revision of our manuscript. We are keen to translate some of them into future studies.
Reviewer 3 (Public review):
This manuscript examines the impact of congenital visual deprivation on the excitatory/inhibitory (E/I) ratio in the visual cortex using Magnetic Resonance Spectroscopy (MRS) and electroencephalography (EEG) in individuals whose sight was restored. Ten individuals with reversed congenital cataracts were compared to age-matched, normally sighted controls, assessing the cortical E/I balance and its interrelationship and to visual acuity. The study reveals that the Glx/GABA ratio in the visual cortex and the intercept and aperiodic signal are significantly altered in those with a history of early visual deprivation, suggesting persistent neurophysiological changes despite visual restoration.
First of all, I would like to disclose that I am not an expert in congenital visual deprivation, nor in MRS. My expertise is in EEG (particularly in the decomposition of periodic and aperiodic activity) and statistical methods.
Although the authors addressed some of the concerns of the previous version, major concerns and flaws remain in terms of methodological and statistical approaches along with the (over)interpretation of the results. Specific concerns include:
(1 3.1 Response to Variability in Visual Deprivation<br />
Rather than listing the advantages and disadvantages of visual deprivation, I recommend providing at least a descriptive analysis of how the duration of visual deprivation influenced the measures of interest. This would enhance the depth and relevance of the discussion.
Although Review 2 and Review 3 (see below) pointed out problems in interpreting multiple correlational analyses in small samples, we addressed this request by reporting such correlations between visual deprivation history and measured EEG/MRS outcomes.
Calculating the correlation between duration of visual deprivation and behavioral or brain measures is, in fact, a common suggestion. The existence of sensitive periods, which are typically assumed to not follow a linear gradual decline of neuroplasticity, does not necessary allow predicting a correlation with duration of blindness. Daphne Maurer has additionally worked on the concept of “sleeper effects” (Maurer et al., 2007), that is, effects on the brain and behavior by early deprivation which are observed only later in life when the function/neural circuits matures.
In accordance with this reasoning, we did not observe a significant correlation between duration of visual deprivation and any of our dependent variables.
(2 3.2) Small Sample Size
The issue of small sample size remains problematic. The justification that previous studies employed similar sample sizes does not adequately address the limitation in the current study. I strongly suggest that the correlation analyses should not feature prominently in the main manuscript or the abstract, especially if the discussion does not substantially rely on these correlations. Please also revisit the recommendations made in the section on statistical concerns.
In the revised manuscript, we explicitly mention that our sample size is not atypical for the special group investigated, but that a replication of our results in larger samples would foster their impact. We only explicitly mention correlations that survived stringent testing for multiple comparisons in the main manuscript.
Given the exploratory nature of the correlations, we have not based the majority of our claims on this analysis.
(3 3.3) Statistical Concerns
While I appreciate the effort of conducting an independent statistical check, it merely validates whether the reported statistical parameters, degrees of freedom (df), and p-values are consistent. However, this does not address the appropriateness of the chosen statistical methods.
We did not intend for the statcheck report to justify the methods used for statistics, which we have done in a separate section with normality and homogeneity testing (Supplementary Material S9), and references to it in the descriptions of the statistical analyses (Methods, Page 13, Lines 326-329 and Page 15, Lines 400-402).
Several points require clarification or improvement:
(4) Correlation Methods: The manuscript does not specify whether the reported correlation analyses are based on Pearson or Spearman correlation.
The depicted correlations are Pearson correlations. We will add this information to the Methods.
(5) Confidence Intervals: Include confidence intervals for correlations to represent the uncertainty associated with these estimates.
We will add the confidence intervals to the second revision of our manuscript.
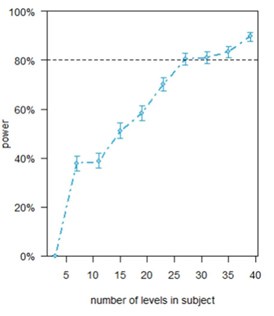
(6) Permutation Statistics: Given the small sample size, I recommend using permutation statistics, as these are exact tests and more appropriate for small datasets.
Our study focuses on a rare population, with a sample size limited by the availability of participants. Our findings provide exploratory insights rather than make strong inferential claims. To this end, we have ensured that our analysis adheres to key statistical assumptions (Shapiro-Wilk as well as Levene’s tests, Supplementary Material S9),and reported our findings with effect sizes, appropriate caution and context.
(7) Adjusted P-Values: Ensure that reported Bonferroni corrected p-values (e.g., p > 0.999) are clearly labeled as adjusted p-values where applicable.
In the revised manuscript, we will change Figure 4 to say ‘adjusted p,’ which we indeed reported.
(8) Figure 2C
Figure 2C still lacks crucial information that the correlation between Glx/GABA ratio and visual acuity was computed solely in the control group (as described in the rebuttal letter). Why was this analysis restricted to the control group? Please provide a rationale.
Figure 2C depicts the correlation between Glx/GABA+ ratio and visual acuity in the congenital cataract reversal group, not the control group. This is mentioned in the Figure 2 legend, as well as in the main text where the figure is referred to (Page 18, Line 475).
The correlation analyses between visual acuity and MRS/EEG measures were only performed in the congenital cataract reversal group since the sighed control group comprised of individuals with vision in the normal range; thus this analyses would not make sense. Table 1 with the individual visual acuities for all participants, including the normally sighted controls, shows the low variance in the latter group.
For variables in which no apiori group differences in variance were predicted, we performed the correlation analyses across groups (see Supplementary Material S12, S15).
We will highlight these motivations more clearly in the Methods of the revised manuscript.
(9 3.4) Interpretation of Aperiodic Signal
Relying on previous studies to interpret the aperiodic slope as a proxy for excitation/inhibition (E/I) does not make the interpretation more robust.
How to interpret aperiodic EEG activity has been subject of extensive investigation. We cite studies which provide evidence from multiple species (monkeys, humans) and measurements (EEG, MEG, ECoG), including studies which pharmacologically manipulated E/I balance.
Whether our findings are robust, in fact, requires a replication study. Importantly, we analyzed the intercept of the aperiodic activity fit as well, and discuss results related to the intercept.
Quote:
“3.4 Interpretation of aperiodic signal:
- Several recent papers demonstrated that the aperiodic signal measured in EEG or ECoG is related to various important aspects such as age, skull thickness, electrode impedance, as well as cognition. Thus, currently, very little is known about the underlying effects which influence the aperiodic intercept and slope. The entire interpretation of the aperiodic slope as a proxy for E/I is based on a computational model and simulation (as described in the Gao et al. paper).
Response: Apart from the modeling work from Gao et al., multiple papers which have also been cited which used ECoG, EEG and MEG and showed concomitant changes in aperiodic activity with pharmacological manipulation of the E/I ratio (Colombo et al., 2019; Molina et al., 2020; Muthukumaraswamy & Liley, 2018). Further, several prior studies have interpreted changes in the aperiodic slope as reflective of changes in the E/I ratio, including studies of developmental groups (Favaro et al., 2023; Hill et al., 2022; McSweeney et al., 2023; Schaworonkow & Voytek, 2021) as well as patient groups (Molina et al., 2020; Ostlund et al., 2021).
- The authors further wrote: We used the slope of the aperiodic (1/f) component of the EEG spectrum as an estimate of E/I ratio (Gao et al., 2017; Medel et al., 2020; Muthukumaraswamy & Liley, 2018). This is a highly speculative interpretation with very little empirical evidence. These papers were conducted with ECoG data (mostly in animals) and mostly under anesthesia. Thus, these studies only allow an indirect interpretation by what the 1/f slope in EEG measurements is actually influenced.
Response: Note that Muthukumaraswamy et al. (2018) used different types of pharmacological manipulations and analyzed periodic and aperiodic MEG activity in humans, in addition to monkey ECoG (Muthukumaraswamy & Liley, 2018). Further, Medel et al. (now published as Medel et al., 2023) compared EEG activity in addition to ECoG data after propofol administration. The interpretation of our results are in line with a number of recent studies in developing (Hill et al., 2022; Schaworonkow & Voytek, 2021) and special populations using EEG. As mentioned above, several prior studies have used the slope of the 1/f component/aperiodic activity as an indirect measure of the E/I ratio (Favaro et al., 2023; Hill et al., 2022; McSweeney et al., 2023; Molina et al., 2020; Ostlund et al., 2021; Schaworonkow & Voytek, 2021), including studies using scalp-recorded EEG from humans.
In the introduction of the revised manuscript, we have made more explicit that this metric is indirect (Page 3, Line 91), (additionally see Discussion, Page 24, Lines 644-645, Page 25, Lines 650-657).
While a full understanding of aperiodic activity needs to be provided, some convergent ideas have emerged. We think that our results contribute to this enterprise, since our study is, to the best of our knowledge, the first which assessed MRS measured neurotransmitter levels and EEG aperiodic activity.“
(10) Additionally, the authors state:
"We cannot think of how any of the exploratory correlations between neurophysiological measures and MRS measures could be accounted for by a difference e.g. in skull thickness."
(11) This could be addressed directly by including skull thickness as a covariate or visualizing it in scatterplots, for instance, by representing skull thickness as the size of the dots.
We are not aware of any study that would justify such an analysis.
Our analyses were based on previous findings in the literature.
Since to the best of our knowledge, no evidence exists that congenital cataracts go together with changes in skull thickness, and that skull thickness might selectively modulate visual cortex Glx/GABA+ but not NAA measures, we decided against following this suggestion.
Notably, the neurotransmitter concentration reported here is after tissue segmentation of the voxel region. The tissue fraction was shown to not differ between groups in the MRS voxels (Supplementary Material S4). The EEG electrode impedance was lowered to <10 kOhm in every participant (Methods, Page 13, Line 344), and preparation was identical across groups.
(12 3.5) Problems with EEG Preprocessing and Analysis
Downsampling: The decision to downsample the data to 60 Hz "to match the stimulation rate" is problematic. This choice conflates subsequent spectral analyses due to aliasing issues, as explained by the Nyquist theorem. While the authors cite prior studies (Schwenk et al., 2020; VanRullen & MacDonald, 2012) to justify this decision, these studies focused on alpha (8-12 Hz), where aliasing is less of a concern compared of analyzing aperiodic signal. Furthermore, in contrast, the current study analyzes the frequency range from 1-20 Hz, which is too narrow for interpreting the aperiodic signal as E/I. Typically, this analysis should include higher frequencies, spanning at least 1-30 Hz or even 1-45 Hz (not 20-40 Hz).
As mentioned in the Methods (Page 15 Line 376) and the previous response, the pop_resample function used by EEGLAB applies an anti-aliasing filter, at half the resampling frequency (as per the Nyquist theorem https://eeglab.org/tutorials/05_Preprocess/resampling.html). The upper cut off of the low pass filter set by EEGlab prior to down sampling (30 Hz) is still far above the frequency of interest in the current study (1-20 Hz), thus allowing us to derive valid results.
Quote:
“- The authors downsampled the data to 60Hz to "to match the stimulation rate". What is the intention of this? Because the subsequent spectral analyses are conflated by this choice (see Nyquist theorem).
Response: This data were collected as part of a study designed to evoke alpha activity with visual white-noise, which ranged in luminance with equal power at all frequencies from 1-60 Hz, restricted by the refresh rate of the monitor on which stimuli were presented (Pant et al., 2023). This paradigm and method was developed by VanRullen and colleagues (Schwenk et al., 2020; Vanrullen & MacDonald, 2012), wherein the analysis requires the same sampling rate between the presented frequencies and the EEG data. The downsampling function used here automatically applies an anti-aliasing filter (EEGLAB 2019) .”
Moreover, the resting-state data were not resampled to 60 Hz. We will make this clearer in the Methods of the revised manuscript.
Our consistent results of group differences across all three EEG conditions, thus, exclude any possibility that they were driven by aliasing artifacts.
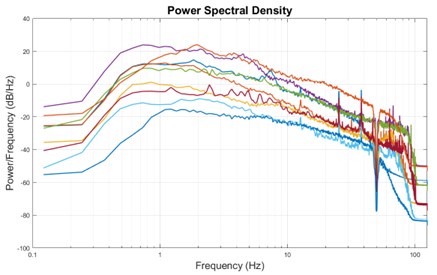
The expected effects of this anti-aliasing filter can be seen in the attached Figure R1, showing an example participant’s spectrum in the 1-30 Hz range (as opposed to the 1-20 Hz plotted in the manuscript), clearly showing a 30-40 dB drop at 30 Hz. Any aliasing due to, for example, remaining line noise, would additionally be visible in this figure (as well as Figure 3) as a peak.
Author response image 1.
Power spectral density of one congenital cataract-reversal (CC) participant in the visual stimulation condition across all channels. The reduced power at 30 Hz shows the effects of the anti-aliasing filter applied by EEGLAB’s pop_resample function.
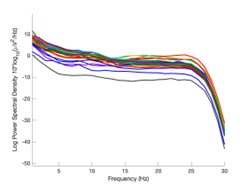
As we stated in the manuscript, and in previous reviews, so far there has been no consensus on the exact range of measuring aperiodic activity. We made a principled decision based on the literature (showing a knee in aperiodic fits of this dataset at 20 Hz) (Medel et al., 2023; Ossandón et al., 2023), data quality (possible contamination by line noise at higher frequencies) and the purpose of the visual stimulation experiment (to look at the lower frequency range by stimulating up to 60 Hz, thereby limiting us to quantifying below 30 Hz), that 1-20 Hz would be the fit range in this dataset.
Quote:
“(3) What's the underlying idea of analyzing two separate aperiodic slopes (20-40Hz and 1-19Hz). This is very unusual to compute the slope between 20-40 Hz, where the SNR is rather low.
"Ossandón et al. (2023), however, observed that in addition to the flatter slope of the aperiodic power spectrum in the high frequency range (20-40 Hz), the slope of the low frequency range (1-19 Hz) was steeper in both, congenital cataract-reversal individuals, as well as in permanently congenitally blind humans."
Response: The present manuscript computed the slope between 1-20 Hz. Ossandón et al. as well as Medel et al. (2023) found a “knee” of the 1/f distribution at 20 Hz and describe further the motivations for computing both slope ranges. For example, Ossandón et al. used a data driven approach and compared single vs. dual fits and found that the latter fitted the data better. Additionally, they found the best fit if a knee at 20 Hz was used. We would like to point out that no standard range exists for the fitting of the 1/f component across the literature and, in fact, very different ranges have been used (Gao et al., 2017; Medel et al., 2023; Muthukumaraswamy & Liley, 2018).“
(13) Baseline Removal: Subtracting the mean activity across an epoch as a baseline removal step is inappropriate for resting-state EEG data. This preprocessing step undermines the validity of the analysis. The EEG dataset has fundamental flaws, many of which were pointed out in the previous review round but remain unaddressed. In its current form, the manuscript falls short of standards for robust EEG analysis. If I were reviewing for another journal, I would recommend rejection based on these flaws.
The baseline removal step from each epoch serves to remove the DC component of the recording and detrend the data. This is a standard preprocessing step (included as an option in preprocessing pipelines recommended by the EEGLAB toolbox, FieldTrip toolbox and MNE toolbox), additionally necessary to improve the efficacy of ICA decomposition (Groppe et al., 2009).
In the previous review round, a clarification of the baseline timing was requested, which we added. Beyond this request, there was no mention of the appropriateness of the baseline removal and/or a request to provide reasons for why it might not undermine the validity of the analysis.
Quote:
“- "Subsequently, baseline removal was conducted by subtracting the mean activity across the length of an epoch from every data point." The actual baseline time segment should be specified.
Response: The time segment was the length of the epoch, that is, 1 second for the resting state conditions and 6.25 seconds for the visual stimulation conditions. This has been explicitly stated in the revised manuscript (Page 13, Line 354).”
Prior work in the time (not frequency) domain on event-related potential (ERP) analysis has suggested that the baselining step might cause spurious effects (Delorme, 2023) (although see (Tanner et al., 2016)). We did not perform ERP analysis at any stage. One recent study suggests spurious group differences in the 1/f signal might be driven by an inappropriate dB division baselining method (Gyurkovics et al., 2021), which we did not perform.
Any effect of our baselining procedure on the FFT spectrum would be below the 1 Hz range, which we did not analyze.
Each of the preprocessing steps in the manuscript match pipelines described and published in extensive prior work. We document how multiple aspects of our EEG results replicate prior findings (Supplementary Material S15, S18, S19), reports of other experimenters, groups and locations, validating that our results are robust.
We therefore reject the claim of methodological flaws in our EEG analyses in the strongest possible terms.
Quote:
“3.5 Problems with EEG preprocessing and analysis:
- It seems that the authors did not identify bad channels nor address the line noise issue (even a problem if a low pass filter of below-the-line noise was applied).
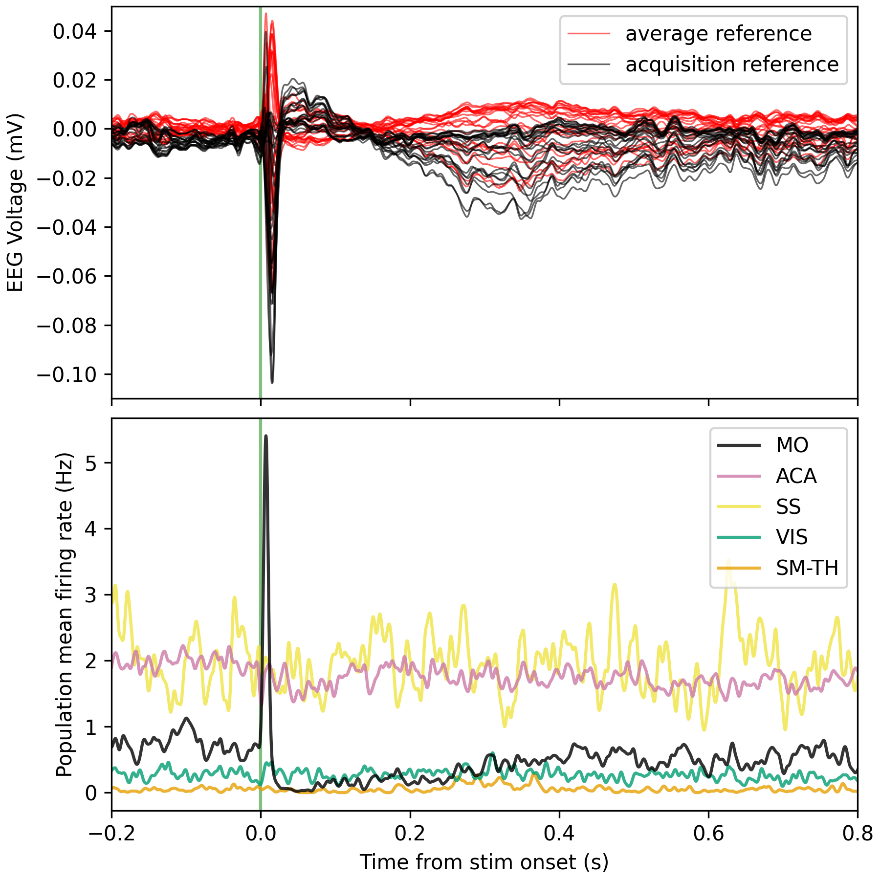
Response: As pointed out in the methods and Figure 1, we only analyzed data from two occipital channels, O1 and O2 neither of which were rejected for any participant. Channel rejection was performed for the larger dataset, published elsewhere (Ossandón et al., 2023; Pant et al., 2023). As control sites we added the frontal channels FP1 and Fp2 (see Supplementary Material S14)
Neither Ossandón et al. (2023) nor Pant et al. (2023) considered frequency ranges above 40 Hz to avoid any possible contamination with line noise. Here, we focused on activity between 0 and 20 Hz, definitely excluding line noise contaminations (Methods, Page 14, Lines 365-367). The low pass filter (FIR, 1-45 Hz) guaranteed that any spill-over effects of line noise would be restricted to frequencies just below the upper cutoff frequency.
Additionally, a prior version of the analysis used spectrum interpolation to remove line noise; the group differences remained stable (Ossandón et al., 2023). We have reported this analysis in the revised manuscript (Page 14, Lines 364-357).
Further, both groups were measured in the same lab, making line noise (~ 50 Hz) as an account for the observed group effects in the 1-20 Hz frequency range highly unlikely. Finally, any of the exploratory MRS-EEG correlations would be hard to explain if the EEG parameters would be contaminated with line noise.
- What was the percentage of segments that needed to be rejected due to the 120μV criteria? This should be reported specifically for EO & EC and controls and patients.
Response: The mean percentage of 1 second segments rejected for each resting state condition and the percentage of 6.25 long segments rejected in each group for the visual stimulation condition have been added to the revised manuscript (Supplementary Material S10), and referred to in the Methods on Page 14, Lines 372-373).
- The authors downsampled the data to 60Hz to "to match the stimulation rate". What is the intention of this? Because the subsequent spectral analyses are conflated by this choice (see Nyquist theorem).
Response: This data were collected as part of a study designed to evoke alpha activity with visual white-noise, which changed in luminance with equal power at all frequencies from 1-60 Hz, restricted by the refresh rate of the monitor on which stimuli were presented (Pant et al., 2023). This paradigm and method was developed by VanRullen and colleagues (Schwenk et al., 2020; VanRullen & MacDonald, 2012), wherein the analysis requires the same sampling rate between the presented frequencies and the EEG data. The downsampling function used here automatically applies an anti-aliasing filter (EEGLAB 2019) .
- "Subsequently, baseline removal was conducted by subtracting the mean activity across the length of an epoch from every data point." The actual baseline time segment should be specified.
The time segment was the length of the epoch, that is, 1 second for the resting state conditions and 6.25 seconds for the visual stimulation conditions. This has now been explicitly stated in the revised manuscript (Page 14, Lines 379-380).<br />
- "We excluded the alpha range (8-14 Hz) for this fit to avoid biasing the results due to documented differences in alpha activity between CC and SC individuals (Bottari et al., 2016; Ossandón et al., 2023; Pant et al., 2023)." This does not really make sense, as the FOOOF algorithm first fits the 1/f slope, for which the alpha activity is not relevant.
Response: We did not use the FOOOF algorithm/toolbox in this manuscript. As stated in the Methods, we used a 1/f fit to the 1-20 Hz spectrum in the log-log space, and subtracted this fit from the original spectrum to obtain the corrected spectrum. Given the pronounced difference in alpha power between groups (Bottari et al., 2016; Ossandón et al., 2023; Pant et al., 2023), we were concerned it might drive differences in the exponent values. Our analysis pipeline had been adapted from previous publications of our group and other labs (Ossandón et al., 2023; Voytek et al., 2015; Waschke et al., 2017).
We have conducted the analysis with and without the exclusion of the alpha range, as well as using the FOOOF toolbox both in the 1-20 Hz and 20-40 Hz ranges (Ossandón et al., 2023). The findings of a steeper slope in the 1-20 Hz range as well as lower alpha power in CC vs SC individuals remained stable. In Ossandón et al., the comparison between the piecewise fits and FOOOF fits led the authors to use the former, as it outperformed the FOOOF algorithm for their data.
- The model fits of the 1/f fitting for EO, EC, and both participant groups should be reported.
Response: In Figure 3 of the manuscript, we depicted the mean spectra and 1/f fits for each group.
In the revised manuscript, we added the fit quality metrics (average R<sup>2</sup> values > 0.91 for each group and condition) (Methods Page 15, Lines 395-396; Supplementary Material S11) and additionally show individual subjects’ fits (Supplementary Material S11).“
(14) The authors mention:
"The EEG data sets reported here were part of data published earlier (Ossandón et al., 2023; Pant et al., 2023)." Thus, the statement "The group differences for the EEG assessments corresponded to those of a larger sample of CC individuals (n=38) " is a circular argument and should be avoided."
The authors addressed this comment and adjusted the statement. However, I do not understand, why not the full sample published earlier (Ossandón et al., 2023) was used in the current study?
The recording of EEG resting state data stated in 2013, while MRS testing could only be set up by the end of 2019. Moreover, not all subjects who qualify for EEG recording qualify for being scanned (e.g. due to MRI safety, claustrophobia)
References
Bottari, D., Troje, N. F., Ley, P., Hense, M., Kekunnaya, R., & Röder, B. (2016). Sight restoration after congenital blindness does not reinstate alpha oscillatory activity in humans. Scientific Reports. https://doi.org/10.1038/srep24683
Colombo, M. A., Napolitani, M., Boly, M., Gosseries, O., Casarotto, S., Rosanova, M., Brichant, J. F., Boveroux, P., Rex, S., Laureys, S., Massimini, M., Chieregato, A., & Sarasso, S. (2019). The spectral exponent of the resting EEG indexes the presence of consciousness during unresponsiveness induced by propofol, xenon, and ketamine. NeuroImage, 189(September 2018), 631–644. https://doi.org/10.1016/j.neuroimage.2019.01.024
Delorme, A. (2023). EEG is better left alone. Scientific Reports, 13(1), 2372. https://doi.org/10.1038/s41598-023-27528-0
Favaro, J., Colombo, M. A., Mikulan, E., Sartori, S., Nosadini, M., Pelizza, M. F., Rosanova, M., Sarasso, S., Massimini, M., & Toldo, I. (2023). The maturation of aperiodic EEG activity across development reveals a progressive differentiation of wakefulness from sleep. NeuroImage, 277. https://doi.org/10.1016/J.NEUROIMAGE.2023.120264
Gao, R., Peterson, E. J., & Voytek, B. (2017). Inferring synaptic excitation/inhibition balance from field potentials. NeuroImage, 158(March), 70–78. https://doi.org/10.1016/j.neuroimage.2017.06.078
Groppe, D. M., Makeig, S., & Kutas, M. (2009). Identifying reliable independent components via split-half comparisons. NeuroImage, 45(4), 1199–1211. https://doi.org/10.1016/j.neuroimage.2008.12.038
Gyurkovics, M., Clements, G. M., Low, K. A., Fabiani, M., & Gratton, G. (2021). The impact of 1/f activity and baseline correction on the results and interpretation of time-frequency analyses of EEG/MEG data: A cautionary tale. NeuroImage, 237. https://doi.org/10.1016/j.neuroimage.2021.118192
Hill, A. T., Clark, G. M., Bigelow, F. J., Lum, J. A. G., & Enticott, P. G. (2022). Periodic and aperiodic neural activity displays age-dependent changes across early-to-middle childhood. Developmental Cognitive Neuroscience, 54, 101076. https://doi.org/10.1016/J.DCN.2022.101076
Maurer, D., Mondloch, C. J., & Lewis, T. L. (2007). Sleeper effects. In Developmental Science. https://doi.org/10.1111/j.1467-7687.2007.00562.x
McSweeney, M., Morales, S., Valadez, E. A., Buzzell, G. A., Yoder, L., Fifer, W. P., Pini, N., Shuffrey, L. C., Elliott, A. J., Isler, J. R., & Fox, N. A. (2023). Age-related trends in aperiodic EEG activity and alpha oscillations during early- to middle-childhood. NeuroImage, 269, 119925. https://doi.org/10.1016/j.neuroimage.2023.119925
Medel, V., Irani, M., Crossley, N., Ossandón, T., & Boncompte, G. (2023). Complexity and 1/f slope jointly reflect brain states. Scientific Reports, 13(1), 21700. https://doi.org/10.1038/s41598-023-47316-0
Molina, J. L., Voytek, B., Thomas, M. L., Joshi, Y. B., Bhakta, S. G., Talledo, J. A., Swerdlow, N. R., & Light, G. A. (2020). Memantine Effects on Electroencephalographic Measures of Putative Excitatory/Inhibitory Balance in Schizophrenia. Biological Psychiatry: Cognitive Neuroscience and Neuroimaging, 5(6), 562–568. https://doi.org/10.1016/j.bpsc.2020.02.004
Muthukumaraswamy, S. D., & Liley, D. T. (2018). 1/F electrophysiological spectra in resting and drug-induced states can be explained by the dynamics of multiple oscillatory relaxation processes. NeuroImage, 179(November 2017), 582–595. https://doi.org/10.1016/j.neuroimage.2018.06.068
Ossandón, J. P., Stange, L., Gudi-Mindermann, H., Rimmele, J. M., Sourav, S., Bottari, D., Kekunnaya, R., & Röder, B. (2023). The development of oscillatory and aperiodic resting state activity is linked to a sensitive period in humans. NeuroImage, 275, 120171. https://doi.org/10.1016/J.NEUROIMAGE.2023.120171
Ostlund, B. D., Alperin, B. R., Drew, T., & Karalunas, S. L. (2021). Behavioral and cognitive correlates of the aperiodic (1/f-like) exponent of the EEG power spectrum in adolescents with and without ADHD. Developmental Cognitive Neuroscience, 48, 100931. https://doi.org/10.1016/j.dcn.2021.100931
Pant, R., Ossandón, J., Stange, L., Shareef, I., Kekunnaya, R., & Röder, B. (2023). Stimulus-evoked and resting-state alpha oscillations show a linked dependence on patterned visual experience for development. NeuroImage: Clinical, 103375. https://doi.org/10.1016/J.NICL.2023.103375
Schaworonkow, N., & Voytek, B. (2021). Longitudinal changes in aperiodic and periodic activity in electrophysiological recordings in the first seven months of life. Developmental Cognitive Neuroscience, 47. https://doi.org/10.1016/j.dcn.2020.100895
Schwenk, J. C. B., VanRullen, R., & Bremmer, F. (2020). Dynamics of Visual Perceptual Echoes Following Short-Term Visual Deprivation. Cerebral Cortex Communications, 1(1). https://doi.org/10.1093/TEXCOM/TGAA012
Tanner, D., Norton, J. J. S., Morgan-Short, K., & Luck, S. J. (2016). On high-pass filter artifacts (they’re real) and baseline correction (it’s a good idea) in ERP/ERMF analysis. Journal of Neuroscience Methods, 266, 166–170. https://doi.org/10.1016/j.jneumeth.2016.01.002
Vanrullen, R., & MacDonald, J. S. P. (2012). Perceptual echoes at 10 Hz in the human brain. Current Biology. https://doi.org/10.1016/j.cub.2012.03.050
Voytek, B., Kramer, M. A., Case, J., Lepage, K. Q., Tempesta, Z. R., Knight, R. T., & Gazzaley, A. (2015). Age-related changes in 1/f neural electrophysiological noise. Journal of Neuroscience, 35(38). https://doi.org/10.1523/JNEUROSCI.2332-14.2015
Waschke, L., Wöstmann, M., & Obleser, J. (2017). States and traits of neural irregularity in the age-varying human brain. Scientific Reports 2017 7:1, 7(1), 1–12. https://doi.org/10.1038/s41598-017-17766-4
The following is the authors’ response to the original reviews.
eLife Assessment
This potentially useful study involves neuro-imaging and electrophysiology in a small cohort of congenital cataract patients after sight recovery and age-matched control participants with normal sight. It aims to characterize the effects of early visual deprivation on excitatory and inhibitory balance in the visual cortex. While the findings are taken to suggest the existence of persistent alterations in Glx/GABA ratio and aperiodic EEG signals, the evidence supporting these claims is incomplete. Specifically, small sample sizes, lack of a specific control cohort, and other methodological limitations will likely restrict the usefulness of the work, with relevance limited to scientists working in this particular subfield.
As pointed out in the public reviews, there are very few human models which allow for assessing the role of early experience on neural circuit development. While the prevalent research in permanent congenital blindness reveals the response and adaptation of the developing brain to an atypical situation (blindness), research in sight restoration addresses the question of whether and how atypical development can be remediated if typical experience (vision) is restored. The literature on the role of visual experience in the development of E/I balance in humans, assessed via Magnetic Resonance Spectroscopy (MRS), has been limited to a few studies on congenital permanent blindness. Thus, we assessed sight recovery individuals with a history of congenital blindness, as limited evidence from other researchers indicated that the visual cortex E/I ratio might differ compared to normally sighted controls.
Individuals with total bilateral congenital cataracts who remained untreated until later in life are extremely rare, particularly if only carefully diagnosed patients are included in a study sample. A sample size of 10 patients is, at the very least, typical of past studies in this population, even for exclusively behavioral assessments. In the present study, in addition to behavioral assessment as an indirect measure of sensitive periods, we investigated participants with two neuroimaging methods (Magnetic Resonance Spectroscopy and electroencephalography) to directly assess the neural correlates of sensitive periods in humans. The electroencephalography data allowed us to link the results of our small sample to findings documented in large cohorts of both, sight recovery individuals and permanently congenitally blind individuals. As pointed out in a recent editorial recommending an “exploration-then-estimation procedure,” (“Consideration of Sample Size in Neuroscience Studies,” 2020), exploratory studies like ours provide crucial direction and specific hypotheses for future work.
We included an age-matched sighted control group recruited from the same community, measured in the same scanner and laboratory, to assess whether early experience is necessary for a typical excitatory/inhibitory (E/I) ratio to emerge in adulthood. The present findings indicate that this is indeed the case. Based on these results, a possible question to answer in future work, with individuals who had developmental cataracts, is whether later visual deprivation causes similar effects. Note that even if visual deprivation at a later stage in life caused similar effects, the current results would not be invalidated; by contrast, they are essential to understand future work on late (permanent or transient) blindness.
Thus, we think that the present manuscript has far reaching implications for our understanding of the conditions under which E/I balance, a crucial characteristic of brain functioning, emerges in humans.
Finally, our manuscript is one of the first few studies that relate MRS neurotransmitter concentrations to parameters of EEG aperiodic activity. Since present research has been using aperiodic activity as a correlate of the E/I ratio, and partially of higher cognitive functions, we think that our manuscript additionally contributes to a better understanding of what might be measured with aperiodic neurophysiological activity.
Public Reviews:<br />
Reviewer #1 (Public Review):
Summary:
In this human neuroimaging and electrophysiology study, the authors aimed to characterize the effects of a period of visual deprivation in the sensitive period on excitatory and inhibitory balance in the visual cortex. They attempted to do so by comparing neurochemistry conditions ('eyes open', 'eyes closed') and resting state, and visually evoked EEG activity between ten congenital cataract patients with recovered sight (CC), and ten age-matched control participants (SC) with normal sight.
First, they used magnetic resonance spectroscopy to measure in vivo neurochemistry from two locations, the primary location of interest in the visual cortex, and a control location in the frontal cortex. Such voxels are used to provide a control for the spatial specificity of any effects because the single-voxel MRS method provides a single sampling location. Using MR-visible proxies of excitatory and inhibitory neurotransmission, Glx and GABA+ respectively, the authors report no group effects in GABA+ or Glx, no difference in the functional conditions 'eyes closed' and 'eyes open'. They found an effect of the group in the ratio of Glx/GABA+ and no similar effect in the control voxel location. They then performed multiple exploratory correlations between MRS measures and visual acuity, and reported a weak positive correlation between the 'eyes open' condition and visual acuity in CC participants.
The same participants then took part in an EEG experiment. The authors selected only two electrodes placed in the visual cortex for analysis and reported a group difference in an EEG index of neural activity, the aperiodic intercept, as well as the aperiodic slope, considered a proxy for cortical inhibition. They report an exploratory correlation between the aperiodic intercept and Glx in one out of three EEG conditions.
The authors report the difference in E/I ratio, and interpret the lower E/I ratio as representing an adaptation to visual deprivation, which would have initially caused a higher E/I ratio. Although intriguing, the strength of evidence in support of this view is not strong. Amongst the limitations are the low sample size, a critical control cohort that could provide evidence for a higher E/I ratio in CC patients without recovered sight for example, and lower data quality in the control voxel.
Strengths of study:
How sensitive period experience shapes the developing brain is an enduring and important question in neuroscience. This question has been particularly difficult to investigate in humans. The authors recruited a small number of sight-recovered participants with bilateral congenital cataracts to investigate the effect of sensitive period deprivation on the balance of excitation and inhibition in the visual brain using measures of brain chemistry and brain electrophysiology. The research is novel, and the paper was interesting and well-written.
Limitations:
(1.1) Low sample size. Ten for CC and ten for SC, and a further two SC participants were rejected due to a lack of frontal control voxel data. The sample size limits the statistical power of the dataset and increases the likelihood of effect inflation.
Applying strict criteria, we only included individuals who were born with no patterned vision in the CC group. The population of individuals who have remained untreated past infancy is small in India, despite a higher prevalence of childhood cataract than Germany. Indeed, from the original 11 CC and 11 SC participants tested, one participant each from the CC and SC group had to be rejected, as their data had been corrupted, resulting in 10 participants in each group.
It was a challenge to recruit participants from this rare group with no history of neurological diagnosis/intake of neuromodulatory medications, who were able and willing to undergo both MRS and EEG. For this study, data collection took more than 2.5 years.
We took care of the validity of our results with two measures; first, we assessed not just MRS, but additionally, EEG measures of E/I ratio. The latter allowed us to link results to a larger population of CC individuals, that is, we replicated the results of a larger group of 28 additional individuals (Ossandón et al., 2023) in our sub-group.
Second, we included a control voxel. As predicted, all group effects were restricted to the occipital voxel.
(1.2) Lack of specific control cohort. The control cohort has normal vision. The control cohort is not specific enough to distinguish between people with sight loss due to different causes and patients with congenital cataracts with co-morbidities. Further data from more specific populations, such as patients whose cataracts have not been removed, with developmental cataracts, or congenitally blind participants, would greatly improve the interpretability of the main finding. The lack of a more specific control cohort is a major caveat that limits a conclusive interpretation of the results.
The existing work on visual deprivation and neurochemical changes, as assessed with MRS, has been limited to permanent congenital blindness. In fact, most of the studies on permanent blindness included only congenitally blind or early blind humans (Coullon et al., 2015; Weaver et al., 2013), or, in separate studies, only late-blind individuals (Bernabeu et al., 2009). Thus, accordingly, we started with the most “extreme” visual deprivation model, sight recovery after congenital blindness. If we had not observed any group difference compared to normally sighted controls, investigating other groups might have been trivial. Based on our results, subsequent studies in late blind individuals, and then individuals with developmental cataracts, can be planned with clear hypotheses.
(1.3) MRS data quality differences. Data quality in the control voxel appears worse than in the visual cortex voxel. The frontal cortex MRS spectrum shows far broader linewidth than the visual cortex (Supplementary Figures). Compared to the visual voxel, the frontal cortex voxel has less defined Glx and GABA+ peaks; lower GABA+ and Glx concentrations, lower NAA SNR values; lower NAA concentrations. If the data quality is a lot worse in the FC, then small effects may not be detectable.
Worse data quality in the frontal than the visual cortex has been repeatedly observed in the MRS literature, attributable to magnetic field distortions (Juchem & Graaf, 2017) resulting from the proximity of the region to the sinuses (recent example: (Rideaux et al., 2022)). Nevertheless, we chose the frontal control region rather than a parietal voxel, given the potential neurochemical changes in multisensory regions of the parietal cortex due to blindness. Such reorganization would be less likely in frontal areas associated with higher cognitive functions. Further, prior MRS studies of the visual cortex have used the frontal cortex as a control region as well (Pitchaimuthu et al., 2017; Rideaux et al., 2022). In the revised manuscript, we more explicitly inform the reader about this data quality difference between regions in the Methods (Pages 11-12, MRS Data Quality/Table 2) and Discussion (Page 25, Lines 644- 647).
Importantly, while in the present study data quality differed between the frontal and visual cortex voxel, it did not differ between groups (Supplementary Material S6).
Further, we checked that the frontal cortex datasets for Glx and GABA+ concentrations were of sufficient quality: the fit error was below 8.31% in both groups (Supplementary Material S3). For reference, Mikkelsen et al. reported a mean GABA+ fit error of 6.24 +/- 1.95% from a posterior cingulate cortex voxel across 8 GE scanners, using the Gannet pipeline. No absolute cutoffs have been proposed for fit errors. However, MRS studies in special populations (I/E ratio assessed in narcolepsy (Gao et al., 2024), GABA concentration assessed in Autism Spectrum Disorder (Maier et al., 2022) have used frontal cortex data with a fit error of <10% to identify differences between cohorts (Gao et al., 2024; Pitchaimuthu et al., 2017). Based on the literature, MRS data from the frontal voxel of the present study would have been of sufficient quality to uncover group differences.
In the revised manuscript, we added the recently published MRS quality assessment form to the supplementary materials (Supplementary Excel File S1). Additionally, we would like to allude to our apriori prediction of group differences for the visual cortex, but not for the frontal cortex voxel. Finally, EEG data quality did not differ between frontal and occipital electrodes; therefore, lower sensitivity of frontal measures cannot easily explain the lack of group differences for frontal measures.
(1.4) Because of the direction of the difference in E/I, the authors interpret their findings as representing signatures of sight improvement after surgery without further evidence, either within the study or from the literature. However, the literature suggests that plasticity and visual deprivation drive the E/I index up rather than down. Decreasing GABA+ is thought to facilitate experience-dependent remodelling. What evidence is there that cortical inhibition increases in response to a visual cortex that is over-sensitised due to congenital cataracts? Without further experimental or literature support this interpretation remains very speculative.
Indeed, higher inhibition was not predicted, which we attempt to reconcile in our discussion section. We base our discussion mainly on the non-human animal literature, which has shown evidence of homeostatic changes after prolonged visual deprivation in the adult brain (Barnes et al., 2015). It is also interesting to note that after monocular deprivation in adult humans, resting GABA+ levels decreased in the visual cortex (Lunghi et al., 2015). Assuming that after delayed sight restoration, adult neuroplasticity mechanisms must be employed, these studies would predict a “balancing” of the increased excitatory drive following sight restoration by a commensurate increase in inhibition (Keck et al., 2017). Additionally, the EEG results of the present study allowed for speculation regarding the underlying neural mechanisms of an altered E/I ratio. The aperiodic EEG activity suggested higher spontaneous spiking (increased intercept) and increased inhibition (steeper aperiodic slope between 1-20 Hz) in CC vs SC individuals (Ossandón et al., 2023).
In the revised manuscript, we have more clearly indicated that these speculations are based primarily on non-human animal work, due to the lack of human studies on the subject (Page 23, Lines 609-613).
(1.5) Heterogeneity in the patient group. Congenital cataract (CC) patients experienced a variety of duration of visual impairment and were of different ages. They presented with co-morbidities (absorbed lens, strabismus, nystagmus). Strabismus has been associated with abnormalities in GABAergic inhibition in the visual cortex. The possible interactions with residual vision and confounds of co-morbidities are not experimentally controlled for in the correlations, and not discussed.
The goal of the present study was to assess whether we would observe changes in E/I ratio after restoring vision at all. We would not have included patients without nystagmus in the CC group of the present study, since it would have been unlikely that they experienced congenital patterned visual deprivation. Amongst diagnosticians, nystagmus or strabismus might not be considered genuine “comorbidities” that emerge in people with congenital cataracts. Rather, these are consequences of congenital visual deprivation, which we employed as diagnostic criteria. Similarly, absorbed lenses are clear signs that cataracts were congenital. As in other models of experience dependent brain development (e.g. the extant literature on congenital permanent blindness, including anophthalmic individuals (Coullon et al., 2015; Weaver et al., 2013), some uncertainty remains regarding whether the (remaining, in our case) abnormalities of the eye, or the blindness they caused, are the factors driving neural changes. In case of people with reversed congenital cataracts, at least the retina is considered to be intact, as they would otherwise not receive cataract removal surgery.
However, we consider it unlikely that strabismus caused the group differences, because the present study shows group differences in the Glx/GABA+ ratio at rest, regardless of eye opening or eye closure, for which strabismus would have caused distinct effects. By contrast, the link between GABA concentration and, for example, interocular suppression in strabismus, have so far been documented during visual stimulation (Mukerji et al., 2022; Sengpiel et al., 2006), and differed in direction depending on the amblyopic vs. non-amblyopic eye. Further, one MRS study did not find group differences in GABA concentration between the visual cortices of 16 amblyopic individuals and sighted controls (Mukerji et al., 2022), supporting that the differences in Glx/GABA+ concentration which we observed were driven by congenital deprivation, and not amblyopia-associated visual acuity or eye movement differences.
In the revised manuscript, we discussed the inclusion criteria in more detail, and the aforementioned reasons why our data remains interpretable (Page 5, Lines 143 – 145, Lines 147-149).
(1.6) Multiple exploratory correlations were performed to relate MRS measures to visual acuity (shown in Supplementary Materials), and only specific ones were shown in the main document. The authors describe the analysis as exploratory in the 'Methods' section. Furthermore, the correlation between visual acuity and E/I metric is weak, and not corrected for multiple comparisons. The results should be presented as preliminary, as no strong conclusions can be made from them. They can provide a hypothesis to test in a future study.
In the revised manuscript, we have clearly indicated that the exploratory correlation analyses are reported to put forth hypotheses for future studies (Page 4, Lines 118-128; Page 5, Lines 132-134; Page 25, Lines 644- 647).
(1.7) P.16 Given the correlation of the aperiodic intercept with age ("Age negatively correlated with the aperiodic intercept across CC and SC individuals, that is, a flattening of the intercept was observed with age"), age needs to be controlled for in the correlation between neurochemistry and the aperiodic intercept. Glx has also been shown to negatively correlate with age.
The correlation between chronological age and aperiodic intercept was observed across groups, but the correlation between Glx and the intercept of the aperiodic EEG activity was seen only in the CC group, even though the SC group was matched for age. Thus, such a correlation was very unlikely to be predominantly driven by an effect of chronological age.
In the revised manuscript, we added the linear regressions with age as a covariate (Supplementary Material S16, referred to in the main Results, Page 21, Lines 534-537), demonstrating the significant relationship between aperiodic intercept and Glx concentration in the CC group.
(1.8) Multiple exploratory correlations were performed to relate MRS to EEG measures (shown in Supplementary Materials), and only specific ones were shown in the main document. Given the multiple measures from the MRS, the correlations with the EEG measures were exploratory, as stated in the text, p.16, and in Figure 4. Yet the introduction said that there was a prior hypothesis "We further hypothesized that neurotransmitter changes would relate to changes in the slope and intercept of the EEG aperiodic activity in the same subjects." It would be great if the text could be revised for consistency and the analysis described as exploratory.
In the revised manuscript, we improved the phrasing (Page 5, Lines 130-132) and consistently reported the correlations as exploratory in the Methods and Discussion. We consider the correlation analyses as exploratory due to our sample size and the absence of prior work. However, we did hypothesize that both MRS and EEG markers would concurrently be altered in CC vs SC individuals.
(1.9) The analysis for the EEG needs to take more advantage of the available data. As far as I understand, only two electrodes were used, yet far more were available as seen in their previous study (Ossandon et al., 2023). The spatial specificity is not established. The authors could use the frontal cortex electrode (FP1, FP2) signals as a control for spatial specificity in the group effects, or even better, all available electrodes and correct for multiple comparisons. Furthermore, they could use the aperiodic intercept vs Glx in SC to evaluate the specificity of the correlation to CC.
The aperiodic intercept and slope did not differ between CC and SC individuals for Fp1 and Fp2, suggesting the spatial specificity of the results. In the revised manuscript, we added this analysis to the Supplementary Material (Supplementary Material S14) and referred to it in our Results (Page 20, Lines 513-514).
Further, Glx concentration in the visual cortex did not correlate with the aperiodic intercept in the SC group (Figure 4), suggesting that this relationship was indeed specific to the CC group.
The data from all electrodes has been analyzed and published in other studies as well (Pant et al., 2023; Ossandón et al., 2023).
Reviewer #2 (Public Review):
Summary:
The manuscript reports non-invasive measures of activity and neurochemical profiles of the visual cortex in congenitally blind patients who recovered vision through the surgical removal of bilateral dense cataracts. The declared aim of the study is to find out how restoring visual function after several months or years of complete blindness impacts the balance between excitation and inhibition in the visual cortex.
Strengths:
The findings are undoubtedly useful for the community, as they contribute towards characterising the many ways this special population differs from normally sighted individuals. The combination of MRS and EEG measures is a promising strategy to estimate a fundamental physiological parameter - the balance between excitation and inhibition in the visual cortex, which animal studies show to be heavily dependent upon early visual experience. Thus, the reported results pave the way for further studies, which may use a similar approach to evaluate more patients and control groups.
Weaknesses:
(2.1) The main issue is the lack of an appropriate comparison group or condition to delineate the effect of sight recovery (as opposed to the effect of congenital blindness). Few previous studies suggested an increased excitation/Inhibition ratio in the visual cortex of congenitally blind patients; the present study reports a decreased E/I ratio instead. The authors claim that this implies a change of E/I ratio following sight recovery. However, supporting this claim would require showing a shift of E/I after vs. before the sight-recovery surgery, or at least it would require comparing patients who did and did not undergo the sight-recovery surgery (as common in the field).
Longitudinal studies would indeed be the best way to test the hypothesis that the lower E/I ratio in the CC group observed by the present study is a consequence of sight restoration.
We have now explicitly stated this in the Limitations section (Page 25, Lines 654-655).
However, longitudinal studies involving neuroimaging are an effortful challenge, particularly in research conducted outside of major developed countries and dedicated neuroimaging research facilities. Crucially, however, had CC and SC individuals, as well as permanently congenitally blind vs SC individuals (Coullon et al., 2015; Weaver et al., 2013), not differed on any neurochemical markers, such a longitudinal study might have been trivial. Thus, in order to justify and better tailor longitudinal studies, cross-sectional studies are an initial step.
(2.2) MR Spectroscopy shows a reduced GLX/GABA ratio in patients vs. sighted controls; however, this finding remains rather isolated, not corroborated by other observations. The difference between patients and controls only emerges for the GLX/GABA ratio, but there is no accompanying difference in either the GLX or the GABA concentrations. There is an attempt to relate the MRS data with acuity measurements and electrophysiological indices, but the explorative correlational analyses do not help to build a coherent picture. A bland correlation between GLX/GABA and visual impairment is reported, but this is specific to the patients' group (N=10) and would not hold across groups (the correlation is positive, predicting the lowest GLX/GABA ratio values for the sighted controls - the opposite of what is found). There is also a strong correlation between GLX concentrations and the EEG power at the lowest temporal frequencies. Although this relation is intriguing, it only holds for a very specific combination of parameters (of the many tested): only with eyes open, only in the patient group.
We interpret these findings differently, that is, in the context of experiments from non-human animals and the larger MRS literature (Page 23, Lines 609-611).
Homeostatic control of E/I balance assumes that the ratio of excitation (reflected here by Glx) and inhibition (reflected here by GABA+) is regulated. Like prior work (Gao et al., 2024, 2024; Narayan et al., 2022; Perica et al., 2022; Steel et al., 2020; Takado et al., 2022; Takei et al., 2016), we assumed that the ratio of Glx/GABA+ is indicative of E/I balance rather than solely the individual neurotransmitter levels. One of the motivations for assessing the ratio vs the absolute concentration is that as per the underlying E/I balance hypothesis, a change in excitation would cause a concomitant change in inhibition, and vice versa, which has been shown in non-human animal work (Fang et al., 2021; Haider et al., 2006; Tao & Poo, 2005) and modeling research (Vreeswijk & Sompolinsky, 1996; Wu et al., 2022). Importantly, our interpretation of the lower E/I ratio is not just from the Glx/GABA+ ratio, but additionally, based on the steeper EEG aperiodic slope (1-20 Hz).
As stated in the Discussion section and Response 1.4, we did not expect to see a lower Glx/GABA+ ratio in CC individuals. We discuss the possible reasons for the direction of the correlation with visual acuity and aperiodic offset during passive visual stimulation, and offer interpretations and (testable) hypotheses.
We interpret the direction of the Glx/GABA+ correlation with visual acuity to imply that patients with highest (compensatory) balancing of the consequences of congenital blindness (hyperexcitation), in light of visual stimulation, are those who recover best. Note, the sighted control group was selected based on their “normal” vision. Thus, clinical visual acuity measures are not expected to sufficiently vary, nor have the resolution to show strong correlations with neurophysiological measures. By contrast, the CC group comprised patients highly varying in visual outcomes, and thus were ideal to investigate such correlations.
This holds for the correlation between Glx and the aperiodic intercept, as well. Previous work has suggested that the intercept of the aperiodic activity is associated with broadband spiking activity in neural circuits (Manning et al., 2009). Thus, an atypical increase of spiking activity during visual stimulation, as indirectly suggested by “old” non-human primate work on visual deprivation (Hyvärinen et al., 1981) might drive a correlation not observed in healthy populations.
In the revised manuscript, we have more clearly indicated in the Discussion that these are possible post-hoc interpretations (Page 23, Lines 584-586; Page 24, Lines 609-620; Page 24, Lines 644-647; Pages 25, Lines 650 - 657). We argue that given the lack of such studies in humans, it is all the more important that extant data be presented completely, even if the direction of the effects are not as expected.
(2.3) For these reasons, the reported findings do not allow us to draw firm conclusions on the relation between EEG parameters and E/I ratio or on the impact of early (vs. late) visual experience on the excitation/inhibition ratio of the human visual cortex.
Indeed, the correlations we have tested between the E/I ratio and EEG parameters were exploratory, and have been reported as such.
We have now made this clear in all the relevant parts of the manuscript (Introduction, Page 5, Lines 132-135; Methods, Page 16, Line 415; Results, Page 21, Figure 4; Discussion, Page 22, Line 568, Page 25, Lines 644-645, Page 25, Lines 650-657).
The goal of our study was not to compare the effects of early vs. late visual experience. The goal was to study whether early visual experience is necessary for a typical E/I ratio in visual neural circuits. We provided clear evidence in favor of this hypothesis. Thus, the present results suggest the necessity of investigating the effects of late visual deprivation. In fact, such research is missing in permanent blindness as well.
Reviewer #3 (Public Review):
This manuscript examines the impact of congenital visual deprivation on the excitatory/inhibitory (E/I) ratio in the visual cortex using Magnetic Resonance Spectroscopy (MRS) and electroencephalography (EEG) in individuals whose sight was restored. Ten individuals with reversed congenital cataracts were compared to age-matched, normally sighted controls, assessing the cortical E/I balance and its interrelationship to visual acuity. The study reveals that the Glx/GABA ratio in the visual cortex and the intercept and aperiodic signal are significantly altered in those with a history of early visual deprivation, suggesting persistent neurophysiological changes despite visual restoration.
My expertise is in EEG (particularly in the decomposition of periodic and aperiodic activity) and statistical methods. I have several major concerns in terms of methodological and statistical approaches along with the (over)interpretation of the results. These major concerns are detailed below.
(3.1) Variability in visual deprivation:
- The document states a large variability in the duration of visual deprivation (probably also the age at restoration), with significant implications for the sensitivity period's impact on visual circuit development. The variability and its potential effects on the outcomes need thorough exploration and discussion.
We work with a rare, unique patient population, which makes it difficult to systematically assess the effects of different visual histories while maintaining stringent inclusion criteria such as complete patterned visual deprivation at birth. Regardless, we considered the large variance in age at surgery and time since surgery as supportive of our interpretation: group differences were found despite the large variance in duration of visual deprivation. Moreover, the existing variance was used to explore possible associations between behavior and neural measures, as well as neurochemical and EEG measures.
In the revised manuscript, we have detailed the advantages (Methods, Page 5, Lines 143 – 145, Lines 147-149; Discussion, Page 26, Lines 677-678) and disadvantages (Discussion, Page 25, Lines 650-657) of our CC sample, with respect to duration of congenital visual deprivation.
(3.2) Sample size:
- The small sample size is a major concern as it may not provide sufficient power to detect subtle effects and/or overestimate significant effects, which then tend not to generalize to new data. One of the biggest drivers of the replication crisis in neuroscience.
We address the small sample size in our Discussion, and make clear that small sample sizes were due to the nature of investigations in special populations. In the revised manuscript, we added the sample sizes of previous studies using MRS in permanently blind individuals (Page 4, Lines 108 - 109). It is worth noting that our EEG results fully align with those of larger samples of congenital cataract reversal individuals (Page 25, Lines 666-676, Supplementary Material S18, S19) (Ossandón et al., 2023), providing us confidence about their validity and reproducibility. Moreover, our MRS results and correlations of those with EEG parameters were spatially specific to occipital cortex measures.
The main problem with the correlation analyses between MRS and EEG measures is that the sample size is simply too small to conduct such an analysis. Moreover, it is unclear from the methods section that this analysis was only conducted in the patient group (which the reviewer assumed from the plots), and not explained why this was done only in the patient group. I would highly recommend removing these correlation analyses.
In the revised manuscript, we have more clearly marked the correlation analyses as exploratory (Introduction, Page 4, Lines 118-128 and Page 5, Lines 132-134; Methods Page 16, Line 415; Discussion Page 22, Line 568, Page 24, Lines 644-645, Page 25, Lines 650-657); note that we do not base most of our discussion on the results of these analyses.
As indicated by Reviewer 1, reporting them allows for deriving more precise hypothesis for future studies. It has to be noted that we investigate an extremely rare population, tested outside of major developed economies and dedicated neuroimaging research facilities. In addition to being a rare patient group, these individuals come from poor communities. Therefore, we consider it justified to report these correlations as exploratory, providing direction for future research.
(3.3) Statistical concerns:
- The statistical analyses, particularly the correlations drawn from a small sample, may not provide reliable estimates (see https://www.sciencedirect.com/science/article/pii/S0092656613000858, which clearly describes this problem).
It would undoubtedly be better to have a larger sample size. We nonetheless think it is of value to the research community to publish this dataset, since 10 multimodal data sets from a carefully diagnosed, rare population, representing a human model for the effects of early experience on brain development, are quite a lot. Sample sizes in prior neuroimaging studies in transient blindness have most often ranged from n = 1 to n = 10. They nevertheless provided valuable direction for future research, and integration of results across multiple studies provides scientific insights.
Identifying possible group differences was the goal of our study, with the correlations being an exploratory analysis, which we have clearly indicated in the methods, results and discussion.
- Statistical analyses for the MRS: The authors should consider some additional permutation statistics, which are more suitable for small sample sizes. The current statistical model (2x2) design ANOVA is not ideal for such small sample sizes. Moreover, it is unclear why the condition (EO & EC) was chosen as a predictor and not the brain region (visual & frontal) or neurochemicals. Finally, the authors did not provide any information on the alpha level nor any information on correction for multiple comparisons (in the methods section). Finally, even if the groups are matched w.r.t. age, the time between surgery and measurement, the duration of visual deprivation, (and sex?), these should be included as covariates as it has been shown that these are highly related to the measurements of interest (especially for the EEG measurements) and the age range of the current study is large.
In our ANOVA models, the neurochemicals were the outcome variables, and the conditions were chosen as predictors based on prior work suggesting that Glx/GABA+ might vary with eye closure (Kurcyus et al., 2018). The study was designed based on a hypothesis of group differences localized to the occipital cortex, due to visual deprivation. The frontal cortex voxel was chosen to indicate whether these differences were spatially specific. Therefore, we conducted separate ANOVAs based on this study design.
We have now clarified the motivation for these conditions in the Introduction (Page 4, Lines 122-125) and the Methods (Page 9, Lines 219-224).
In the revised manuscript, we added the rationale for parametric analyses for our outcomes (Shapiro-Wilk as well as Levene’s tests, Supplementary Material S9). Note that in the Supplementary Materials (S12, S14), we have reported the correlations between visual history metrics and MRS/EEG outcomes, thereby investigating whether the variance in visual history might have driven these results. Specifically, we found a (negative) correlation between visual cortex Glx/GABA+ concentration during eye closure and the visual acuity in the CC group (Figure 2c). None of the other exploratory correlations between MRS/EEG outcomes vs time since surgery, duration of blindness or visual acuity were significant in the CC group (Supplementary Material S12, S15).
The alpha level used for the ANOVA models specified in the Methods section was 0.05. The alpha level for the exploratory analyses reported in the main manuscript was 0.008, after correcting for (6) multiple comparisons using the Bonferroni correction, also specified in the Methods. Note that the p-values following correction are expressed as multiplied by 6, due to most readers assuming an alpha level of 0.05 (see response regarding large p-values).
We used a control group matched for age, recruited and tested in the same institutes, using the same setup. We feel that we followed the gold standards for recruiting a healthy control group for a patient group.
- EEG statistical analyses: The same critique as for the MRS statistical analyses applies to the EEG analysis. In addition: was the 2x3 ANOVA conducted for EO and EC independently? This seems to be inconsistent with the approach in the MRS analyses, in which the authors chose EO & EC as predictors in their 2x2 ANOVA.
The 2x3 ANOVA was not conducted independently for the eyes open/eyes closed condition. The ANOVA conducted on the EEG metrics was 2x3 because it had two groups (CC, SC) and three conditions (eyes open (EO), eyes closed (EC) and visual stimulation (LU)) as predictors.
- Figure 4: The authors report a p-value of >0.999 with a correlation coefficient of -0.42 with a sample size of 10 subjects. This can't be correct (it should be around: p = 0.22). All statistical analyses should be checked.
As specified in the Methods and Figure legend, the reported p values in Figure 4 have been corrected using the Bonferroni correction, and therefore multiplied by the number of comparisons, leading to the seemingly large values.
Additionally, to check all statistical analyses, we put the manuscript through an independent Statistics Check (Nuijten & Polanin, 2020) (https://michelenuijten.shinyapps.io/statcheck-web/) and have uploaded the consistency report with the revised Supplementary Material (Supplementary Report 1).
- Figure 2c. Eyes closed condition: The highest score of the *Glx/GABA ratio seems to be ~3.6. In subplot 2a, there seem to be 3 subjects that show a Glx/GABA ratio score > 3.6. How can this be explained? There is also a discrepancy for the eyes-closed condition.
The three subjects that show the Glx/GABA+ ratio > 3.6 in subplot 2a are in the SC group, whereas the correlations plotted in figure 2c are only for the CC group, where the highest score is indeed ~3.6.
(3.4) Interpretation of aperiodic signal:
- Several recent papers demonstrated that the aperiodic signal measured in EEG or ECoG is related to various important aspects such as age, skull thickness, electrode impedance, as well as cognition. Thus, currently, very little is known about the underlying effects which influence the aperiodic intercept and slope. The entire interpretation of the aperiodic slope as a proxy for E/I is based on a computational model and simulation (as described in the Gao et al. paper).
Apart from the modeling work from Gao et al., multiple papers which have also been cited which used ECoG, EEG and MEG and showed concomitant changes in aperiodic activity with pharmacological manipulation of the E/I ratio (Colombo et al., 2019; Molina et al., 2020; Muthukumaraswamy & Liley, 2018). Further, several prior studies have interpreted changes in the aperiodic slope as reflective of changes in the E/I ratio, including studies of developmental groups (Favaro et al., 2023; Hill et al., 2022; McSweeney et al., 2023; Schaworonkow & Voytek, 2021) as well as patient groups (Molina et al., 2020; Ostlund et al., 2021).
In the revised manuscript, we have cited those studies not already included in the Introduction (Page 3, Lines 92-94).
- Especially the aperiodic intercept is a very sensitive measure to many influences (e.g. skull thickness, electrode impedance...). As crucial results (correlation aperiodic intercept and MRS measures) are facing this problem, this needs to be reevaluated. It is safer to make statements on the aperiodic slope than intercept. In theory, some of the potentially confounding measures are available to the authors (e.g. skull thickness can be computed from T1w images; electrode impedances are usually acquired alongside the EEG data) and could be therefore controlled.
All electrophysiological measures indeed depend on parameters such as skull thickness and electrode impedance. As in the extant literature using neurophysiological measures to compare brain function between patient and control groups, we used a control group matched in age/sex, recruited in the same region, tested with the same devices, and analyzed with the same analysis pipeline. For example, impedance was kept below 10 kOhm for all subjects.
This is now mentioned in the Methods, Page 13, Line 344.
There is no evidence available suggesting that congenital cataracts are associated with changes in skull thickness that would cause the observed pattern of group results. Moreover, we cannot think of how any of the exploratory correlations between neurophysiological measures and MRS measures could be accounted for by a difference e.g. in skull thickness.
- The authors wrote: "Higher frequencies (such as 20-40 Hz) have been predominantly associated with local circuit activity and feedforward signaling (Bastos et al., 2018; Van Kerkoerle et al., 2014); the increased 20-40 Hz slope may therefore signal increased spontaneous spiking activity in local networks. We speculate that the steeper slope of the aperiodic activity for the lower frequency range (1-20 Hz) in CC individuals reflects the concomitant increase in inhibition." The authors confuse the interpretation of periodic and aperiodic signals. This section refers to the interpretation of the periodic signal (higher frequencies). This interpretation cannot simply be translated to the aperiodic signal (slope).
Prior work has not always separated the aperiodic and periodic components, making it unclear what might have driven these effects in our data. The interpretation of the higher frequency range was intended to contrast with the interpretations of lower frequency range, in order to speculate as to why the two aperiodic fits might go in differing directions. Note that Ossandón et al. reported highly similar results (group differences for CC individuals and for permanently congenitally blind humans) for the aperiodic activity between 20-40 Hz and oscillatory activity in the gamma range.
In the revised Discussion, we removed this section. We primarily interpret the increased offset and prior findings from fMRI-BOLD data (Raczy et al., 2023) as an increase in broadband neuronal firing.
- The authors further wrote: We used the slope of the aperiodic (1/f) component of the EEG spectrum as an estimate of E/I ratio (Gao et al., 2017; Medel et al., 2020; Muthukumaraswamy & Liley, 2018). This is a highly speculative interpretation with very little empirical evidence. These papers were conducted with ECoG data (mostly in animals) and mostly under anesthesia. Thus, these studies only allow an indirect interpretation by what the 1/f slope in EEG measurements is actually influenced.
Note that Muthukumaraswamy et al. (2018) used different types of pharmacological manipulations and analyzed periodic and aperiodic MEG activity in humans, in addition to monkey ECoG (Muthukumaraswamy & Liley, 2018). Further, Medel et al. (now published as Medel et al., 2023) compared EEG activity in addition to ECoG data after propofol administration. The interpretation of our results are in line with a number of recent studies in developing (Hill et al., 2022; Schaworonkow & Voytek, 2021) and special populations using EEG. As mentioned above, several prior studies have used the slope of the 1/f component/aperiodic activity as an indirect measure of the E/I ratio (Favaro et al., 2023; Hill et al., 2022; McSweeney et al., 2023; Molina et al., 2020; Ostlund et al., 2021; Schaworonkow & Voytek, 2021), including studies using scalp-recorded EEG from humans.
In the introduction of the revised manuscript, we have made more explicit that this metric is indirect (Page 3, Line 91), (additionally see Discussion, Page 24, Lines 644-645, Page 25, Lines 650-657).
While a full understanding of aperiodic activity needs to be provided, some convergent ideas have emerged. We think that our results contribute to this enterprise, since our study is, to the best of our knowledge, the first which assessed MRS measured neurotransmitter levels and EEG aperiodic activity.
(3.5) Problems with EEG preprocessing and analysis:
- It seems that the authors did not identify bad channels nor address the line noise issue (even a problem if a low pass filter of below-the-line noise was applied).
As pointed out in the methods and Figure 1, we only analyzed data from two occipital channels, O1 and O2 neither of which were rejected for any participant. Channel rejection was performed for the larger dataset, published elsewhere (Ossandón et al., 2023; Pant et al., 2023). As control sites we added the frontal channels FP1 and Fp2 (see Supplementary Material S14)
Neither Ossandón et al. (2023) nor Pant et al. (2023) considered frequency ranges above 40 Hz to avoid any possible contamination with line noise. Here, we focused on activity between 0 and 20 Hz, definitely excluding line noise contaminations (Methods, Page 14, Lines 365-367). The low pass filter (FIR, 1-45 Hz) guaranteed that any spill-over effects of line noise would be restricted to frequencies just below the upper cutoff frequency.
Additionally, a prior version of the analysis used spectrum interpolation to remove line noise; the group differences remained stable (Ossandón et al., 2023). We have reported this analysis in the revised manuscript (Page 14, Lines 364-357).
Further, both groups were measured in the same lab, making line noise (~ 50 Hz) as an account for the observed group effects in the 1-20 Hz frequency range highly unlikely. Finally, any of the exploratory MRS-EEG correlations would be hard to explain if the EEG parameters would be contaminated with line noise.
- What was the percentage of segments that needed to be rejected due to the 120μV criteria? This should be reported specifically for EO & EC and controls and patients.
The mean percentage of 1 second segments rejected for each resting state condition and the percentage of 6.25 long segments rejected in each group for the visual stimulation condition have been added to the revised manuscript (Supplementary Material S10), and referred to in the Methods on Page 14, Lines 372-373).
- The authors downsampled the data to 60Hz to "to match the stimulation rate". What is the intention of this? Because the subsequent spectral analyses are conflated by this choice (see Nyquist theorem).
This data were collected as part of a study designed to evoke alpha activity with visual white-noise, which changed in luminance with equal power at all frequencies from 1-60 Hz, restricted by the refresh rate of the monitor on which stimuli were presented (Pant et al., 2023). This paradigm and method was developed by VanRullen and colleagues (Schwenk et al., 2020; VanRullen & MacDonald, 2012), wherein the analysis requires the same sampling rate between the presented frequencies and the EEG data. The downsampling function used here automatically applies an anti-aliasing filter (EEGLAB 2019) .
- "Subsequently, baseline removal was conducted by subtracting the mean activity across the length of an epoch from every data point." The actual baseline time segment should be specified.
The time segment was the length of the epoch, that is, 1 second for the resting state conditions and 6.25 seconds for the visual stimulation conditions. This has now been explicitly stated in the revised manuscript (Page 14, Lines 379-380).
- "We excluded the alpha range (8-14 Hz) for this fit to avoid biasing the results due to documented differences in alpha activity between CC and SC individuals (Bottari et al., 2016; Ossandón et al., 2023; Pant et al., 2023)." This does not really make sense, as the FOOOF algorithm first fits the 1/f slope, for which the alpha activity is not relevant.
We did not use the FOOOF algorithm/toolbox in this manuscript. As stated in the Methods, we used a 1/f fit to the 1-20 Hz spectrum in the log-log space, and subtracted this fit from the original spectrum to obtain the corrected spectrum. Given the pronounced difference in alpha power between groups (Bottari et al., 2016; Ossandón et al., 2023; Pant et al., 2023), we were concerned it might drive differences in the exponent values. Our analysis pipeline had been adapted from previous publications of our group and other labs (Ossandón et al., 2023; Voytek et al., 2015; Waschke et al., 2017).
We have conducted the analysis with and without the exclusion of the alpha range, as well as using the FOOOF toolbox both in the 1-20 Hz and 20-40 Hz ranges (Ossandón et al., 2023). The findings of a steeper slope in the 1-20 Hz range as well as lower alpha power in CC vs SC individuals remained stable. In Ossandón et al., the comparison between the piecewise fits and FOOOF fits led the authors to use the former, as it outperformed the FOOOF algorithm for their data.
- The model fits of the 1/f fitting for EO, EC, and both participant groups should be reported.
In Figure 3 of the manuscript, we depicted the mean spectra and 1/f fits for each group.
In the revised manuscript, we added the fit quality metrics (average R<sup>2</sup> values > 0.91 for each group and condition) (Methods Page 15, Lines 395-396; Supplementary Material S11) and additionally show individual subjects’ fits (Supplementary Material S11).
(3.6) Validity of GABA measurements and results:
- According the a newer study by the authors of the Gannet toolbox (https://analyticalsciencejournals.onlinelibrary.wiley.com/doi/abs/10.1002/nbm.5076), the reliability and reproducibility of the gamma-aminobutyric acid (GABA) measurement can vary significantly depending on acquisition and modeling parameter. Thus, did the author address these challenges?
We took care of data quality while acquiring MRS data by ensuring appropriate voxel placement and linewidth prior to scanning (Page 9, Lines 229-237). We now address this explicitly in the Methods in the “MRS Data Quality” section. Acquisition as well as modeling parameters were constant for both groups, so they cannot have driven group differences.
The linked article compares the reproducibility of GABA measurement using Osprey (Oeltzschner et al., 2020), which was released in 2020 and uses linear combination modeling to fit the peak, as opposed to Gannet’s simple peak fitting (Hupfeld et al., 2024). The study finds better test-retest reliability for Osprey compared to Gannet’s method.
As the present work was conceptualized in 2018, we used Gannet 3.0, which was the state-of-the-art edited-spectrum analysis toolbox at the time, and still is widely used.
In the revised manuscript, we re-analyzed the data using linear combination modeling with Osprey (Oeltzschner et al., 2020), and reported that the main findings remained the same, i.e. the Glx/GABA+ concentration ratio was lower in the visual cortex of congenital cataract reversal individuals compared to normally sighted controls, regardless of whether participants were scanned with eyes open or with eyes closed. Further, NAA concentration did not differ between groups (Supplementary Material S3). Thus, we demonstrate that our findings were robust to analysis pipelines, and state this in the Methods (Page 9, Lines 242-246) and Results (Page 19, Lines 464-467).
- Furthermore, the authors wrote: "We confirmed the within-subject stability of metabolite quantification by testing a subset of the sighted controls (n=6) 2-4 weeks apart. Looking at the supplementary Figure 5 (which would be rather plotted as ICC or Blant-Altman plots), the within-subject stability compared to between-subject variability seems not to be great. Furthermore, I don't think such a small sample size qualifies for a rigorous assessment of stability.
Indeed, we did not intend to provide a rigorous assessment of within-subject stability. Rather, we aimed to confirm that data quality/concentration ratios did not systematically differ between the same subjects tested longitudinally; driven, for example, by scanner heating or time of day. As with the phantom testing, we attempted to give readers an idea of the quality of the data, as they were collected from a primarily clinical rather than a research site.
In the revised manuscript, we have removed the statement regarding stability and the associated section.
- "Why might an enhanced inhibitory drive, as indicated by the lower Glx/GABA ratio" Is this interpretation really warranted, as the results of the group differences in the Glx/GABA ratio seem to be rather driven by a decreased Glx concentration in CC rather than an increased GABA (see Figure 2).
We used the Glx/GABA+ ratio as a measure, rather than individual Glx or GABA+ concentration, which did not significantly differ between groups. As detailed in Response 2.2, we think this metric aligns better with an underlying E/I balance hypothesis and has been used in many previous studies (Gao et al., 2024; Liu et al., 2015; Narayan et al., 2022; Perica et al., 2022).
Our interpretation of an enhanced inhibitory drive additionally comes from the combination of aperiodic EEG (1-20 Hz) and MRS measures, which, when considered together, are consistent with a decreased E/I ratio.
In the revised manuscript, we have rewritten the Discussion and removed this section.
- Glx concentration predicted the aperiodic intercept in CC individuals' visual cortices during ambient and flickering visual stimulation. Why specifically investigate the Glx concentration, when the paper is about E/I ratio?
As stated in the methods, we exploratorily assessed the relationship between all MRS parameters (Glx, GABA+ and Glx/GABA+ ratio) with the aperiodic parameters (slope, offset), and corrected for multiple comparisons accordingly. We think this is a worthwhile analysis considering the rarity of the dataset/population (see 1.2, 1.6, 2.1 and Reviewer 1’s comments about future hypotheses). We only report the Glx – aperiodic intercept correlation in the main manuscript as it survived correction for multiple comparisons.
(3.7) Interpretation of the correlation between MRS measurements and EEG aperiodic signal:
- The authors wrote: "The intercept of the aperiodic activity was highly correlated with the Glx concentration during rest with eyes open and during flickering stimulation (also see Supplementary Material S11). Based on the assumption that the aperiodic intercept reflects broadband firing (Manning et al., 2009; Winawer et al., 2013), this suggests that the Glx concentration might be related to broadband firing in CC individuals during active and passive visual stimulation." These results should not be interpreted (or with very caution) for several reasons (see also problem with influences on aperiodic intercept and small sample size). This is a result of the exploratory analyses of correlating every EEG parameter with every MRS parameter. This requires well-powered replication before any interpretation can be provided. Furthermore and importantly: why should this be specifically only in CC patients, but not in the SC control group?
We have indicated clearly in all parts of the manuscript that these correlations are presented as exploratory. Further, we interpret the Glx-aperiodic offset correlation, and none of the others, as it survived the Bonferroni correction for multiple comparisons. We offer a hypothesis in the Discussion as to why such a correlation might exist in the CC but not the SC group (see response 2.2), and do not speculate further.
(3.8) Language and presentation:
- The manuscript requires language improvements and correction of numerous typos. Over-simplifications and unclear statements are present, which could mislead or confuse readers (see also interpretation of aperiodic signal).
In the revised manuscript, we have checked that speculations are clearly marked, and typos are removed.
- The authors state that "Together, the present results provide strong evidence for experience-dependent development of the E/I ratio in the human visual cortex, with consequences for behavior." The results of the study do not provide any strong evidence, because of the small sample size and exploratory analyses approach and not accounting for possible confounding factors.
We disagree with this statement and allude to convergent evidence of both MRS and neurophysiological measures. The latter link to corresponding results observed in a larger sample of CC individuals (Ossandón et al., 2023). In the revised manuscript, we have rephrased the statement as “to provide initial evidence” (Page 22, Line 676).
- "Our results imply a change in neurotransmitter concentrations as a consequence of *restoring* vision following congenital blindness." This is a speculative statement to infer a causal relationship on cross-sectional data.
As mentioned under 2.1, we conducted a cross-sectional study which might justify future longitudinal work. In order to advance science, new testable hypotheses were put forward at the end of a manuscript.
In the revised manuscript, we rephrased the sentence and added “might imply” to better indicate the hypothetical character of this idea (Page 22, Lines 586-587).
- In the limitation section, the authors wrote: "The sample size of the present study is relatively high for the rare population , but undoubtedly, overall, rather small." This sentence should be rewritten, as the study is plein underpowered. The further justification "We nevertheless think that our results are valid. Our findings neurochemically (Glx and GABA+ concentration), and anatomically (visual cortex) specific. The MRS parameters varied with parameters of the aperiodic EEG activity and visual acuity. The group differences for the EEG assessments corresponded to those of a larger sample of CC individuals (n=38) (Ossandón et al., 2023), and effects of chronological age were as expected from the literature." These statements do not provide any validation or justification of small samples. Furthermore, the current data set is a subset of an earlier published paper by the same authors "The EEG data sets reported here were part of data published earlier (Ossandón et al., 2023; Pant et al., 2023)." Thus, the statement "The group differences for the EEG assessments corresponded to those of a larger sample of CC individuals (n=38) " is a circular argument and should be avoided.
Our intention was not to justify having a small sample, but to justify why we think the results might be valid as they align with/replicate existing literature.
In the revised manuscript, we added a figure showing that the EEG results of the 10 subjects considered here correspond to those of the 28 other subjects of Ossandón et al (Supplementary Material S18). We adapted the text accordingly, clearly stating that the pattern of EEG results of the ten subjects reported here replicate those of the 28 additional subjects of Ossandón et al. (2023) (Page 25, Lines 671-672).
References (Public Review)
Barnes, S. J., Sammons, R. P., Jacobsen, R. I., Mackie, J., Keller, G. B., & Keck, T. (2015). Subnetwork-specific homeostatic plasticity in mouse visual cortex in vivo. Neuron, 86(5), 1290–1303. https://doi.org/10.1016/J.NEURON.2015.05.010
Bernabeu, A., Alfaro, A., García, M., & Fernández, E. (2009). Proton magnetic resonance spectroscopy (1H-MRS) reveals the presence of elevated myo-inositol in the occipital cortex of blind subjects. NeuroImage, 47(4), 1172–1176. https://doi.org/10.1016/j.neuroimage.2009.04.080
Bottari, D., Troje, N. F., Ley, P., Hense, M., Kekunnaya, R., & Röder, B. (2016). Sight restoration after congenital blindness does not reinstate alpha oscillatory activity in humans. Scientific Reports. https://doi.org/10.1038/srep24683
Colombo, M. A., Napolitani, M., Boly, M., Gosseries, O., Casarotto, S., Rosanova, M., Brichant, J. F., Boveroux, P., Rex, S., Laureys, S., Massimini, M., Chieregato, A., & Sarasso, S. (2019). The spectral exponent of the resting EEG indexes the presence of consciousness during unresponsiveness induced by propofol, xenon, and ketamine. NeuroImage, 189(September 2018), 631–644. https://doi.org/10.1016/j.neuroimage.2019.01.024
Consideration of Sample Size in Neuroscience Studies. (2020). Journal of Neuroscience, 40(21), 4076–4077. https://doi.org/10.1523/JNEUROSCI.0866-20.2020
Coullon, G. S. L., Emir, U. E., Fine, I., Watkins, K. E., & Bridge, H. (2015). Neurochemical changes in the pericalcarine cortex in congenital blindness attributable to bilateral anophthalmia. Journal of Neurophysiology. https://doi.org/10.1152/jn.00567.2015
Fang, Q., Li, Y. T., Peng, B., Li, Z., Zhang, L. I., & Tao, H. W. (2021). Balanced enhancements of synaptic excitation and inhibition underlie developmental maturation of receptive fields in the mouse visual cortex. Journal of Neuroscience, 41(49), 10065–10079. https://doi.org/10.1523/JNEUROSCI.0442-21.2021
Favaro, J., Colombo, M. A., Mikulan, E., Sartori, S., Nosadini, M., Pelizza, M. F., Rosanova, M., Sarasso, S., Massimini, M., & Toldo, I. (2023). The maturation of aperiodic EEG activity across development reveals a progressive differentiation of wakefulness from sleep. NeuroImage, 277. https://doi.org/10.1016/J.NEUROIMAGE.2023.120264
Gao, Y., Liu, Y., Zhao, S., Liu, Y., Zhang, C., Hui, S., Mikkelsen, M., Edden, R. A. E., Meng, X., Yu, B., & Xiao, L. (2024). MRS study on the correlation between frontal GABA+/Glx ratio and abnormal cognitive function in medication-naive patients with narcolepsy. Sleep Medicine, 119, 1–8. https://doi.org/10.1016/j.sleep.2024.04.004
Haider, B., Duque, A., Hasenstaub, A. R., & McCormick, D. A. (2006). Neocortical network activity in vivo is generated through a dynamic balance of excitation and inhibition. Journal of Neuroscience. https://doi.org/10.1523/JNEUROSCI.5297-05.2006
Hill, A. T., Clark, G. M., Bigelow, F. J., Lum, J. A. G., & Enticott, P. G. (2022). Periodic and aperiodic neural activity displays age-dependent changes across early-to-middle childhood. Developmental Cognitive Neuroscience, 54, 101076. https://doi.org/10.1016/J.DCN.2022.101076
Hupfeld, K. E., Zöllner, H. J., Hui, S. C. N., Song, Y., Murali-Manohar, S., Yedavalli, V., Oeltzschner, G., Prisciandaro, J. J., & Edden, R. A. E. (2024). Impact of acquisition and modeling parameters on the test–retest reproducibility of edited GABA+. NMR in Biomedicine, 37(4), e5076. https://doi.org/10.1002/nbm.5076
Hyvärinen, J., Carlson, S., & Hyvärinen, L. (1981). Early visual deprivation alters modality of neuronal responses in area 19 of monkey cortex. Neuroscience Letters, 26(3), 239–243. https://doi.org/10.1016/0304-3940(81)90139-7
Juchem, C., & Graaf, R. A. de. (2017). B0 magnetic field homogeneity and shimming for in vivo magnetic resonance spectroscopy. Analytical Biochemistry, 529, 17–29. https://doi.org/10.1016/j.ab.2016.06.003
Keck, T., Hübener, M., & Bonhoeffer, T. (2017). Interactions between synaptic homeostatic mechanisms: An attempt to reconcile BCM theory, synaptic scaling, and changing excitation/inhibition balance. Current Opinion in Neurobiology, 43, 87–93. https://doi.org/10.1016/J.CONB.2017.02.003
Kurcyus, K., Annac, E., Hanning, N. M., Harris, A. D., Oeltzschner, G., Edden, R., & Riedl, V. (2018). Opposite Dynamics of GABA and Glutamate Levels in the Occipital Cortex during Visual Processing. Journal of Neuroscience, 38(46), 9967–9976. https://doi.org/10.1523/JNEUROSCI.1214-18.2018
Liu, B., Wang, G., Gao, D., Gao, F., Zhao, B., Qiao, M., Yang, H., Yu, Y., Ren, F., Yang, P., Chen, W., & Rae, C. D. (2015). Alterations of GABA and glutamate-glutamine levels in premenstrual dysphoric disorder: A 3T proton magnetic resonance spectroscopy study. Psychiatry Research - Neuroimaging, 231(1), 64–70. https://doi.org/10.1016/J.PSCYCHRESNS.2014.10.020
Lunghi, C., Berchicci, M., Morrone, M. C., & Russo, F. D. (2015). Short‐term monocular deprivation alters early components of visual evoked potentials. The Journal of Physiology, 593(19), 4361. https://doi.org/10.1113/JP270950
Maier, S., Düppers, A. L., Runge, K., Dacko, M., Lange, T., Fangmeier, T., Riedel, A., Ebert, D., Endres, D., Domschke, K., Perlov, E., Nickel, K., & Tebartz van Elst, L. (2022). Increased prefrontal GABA concentrations in adults with autism spectrum disorders. Autism Research, 15(7), 1222–1236. https://doi.org/10.1002/aur.2740
Manning, J. R., Jacobs, J., Fried, I., & Kahana, M. J. (2009). Broadband shifts in local field potential power spectra are correlated with single-neuron spiking in humans. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 29(43), 13613–13620. https://doi.org/10.1523/JNEUROSCI.2041-09.2009
McSweeney, M., Morales, S., Valadez, E. A., Buzzell, G. A., Yoder, L., Fifer, W. P., Pini, N., Shuffrey, L. C., Elliott, A. J., Isler, J. R., & Fox, N. A. (2023). Age-related trends in aperiodic EEG activity and alpha oscillations during early- to middle-childhood. NeuroImage, 269, 119925. https://doi.org/10.1016/j.neuroimage.2023.119925
Medel, V., Irani, M., Crossley, N., Ossandón, T., & Boncompte, G. (2023). Complexity and 1/f slope jointly reflect brain states. Scientific Reports, 13(1), 21700. https://doi.org/10.1038/s41598-023-47316-0
Molina, J. L., Voytek, B., Thomas, M. L., Joshi, Y. B., Bhakta, S. G., Talledo, J. A., Swerdlow, N. R., & Light, G. A. (2020). Memantine Effects on Electroencephalographic Measures of Putative Excitatory/Inhibitory Balance in Schizophrenia. Biological Psychiatry: Cognitive Neuroscience and Neuroimaging, 5(6), 562–568. https://doi.org/10.1016/j.bpsc.2020.02.004
Mukerji, A., Byrne, K. N., Yang, E., Levi, D. M., & Silver, M. A. (2022). Visual cortical γ−aminobutyric acid and perceptual suppression in amblyopia. Frontiers in Human Neuroscience, 16. https://doi.org/10.3389/fnhum.2022.949395
Muthukumaraswamy, S. D., & Liley, D. T. (2018). 1/F electrophysiological spectra in resting and drug-induced states can be explained by the dynamics of multiple oscillatory relaxation processes. NeuroImage, 179(November 2017), 582–595. https://doi.org/10.1016/j.neuroimage.2018.06.068
Narayan, G. A., Hill, K. R., Wengler, K., He, X., Wang, J., Yang, J., Parsey, R. V., & DeLorenzo, C. (2022). Does the change in glutamate to GABA ratio correlate with change in depression severity? A randomized, double-blind clinical trial. Molecular Psychiatry, 27(9), 3833—3841. https://doi.org/10.1038/s41380-022-01730-4
Nuijten, M. B., & Polanin, J. R. (2020). “statcheck”: Automatically detect statistical reporting inconsistencies to increase reproducibility of meta-analyses. Research Synthesis Methods, 11(5), 574–579. https://doi.org/10.1002/jrsm.1408
Oeltzschner, G., Zöllner, H. J., Hui, S. C. N., Mikkelsen, M., Saleh, M. G., Tapper, S., & Edden, R. A. E. (2020). Osprey: Open-source processing, reconstruction & estimation of magnetic resonance spectroscopy data. Journal of Neuroscience Methods, 343, 108827. https://doi.org/10.1016/j.jneumeth.2020.108827
Ossandón, J. P., Stange, L., Gudi-Mindermann, H., Rimmele, J. M., Sourav, S., Bottari, D., Kekunnaya, R., & Röder, B. (2023). The development of oscillatory and aperiodic resting state activity is linked to a sensitive period in humans. NeuroImage, 275, 120171. https://doi.org/10.1016/J.NEUROIMAGE.2023.120171
Ostlund, B. D., Alperin, B. R., Drew, T., & Karalunas, S. L. (2021). Behavioral and cognitive correlates of the aperiodic (1/f-like) exponent of the EEG power spectrum in adolescents with and without ADHD. Developmental Cognitive Neuroscience, 48, 100931. https://doi.org/10.1016/j.dcn.2021.100931
Pant, R., Ossandón, J., Stange, L., Shareef, I., Kekunnaya, R., & Röder, B. (2023). Stimulus-evoked and resting-state alpha oscillations show a linked dependence on patterned visual experience for development. NeuroImage: Clinical, 103375. https://doi.org/10.1016/J.NICL.2023.103375
Perica, M. I., Calabro, F. J., Larsen, B., Foran, W., Yushmanov, V. E., Hetherington, H., Tervo-Clemmens, B., Moon, C.-H., & Luna, B. (2022). Development of frontal GABA and glutamate supports excitation/inhibition balance from adolescence into adulthood. Progress in Neurobiology, 219, 102370. https://doi.org/10.1016/j.pneurobio.2022.102370
Pitchaimuthu, K., Wu, Q. Z., Carter, O., Nguyen, B. N., Ahn, S., Egan, G. F., & McKendrick, A. M. (2017). Occipital GABA levels in older adults and their relationship to visual perceptual suppression. Scientific Reports, 7(1). https://doi.org/10.1038/S41598-017-14577-5
Rideaux, R., Ehrhardt, S. E., Wards, Y., Filmer, H. L., Jin, J., Deelchand, D. K., Marjańska, M., Mattingley, J. B., & Dux, P. E. (2022). On the relationship between GABA+ and glutamate across the brain. NeuroImage, 257, 119273. https://doi.org/10.1016/J.NEUROIMAGE.2022.119273
Schaworonkow, N., & Voytek, B. (2021). Longitudinal changes in aperiodic and periodic activity in electrophysiological recordings in the first seven months of life. Developmental Cognitive Neuroscience, 47. https://doi.org/10.1016/j.dcn.2020.100895
Schwenk, J. C. B., VanRullen, R., & Bremmer, F. (2020). Dynamics of Visual Perceptual Echoes Following Short-Term Visual Deprivation. Cerebral Cortex Communications, 1(1). https://doi.org/10.1093/TEXCOM/TGAA012
Sengpiel, F., Jirmann, K.-U., Vorobyov, V., & Eysel, U. T. (2006). Strabismic Suppression Is Mediated by Inhibitory Interactions in the Primary Visual Cortex. Cerebral Cortex, 16(12), 1750–1758. https://doi.org/10.1093/cercor/bhj110
Steel, A., Mikkelsen, M., Edden, R. A. E., & Robertson, C. E. (2020). Regional balance between glutamate+glutamine and GABA+ in the resting human brain. NeuroImage, 220. https://doi.org/10.1016/J.NEUROIMAGE.2020.117112
Takado, Y., Takuwa, H., Sampei, K., Urushihata, T., Takahashi, M., Shimojo, M., Uchida, S., Nitta, N., Shibata, S., Nagashima, K., Ochi, Y., Ono, M., Maeda, J., Tomita, Y., Sahara, N., Near, J., Aoki, I., Shibata, K., & Higuchi, M. (2022). MRS-measured glutamate versus GABA reflects excitatory versus inhibitory neural activities in awake mice. Journal of Cerebral Blood Flow & Metabolism, 42(1), 197. https://doi.org/10.1177/0271678X211045449
Takei, Y., Fujihara, K., Tagawa, M., Hironaga, N., Near, J., Kasagi, M., Takahashi, Y., Motegi, T., Suzuki, Y., Aoyama, Y., Sakurai, N., Yamaguchi, M., Tobimatsu, S., Ujita, K., Tsushima, Y., Narita, K., & Fukuda, M. (2016). The inhibition/excitation ratio related to task-induced oscillatory modulations during a working memory task: A multtimodal-imaging study using MEG and MRS. NeuroImage, 128, 302–315. https://doi.org/10.1016/J.NEUROIMAGE.2015.12.057
Tao, H. W., & Poo, M. M. (2005). Activity-dependent matching of excitatory and inhibitory inputs during refinement of visual receptive fields. Neuron, 45(6), 829–836. https://doi.org/10.1016/J.NEURON.2005.01.046
Vanrullen, R., & MacDonald, J. S. P. (2012). Perceptual echoes at 10 Hz in the human brain. Current Biology. https://doi.org/10.1016/j.cub.2012.03.050
Voytek, B., Kramer, M. A., Case, J., Lepage, K. Q., Tempesta, Z. R., Knight, R. T., & Gazzaley, A. (2015). Age-related changes in 1/f neural electrophysiological noise. Journal of Neuroscience, 35(38). https://doi.org/10.1523/JNEUROSCI.2332-14.2015
Vreeswijk, C. V., & Sompolinsky, H. (1996). Chaos in neuronal networks with balanced excitatory and inhibitory activity. Science, 274(5293), 1724–1726. https://doi.org/10.1126/SCIENCE.274.5293.1724
Waschke, L., Wöstmann, M., & Obleser, J. (2017). States and traits of neural irregularity in the age-varying human brain. Scientific Reports 2017 7:1, 7(1), 1–12. https://doi.org/10.1038/s41598-017-17766-4
Weaver, K. E., Richards, T. L., Saenz, M., Petropoulos, H., & Fine, I. (2013). Neurochemical changes within human early blind occipital cortex. Neuroscience. https://doi.org/10.1016/j.neuroscience.2013.08.004
Wu, Y. K., Miehl, C., & Gjorgjieva, J. (2022). Regulation of circuit organization and function through inhibitory synaptic plasticity. Trends in Neurosciences, 45(12), 884–898. https://doi.org/10.1016/J.TINS.2022.10.006
Recommendations for the Authors:
Reviewer #1 (Recommendations for The Authors):
Thank you for the interesting submission. I have inserted my comments to the authors here. Some of them will be more granular comments related to the concerns raised in the public review.
(1) Introduction:
Could you please justify the rationale for using eyes open and eyes closed in the MRS condition, and the use of the three different conditions in the EEG experiment? If these resulted in negative findings, then the implications should be discussed.
Previous work with MRS in sighted individuals has suggested that eye opening in darkness results in a decrease of visual cortex GABA+ concentration, while visual stimulation results in an increase of Glx concentration, compared to a baseline concentration at eye closure (Kurcyus et al., 2018). Moreover visual stimulation/eye opening is known to result in an alpha desynchronization (Adrian & Matthews, 1934).
While previous work of our group has shown significantly reduced alpha oscillatory activity in congenital cataract reversal individual, desynchronization following eye opening was indistinguishable when compared to normally sighted controls (Ossandón et al., 2023; Pant et al., 2023).
Thus, we decided to include both conditions to test whether a similar pattern of results would emerge for GABA+/Glx concentration.
We added our motivation to the Introduction of the revised manuscript (Page 4, Lines 122-125) along with the Methods (Page 9, Lines 219-223).
It does not become clear from the introduction why a higher intercept is predicted in the EEG measure. The rationale for this hypothesis needs to be explained better.
Given the prior findings suggesting an increased E/I ratio in CC individuals and the proposed link between neuronal firing (Manning et al., 2009) and the aperiodic intercept, we expected a higher intercept for the CC compared to the SC group.
We have now added this explanation to the Introduction (Page 4, Lines 126-128).
(2) Participants
Were participants screened for common MRS exclusion criteria such as history of psychiatric conditions or antidepressant medication, which could alter neurochemistry? If not, then this needs to be pointed out.
All participants were clinically screened at the LV Prasad Eye Institute, and additionally self-reported no neurological or psychiatric conditions or medications. Moreover, all subjects were screened based exclusion criteria for being scanned using the standard questionnaire of the radiology center.
We have now made this clear in the Methods (Page 7, Lines 168-171).
Table 1 needs to show the age of the participant, which can only be derived by adding the columns 'duration of deprivation' and 'time since surgery'. Table 1 also needs to include the controls.
We have accordingly modified Table 1 in the revised manuscript and added age for the patients as well as the controls (Table 1, Pages 6-7).
The control cohort is not specific enough to exclude reduced visual acuity, or co-morbidities, as the primary driver of the differences between groups. Ideally, a cohort with developmental cataracts is recruited. Normally sighted participants as a control cohort cannot distinguish between different types of sight loss, or stages of plasticity.
The goal of this study was not to distinguish between different types of sight loss or stages of plasticity. We aimed to assess whether the most extreme forms of visual deprivation (i.e. congenital and total patterned vision loss) affected the E/I ratio. Low visual acuity and nystagmus are genuine diagnostic criteria (Methods, Page 5, Lines 142-145). Visual acuity cannot solely explain the current findings, since the MRS data were acquired both with eyes closed or diffuse visual stimulation in a dimly lit room, without any visual task.
With the awareness of the present results, we consider it worthwhile for the future to investigate additional groups such as developmental cataract-reversal individuals, to narrow down the contribution of the age of onset and degree of visual deprivation to the observed group differences.
(3) Data collection and analysis
- More detail is needed: how long were the sessions, how long was each part?
We have added this information on Page 7, Lines 178-181 of the Methods. MRS scanning took between 45 and 60 minutes, EEG testing took 20 minutes excluding the time for capping, and visual acuity testing took 3-5 minutes.
- It should be mentioned here that the EEG data is a reanalysis of a subset of legacy data, published previously in Ossandón et al., 2023; Pant et al., 2023.
In the revised manuscript, we explicitly state at the beginning of the “Electrophysiology recordings” section of the Methods (Page 13, Lines 331-334) that the EEG datasets were a subset of previously published data.
(4) MRS Spectroscopy
- Please fill out the minimum reporting standards form (Lin et al., 2021), or report all the requested measures in the main document https://pubmed.ncbi.nlm.nih.gov/33559967/
We have now filled out this form and added it as Supplementary Material (Supplementary Excel File 1). Additionally, all the requested information has been moved to the Methods section of the main document (MRS Data Quality, Pages 10-12).
- Information on how the voxels were placed is missing. The visual cortex voxel is not angled parallel to the calcarine, as is a common way to capture processing in the early visual cortex. Describe in the paper what the criteria for successful placement were, and how was it ensured that non-brain tissue was avoided in a voxel of this size.
Voxel placement was optimized in each subject to avoid the meninges, ventricles, skull and subcortical structures, ensured by examining the voxel region across slices in the acquired T1 volume for each subject. Saturation bands were placed to nullify the skull signal during MRS acquisition, at the anterior (frontal) and posterior (visual) edge of the voxel for every subject. Due to limitations in the clinical scanner rotated/skewed voxels were not possible, and thus voxels were not always located precisely parallel to the calcarine.
We have added this information to Page 9 (Lines 229-237) of the revised manuscript.
- Figure 1. shows voxels that are very close to the edge of the brain (frontal cortex) or to the tentorium (visual cortex). Could the authors please calculate the percentage overlap between the visual cortex MRS voxel and the visual cortex, and compare them across groups to ensure that there is no between-group bias from voxel placement?
We have now added the requested analysis to Supplementary Material S2 and referred to it in the main manuscript on Page 9, Lines 236-237.
Briefly, the percentage overlap with areas V1-V6 in every individual subject’s visual cortex voxel was 60% or more; the mean overlap in the CC group was 67% and the SC group 70%. The percentage overlap did not differ between groups ( t-test (t(18) = -1.14, p = 0.269)).
- Figure 1. I would recommend displaying data on a skull-stripped image to avoid identifying information from the participant's T1 profile.
We have now replaced the images in Figure 1 with skull-stripped images. Note that images from SPM12 were used instead of GannetCoregister, as GannetCoregister only displays images with the skull.
- Please show more rigor with the MRS quality measures. Several examples of inconsistency and omissions are below.
• SNR was quantified and shows a difference in SNR between voxel positions, with lower SNR in the frontal cortex. No explanation or discussion of the difference was provided.
• Looking at S1, the linewidth of NAA seems to be a lot broader in the frontal cortex than in the visual cortex. The figures suggest that acquisition quality was very different between voxel locations, making the comparison difficult.
• Linewidth of NAA is a generally agreed measure of shim quality in megapress acquisitions (Craven et al., 2022).
The data quality difference between the frontal and visual cortices has been observed in the literature (Juchem & Graaf, 2017; Rideaux et al., 2022). We nevertheless chose a frontal cortex voxel as control site instead of the often-chosen sensorimotor cortex. The main motivation was to avoid any cortical region linked to sensory processing since crossmodal compensation as a consequence of visual deprivation is a well-documented phenomenon.
We now make this clearer in the Methods (Page 11, Lines 284 – 299), in the Discussion/Limitations (Page 25, Lines 662 - 665).
- To get a handle on the data quality, I would recommend that the authors display their MRS quality measures in a separate section 'MRS quality measure', including NAA linewidth, NAA SNR, GABA+ CRLB, Glx CRLB, and test for the main effects and interaction of voxel location (VC, FC) and group (SC, CC) and discuss any discrepancies.
We have moved all the quality metric values for GABA+, Glx and NAA from the supplement to the Methods section (see Table 2), and added the requested section titled “MRS Data quality.”
We have conducted the requested analyses and reported them in Supplementary Material S6: there was a strong effect of region confirming that data quality was better in the visual than frontal region. We have referred to this in the main manuscript on Page 11, Line 299.
In the revised manuscript, we discuss the data quality in the frontal cortex, and how we ensured it was comparable to prior work. Moreover, there were no significant group effects, or group-by-region interactions, suggesting that group differences observed for the visual cortex voxel cannot be accounted for by differences in data quality. We now included a section on data quality, both in the Methods (Page 11, Lines 284 – 299), and the limitations section of the Discussion (Page 25, Lines 662 - 665).
Please clarify the MRS acquisition, "Each MEGA- PRESS scan lasted for 8 minutes and was acquired with the following specifications: TR = 2000 ms, TE = 68 ms, Voxel size = 40 mm x 30 mm x 25mm, 192 averages (each consists of two TRs). "192 averages x 2 TRs x 2s TR = 12.8 min, not 8 min, apologies if I have misunderstood these details.
We have corrected this error in the revised manuscript and stated the parameters more clearly – there were a total of 256 averages, resulting in an (256 repetitions with 1 TR * 2 s/60) 8.5-minute scan (Page 8, Lines 212-213).
- What was presented to participants in the eyes open MRS? Was it just normal room illumination or was it completely dark? Please add details to your methods.
The scans were conducted in regular room illumination, with no visual stimulation.
We have now clarified this on Page 9 (Lines 223-224) of the Methods.
(5) MRS analysis
How was the tissue fraction correction performed? Please add or refer to the exact equation from Harris et al., 2015.
We have clarified that the reported GABA+/Glx values are water-normalized alpha corrected values (Page 10, Line 249), and cited Harris et al., 2015 on Page 10 (Line 251) of the Methods.
(6) Statistical approach
How was the sample size determined? Please add your justification for the sample size
We collected as many qualifying patients as we were able to recruit for this study within 2.5 years of data collection (commencing August 2019, ending February 2022), given the constraints of the patient population and the pandemic. We have now made this clear in the Discussion (Page 25, Lines 650-652).
Please report the tests for normality.
We have now reported the Shapiro-Wilk test results for normality as well as Levene’s test for homogeneity of variance between groups for every dependent variable in our dataset in Supplementary Material S9, and added references to it in the descriptions of the statistical analyses (Methods, Page13, Lines 326-329 and Page 15, Lines 400-402).
Calculate the Bayes Factor where possible.
As our analyses are all frequentist, instead of re-analyzing the data within a Bayesian framework, we added partial eta squared values for all the reported ANOVAs (η<sub>p</sub><sup>²</sup>) for readers to get an idea of the effect size (Results).
I recommend partial correlations to control for the influence of age, duration, and time of surgery, rather than separate correlations.
Given the combination of small sample size and the expected multicollinearity in our variables (duration of blindness, for example, would be expected to correlate with age, as well as visual acuity post-surgery), partial correlations could not be calculated on this data.
We are aware of the limits of correlational analyses. Given the unique data set of a rare population we had exploratorily planned to relate behavioral, EEG and MRS parameters by calculating correlations. Since no similar data existed when we started (and to the best of our knowledge our data set is still unique), these correlation analyses were explorative, but the most transparent to run.
We have now clearly outlined these limitations in our Introduction (Page 5, Lines 133-135), Methods (Page 15, Lines 408-410) and Discussion section (Page 24, Line 634, Page 25, Lines 652-65) to ensure that the results are interpreted with appropriate caution.
(7) Visual acuity
Is the VA monocular average, from the dominant eye, or bilateral?
We have now clarified that the VA reported here is bilateral (Methods, Page 7 Line 165 and Page 15, Line 405). Bilateral visual acuity in congenital cataract-reversal individuals typically corresponds to the visual acuity of the best eye.
It is mentioned here that correlations with VA are exploratory, please be consistent as the introduction mentions that there was a hypothesis that you sought to test.
We have now accordingly modified the Introduction (Page 5, Lines 133-135) and added the appropriate caveats in the discussion with regards to interpretations (Page 25, Lines 652-665).
(8) Correlation analyses between MRS and EEG
It is mentioned here that correlations between EEG and MRS are exploratory, please consistently point out the exploratory nature, as these results are preliminary and should not be overinterpreted ("We did not have prior hypotheses as to the best of our knowledge no extant literature has tested the correlation between aperiodic EEG activity and MRS measures of GABA+,Glx and Glx/GABA+." ).
In the revised manuscript, we explicitly state the reported associations between EEG (aperiodic component) and MRS parameters allow for putting forward directed / more specific hypotheses for future studies (Introduction, Page 5, Lines 133-135; Methods, Page 15, Line 415. Discussion, Page 25, Lines 644-645 and Lines 652-665).
(9) Results
Figure 2 uses the same y-axis for the visual cortex and frontal cortex to facilitate a comparison between the two locations. Comparing Figure 2 a with b demonstrates poorer spectral peaks and reduced amplitudes. Lower spectral quality in the frontal cortex voxel could contribute to the absence of a group effect in the control voxel location. The major caveat that spectral quality differs between voxels needs to be pointed out and the limitations thereof discussed.
We have now explicitly pointed out this issue in the Methods (MRS Data Quality, Supplementary Material S6) and Discussion in the Limitations section (Page 25, Lines 662-665). While data quality was lower for the frontal compared to the visual cortex voxels, as has been observed previously (Juchem & Graaf, 2017; Rideaux et al., 2022), this was not an issue for the EEG recordings. Thus, lower sensitivity of frontal measures cannot easily explain the lack of group differences for frontal measures. Crucially, data quality did not differ between groups.
The results in 2c are the result of multiple correlations with metabolite values ("As in previous studies, we ran a number of exploratory correlation analyses between GABA+, Glx, and Glx/GABA+ concentrations, and visual acuity at the date of testing, duration of visual deprivation, and time since surgery respectively in the CC group"), it seems at least six for the visual acuity measure (VA vs Glx, VA vs GABA+, VA vs Glx/GABA+ x 2 conditions). While the trends are interesting, they should be interpreted with caution because of the exploratory nature, small sample size, the lack of multiple comparison correction, and the influence of two extreme data points. The authors should not overinterpret these results and should point out the need for replication.
See response to (6) last section, which we copy here for convenience:
We are aware of the limits of correlational analyses. Given the unique data set of a rare population we exploratorily related behavioral, EEG and MRS parameters by calculating correlations. Since no similar data existed when we started (and to the best of our knowledge our data set is still unique), these correlation analyses were explorative, but the most transparent to run.
We have now clearly outlined these limitations in our Discussion section to ensure that the results are interpreted with appropriate caution (Discussion, Page 25, Lines 644-645 and Lines 652-665).
(10) Discussion:
Please explain the decrease in E/I balance from MRS in view of recent findings on an increase in E/I balance in CC using RSN-fMRI (Raczy et al., 2022) and EEG (Ossandon et al. 2023).
We have edited our Abstract (Page 1-2, Lines 31-35) and Discussion (Page 23, Lines 584-590; Page 24, Lines 613-620). In brief, we think our results reflect a homeostatic regulation of E/I balance, that is, an increase in inhibition due to an increase in stimulus driven excitation following sight restoration.
Names limitations but does nothing to mitigate concerns about spatial specificity. The limitations need to be rewritten to include differences in SNR between the visual cortex and frontal lobe. Needs to include caveats of small samples, including effect inflation.
We have now discussed the data quality differences between the visual and frontal cortex voxel in MRS data quality, which we find irrespective of group (MRS Data Quality, Supplementary Material S6). We also reiterate why this might not explain our results; data quality was comparable to prior studies which have found group differences in frontal cortex (Methods Page 11, Lines 284 – 299), and data quality did not differ between groups. Further, EEG data quality did not differ across frontal and occipital regions, but group differences in EEG datasets were localized to the occipital cortex.
Reviewer #2 (Recommendations for The Authors):
To address the main weakness, the authors could consider including data from a third group, of congenitally blind individuals. Including this would go a very long way towards making the findings interpretable and relating them to the rest of the literature.
Unfortunately, recruitment of these groups was not possible due to the pandemic. Indeed, we would consider a pre- vs post- surgery approach the most suitable design in the future, which, however, will require several years to be completed. Such time and resource intensive longitudinal studies are justified by the present cross-sectional results.
We have explicitly stated our contribution and need for future studies in the Limitations section of the Discussion (Page 25, Lines 650-657).
Analysing the amplitude of alpha rhythms, as well as the other "aperiodic" components, would be useful to relate the profile of the tested patients with previous studies. Visual inspection of Figure 3 suggests that alpha power with eyes closed is not reduced in the patients' group compared to the controls. This would be inconsistent with previous studies (including research from the same group) and it could suggest that the small selected sample is not really representative of the sight-recovery population - certainly one of the most heterogeneous study populations. This further highlights the difficulty of drawing conclusions on the effects of visual experience merely based on this N=10 set of patients.
Alpha power was indeed reduced in the present subsample of 10 CC individuals (Supplementary Material S19). A possible source of the confusion (that the graphs of the CC and SC group look so similar for the EC condition in Figure 3) likely is that the spectra are shown with aperiodic components not yet removed, and scales to accommodate very different alpha power values. As documented in Supplementary Material S18 and S19, alpha power and the aperiodic intercept/slope results of the resting state data in the present 10 CC individuals correspond to the results from a larger sample of CC individuals (n = 28) in Ossandón et al., 2023. We explicitly highlight this “replication” in the main manuscript (Page 25 -26, Lines 671-676). Thus, the present sub-sample of CC individuals are representative for their population.
To further characterise the MRS results, the authors may consider an alternative normalisation scheme. It is not clear whether the lack of significant GABA and GLX differences in the face of a significant group difference in the GLX/GABA ratio is due to the former measures being noisier since taking the ratio between two metabolites often helps reduce inter-individual variability and thereby helps revealing group differences. It remains an open question whether the GABA or GLX concentrations would show significant group differences after appropriate normalisation (e.g. NAA?).
We repeated the analysis with Creatine-normalized values of GABA+ and Glx, and the main results i.e. reduced Glx/GABA+ concentration in the visual cortex of CC vs SC individuals, and no such difference in the frontal cortex, remained the same (Supplementary Material S5).
Further, we re-analyzed the data using Osprey, an open-source toolbox that uses linear combination modeling, and found once more that our results did not change (Supplementary Material S3). We refer to these findings in the Methods (Page 10, Lines 272-275) and Results (Page 10, Lines 467-471) of the main manuscript.
In fact, the Glx concentration in the visual cortex of CC vs SC individuals was significantly decreased when Cr-normalized values were used (which was not significant in the original analysis). However, we do not interpret this result as it was not replicated with the water-normalized values from Gannet or Osprey.
I suggest revising the discussion to present a more balanced picture of the existent evidence of the relation between E/I and EEG indices. Although there is evidence that the 1/f slope changes across development, in a way that could be consistent with a higher slope reflecting more immature and excitable tissue, the link with cortical E/I is far from established, especially when referring to specific EEG indices (intercept vs. slope, measured in lower vs. higher frequency ranges).
We have revised the Introduction (Page 4, Line 91, Lines 101-102) and Discussion (Page 22, Lines 568-569, Page 24, Lines 645-647 and Lines 654-657) in the manuscript accordingly; we allude to the fact that the links between cortical E/I and aperiodic EEG indices have not yet been unequivocally established in the literature.
Minor:
- The authors estimated NAA concentration with different software than the one used to estimate GLX and GABA; this examined the OFF spectra only; I suggest that the authors consider running their analysis with LCModel, which would allow a straightforward approach to estimate concentrations of all three metabolites from the same edited spectrum and automatically return normalised concentrations as well as water-related ones.
We re-analyzed all of the MRS datasets using Osprey, which uses linear combination modelling and has shown quantification results similar to LCModel for NAA (Oeltzschner et al., 2020). The results of a lower Glx/GABA+ concentration in the visual cortex of CC vs SC individuals, and no difference in NAA concentration, were replicated using this pipeline.
We have now added these analyses to the Supplementary Material S3 and referred to them in the Methods (Page 9, Lines 242-246) and Results (Page 18, Lines 464-467).
- Of course the normalisation used to estimate GABA and GLX values is completely irrelevant when the two values are expressed as ratio GLX/GABA - this may be reflected in the text ("water normalised GLX/GABA concentration" should read "GLX/GABA concentration" instead).
We have adapted the text on Page 16 (Line 431) and have ensured that throughout the manuscript the use of “water-normalized” is in reference to Glx or GABA+ concentration, and not the ratio.
- Please specify which equation was used for tissue correction - is it alpha-correction?
We have clarified that the reported GABA+/Glx values are water-normalized alpha corrected values (Page 10, Line 249), and cited Harris et al., 2015 on Page 10 (Line 251) of the Methods.
- Since ANOVA was used, the assumption is that values are normally distributed. Please report evidence supporting this assumption.
We have now reported the Shapiro-Wilk test results for normality as well as Levene’s test for homogeneity of variance between groups for every dependent variable in our dataset in Supplementary Material S9, and added references to it in the Methods (Page 13, Lines 326-329 and Page 15, Lines 400-402).
Reviewer #3 (Recommendations for The Authors):
In addition to addressing major comments listed in my Public Review, I have the following, more granular comments, which should also be addressed:
(1) The paper's structure could be improved by presenting visual acuity data before diving into MRS and EEG results to better contextualize the findings.
We now explicitly state in the Methods (Page 5, Line 155) that lower visual acuity is expected in a cohort of CC individuals with long lasting congenital visual deprivation.
We have additionally included a plot of visual acuities of the two groups (Supplementary Material S1).
(2) The paper should better explain the differences between CC for which sight is restored and congenitally blind patients. The authors write in the introduction that there are sensitive periods/epochs during the lifespan for the development of local inhibitory neural circuits. and "Human neuroimaging studies have similarly demonstrated that visual experience during the first weeks and months of life is crucial for the development of visual circuits. If human infants born with dense bilateral cataracts are treated later than a few weeks from birth, they suffer from a permanent reduction of not only visual acuity (Birch et al., 1998; Khanna et al., 2013) and stereovision (Birch et al., 1993; Tytla et al., 1993) but additionally from impairments in higher-level visual functions, such as face perception (Le Grand et al., 2001; Putzar et al., 2010; Röder et al., 2013)...".
Thus it seems that the current participants (sight restored after a sensitive period) seem to be similarly affected by the development of the local inhibitory circuits as congenitally blind. To assess the effect of plasticity and sight restoration longitudinal data would be necessary.
In the Introduction (Page 2, Lines 59-64; Page 3, Lines 111-114) we added that in order to identify sensitive periods e.g. for the elaboration of visual neural circuits, sight recovery individuals need to be investigated. The study of permanently blind individuals allows for investigating the role of experience (whether sight is necessary to introduce the maturation of visual neural circuits), but not whether visual input needs to be available at early epochs in life (i.e. whether sight restoration following congenital blindness could nevertheless lead to the development of visual circuits).
This is indeed the conclusion we make in the Discussion section. We have now highlighted the need for longitudinal assessments in the Discussion (Page 25, Lines 654-656).
(3) What's the underlying idea of analyzing two separate aperiodic slopes (20-40Hz and 1-19Hz). This is very unusual to compute the slope between 20-40 Hz, where the SNR is rather low.
"Ossandón et al. (2023), however, observed that in addition to the flatter slope of the aperiodic power spectrum in the high frequency range (20-40 Hz), the slope of the low frequency range (1-19 Hz) was steeper in both, congenital cataract-reversal individuals, as well as in permanently congenitally blind humans."
The present manuscript computed the slope between 1-20 Hz. Ossandón et al. as well as Medel et al. (2023) found a “knee” of the 1/f distribution at 20 Hz and describe further the motivations for computing both slope ranges. For example, Ossandón et al. used a data driven approach and compared single vs. dual fits and found that the latter fitted the data better. Additionally, they found the best fit if a knee at 20 Hz was used. We would like to point out that no standard range exists for the fitting of the 1/f component across the literature and, in fact, very different ranges have been used (Gao et al., 2017; Medel et al., 2023; Muthukumaraswamy & Liley, 2018).
(4) "For this scan, participants were instructed to keep their eyes closed and stay as still as possible." Why should it be important to have the eyes closed during a T1w data acquisition? This statement at this location does not make sense.
To avoid misunderstandings, we removed this statement in this context.
(5) "Two SC subjects did not complete the frontal cortex scan for the EO condition and were excluded from the statistical comparisons of frontal cortex neurotransmitter concentrations."<br />
Why did the authors not conduct whole-brain MRS, which seems to be on the market for quite some time (e.g. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3590062/) ?
Similar to previous work (Coullon et al., 2015; Weaver et al., 2013) our hypothesis was related to the visual cortex, and we chose the frontal cortex voxel as a control. This has now been clarified in the Introduction (Page 4, Lines 103-114), Methods (Page 9, Lines 225-227) and Discussion (Page 25, Lines 662-665).
(6) In "....during visual stimulation with stimuli that changed in luminance (LU) (Pant et al., 2023)." the authors should provide a link on the visual stimulation, which is provided further below
In the revised manuscript, we have moved up the description of the visual stimulation (Page 13, Line 336).
(7) "During the EO condition, participants were asked to fixate on a blank screen." This is not really possible. Typically, resting state EO conditions include a fixation cross, as the participants would not be able to fixate on a blank screen and move their eyes, which would impact the recordings.
We have now rephrased this as “look towards” with the goal of avoiding eye movements (Page 14, Line 347).
(8) "Components corresponding to horizontal or vertical eye movements were identified via visual inspection and removed (Plöchl et al., 2012)." It is unclear what the Plöchl reference should serve for. Is the intention of the authors to state that manual (and subjective) visual inspection of the ICA components is adequate? I would recommend removing this reference.
The intention was to provide the basis for classification during the visual inspection, as opposed to an automated method such as ICLabel.
We stated this clearly in the revised manuscript (Page 14 Lines 368-370).
(9) "The datasets were divided into 6.25 s long epochs corresponding to each trial." This is a bit inaccurate, as the trial also included some motor response task. Thus, I assume the 6.25 s are related to the visual stimulation.
We have modified the sentence accordingly (Page 15, Line 378).
(10) Figure 2. a & b. Just an esthetic suggestion: I would recommend removing the lines between the EC and EO conditions, as they suggest some longitudinal changes. Unless it is important to highlight the changes between EC and EO within each subject.
In fact, EC vs. EO was a within-subject factor with expected changes for the EEG and possible changes in the MRS parameters. To allow the reader to track changes due to EC vs. EO for individual subjects (rather than just comparing the change in the mean scores), we use lines.
(11) Figure 3A: I would plot the same y-axis range for both groups to make it more comparable.
We have changed Figure 3A accordingly.
(12) " flattening of the intercept" replaces flattening, as it is too related to slope.
We have replaced “flattening” with “reduction” (Page 20, Line 517).
(13) The plotting of only the significant correlation between MRS measures and EEG measures seems to be rather selective reporting. For this type of exploratory analysis, I would recommend plotting all of the scatter plots and moving the entire exploratory analysis to the supplementary (as this provides the smallest evidence of the results).
We have made clear in the Methods (Page 16, Lines 415-426), Results and Discussion (page 24, Lines 644-645), as well as in the Supplementary material, that the reason for only reporting the significant correlation was that this correlation survived correction for multiple comparisons, while all other correlations did not. We additionally explicitly allude to the Supplementary Material where the plots for all correlations are shown (Results, Page 21, Lines 546-552).
(14) "Here, we speculate that due to limited structural plasticity after a phase of congenital blindness, the neural circuits of CC individuals, which had adapted to blindness after birth, employ available, likely predominantly physiological plasticity mechanisms (Knudsen, 1998; Mower et al., 1985; Röder et al., 2021), in order to re-adapt to the newly available visual excitation following sight restoration."
I don't understand the logic here. The CC individuals are congenitally blind, thus why should there be any physiological plasticity mechanism to adapt to blindness, if they were blind at birth?
With “adapt to blindness” we mean adaptation of a brain to an atypical or unexpected condition when taking an evolutionary perspective (i.e. the lack of vision). We have made this clear in the revised manuscript (Introduction, Page 4, Lines 111-114; Discussion, Page 23, Lines 584-591).
(15) "An overall reduction in Glx/GABA ratio would counteract the aforementioned adaptations to congenital blindness, e.g. a lower threshold for excitation, which might come with the risk of runaway excitation in the presence of restored visually-elicited excitation."
This could be tested by actually investigating the visual excitation by visual stimulation studies.
The visual stimulation condition in the EEG experiment of the present study found a higher aperiodic intercept in CC compared to SC individuals. Given the proposed link between the intercept and spontaneous neural firing (Manning et al., 2009), we interpreted the higher intercept in CC individuals as increased broadband neural firing during visual stimulation (Results Figure 3; Discussion Page 24, Lines 635-640). This idea is compatible with enhanced BOLD responses during an EO condition in CC individuals (Raczy et al., 2022). Future work should systematically manipulate visual stimulation to test this idea.
(16) As the authors also collected T1w images, the hypothesis of increased visual cortex thickness in CC. Was this investigated?
This hypothesis was investigated in a separate publication which included this subset of participants (Hölig et al., 2023), and found increased visual cortical thickness in the CC group. We refer to this publication, and related work (Feng et al., 2021) in the present manuscript.
(17) The entire discussion of age should be omitted, as the current data set is too small to assess age effects.
We have removed this section and just allude to the fact that we replicated typical age trends to underline the validity of the present data (Page 26, Lines 675-676).
(18) Table1: should include the age and the age at the time point of surgery.
We added age to the revised Table 1. We clarified that in CC individuals, duration of blindness is the same as age at the time point of surgery (Page 6, Line 163).
(19) Why no group comparisons of visual acuity are reported?
Lower visual acuity in CC than SC individuals is a well-documented fact.
We have now added the visual acuity plots for readers (Supplementary Material S1, referred to in the Methods, Page 5, Line 155) which highlight this common finding.
References (Recommendations to the Authors)
Adrian, E. D., & Matthews, B. H. C. (1934). The berger rhythm: Potential changes from the occipital lobes in man. Brain. https://doi.org/10.1093/brain/57.4.355
Coullon, G. S. L., Emir, U. E., Fine, I., Watkins, K. E., & Bridge, H. (2015). Neurochemical changes in the pericalcarine cortex in congenital blindness attributable to bilateral anophthalmia. Journal of Neurophysiology. https://doi.org/10.1152/jn.00567.2015
Feng, Y., Collignon, O., Maurer, D., Yao, K., & Gao, X. (2021). Brief postnatal visual deprivation triggers long-lasting interactive structural and functional reorganization of the human cortex. Frontiers in Medicine, 8, 752021. https://doi.org/10.3389/FMED.2021.752021/BIBTEX
Gao, R., Peterson, E. J., & Voytek, B. (2017). Inferring synaptic excitation/inhibition balance from field potentials. NeuroImage, 158(March), 70–78. https://doi.org/10.1016/j.neuroimage.2017.06.078
Hölig, C., Guerreiro, M. J. S., Lingareddy, S., Kekunnaya, R., & Röder, B. (2023). Sight restoration in congenitally blind humans does not restore visual brain structure. Cerebral Cortex, 33(5), 2152–2161. https://doi.org/10.1093/CERCOR/BHAC197
Juchem, C., & Graaf, R. A. de. (2017). B0 magnetic field homogeneity and shimming for in vivo magnetic resonance spectroscopy. Analytical Biochemistry, 529, 17–29. https://doi.org/10.1016/j.ab.2016.06.003
Kurcyus, K., Annac, E., Hanning, N. M., Harris, A. D., Oeltzschner, G., Edden, R., & Riedl, V. (2018). Opposite Dynamics of GABA and Glutamate Levels in the Occipital Cortex during Visual Processing. Journal of Neuroscience, 38(46), 9967–9976. https://doi.org/10.1523/JNEUROSCI.1214-18.2018
Manning, J. R., Jacobs, J., Fried, I., & Kahana, M. J. (2009). Broadband shifts in local field potential power spectra are correlated with single-neuron spiking in humans. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 29(43), 13613–13620. https://doi.org/10.1523/JNEUROSCI.2041-09.2009
Medel, V., Irani, M., Crossley, N., Ossandón, T., & Boncompte, G. (2023). Complexity and 1/f slope jointly reflect brain states. Scientific Reports, 13(1), 21700. https://doi.org/10.1038/s41598-023-47316-0
Muthukumaraswamy, S. D., & Liley, D. T. (2018). 1/F electrophysiological spectra in resting and drug-induced states can be explained by the dynamics of multiple oscillatory relaxation processes. NeuroImage, 179(November 2017), 582–595. https://doi.org/10.1016/j.neuroimage.2018.06.068
Oeltzschner, G., Zöllner, H. J., Hui, S. C. N., Mikkelsen, M., Saleh, M. G., Tapper, S., & Edden, R. A. E. (2020). Osprey: Open-source processing, reconstruction & estimation of magnetic resonance spectroscopy data. Journal of Neuroscience Methods, 343, 108827. https://doi.org/10.1016/j.jneumeth.2020.108827
Ossandón, J. P., Stange, L., Gudi-Mindermann, H., Rimmele, J. M., Sourav, S., Bottari, D., Kekunnaya, R., & Röder, B. (2023). The development of oscillatory and aperiodic resting state activity is linked to a sensitive period in humans. NeuroImage, 275, 120171. https://doi.org/10.1016/J.NEUROIMAGE.2023.120171
Pant, R., Ossandón, J., Stange, L., Shareef, I., Kekunnaya, R., & Röder, B. (2023). Stimulus-evoked and resting-state alpha oscillations show a linked dependence on patterned visual experience for development. NeuroImage: Clinical, 103375. https://doi.org/10.1016/J.NICL.2023.103375
Raczy, K., Holig, C., Guerreiro, M. J. S., Lingareddy, S., Kekunnaya, R., & Roder, B. (2022). Typical resting-state activity of the brain requires visual input during an early sensitive period. Brain Communications, 4(4). https://doi.org/10.1093/BRAINCOMMS/FCAC146
Rideaux, R., Ehrhardt, S. E., Wards, Y., Filmer, H. L., Jin, J., Deelchand, D. K., Marjańska, M., Mattingley, J. B., & Dux, P. E. (2022). On the relationship between GABA+ and glutamate across the brain. NeuroImage, 257, 119273. https://doi.org/10.1016/J.NEUROIMAGE.2022.119273
Weaver, K. E., Richards, T. L., Saenz, M., Petropoulos, H., & Fine, I. (2013). Neurochemical changes within human early blind occipital cortex. Neuroscience. https://doi.org/10.1016/j.neuroscience.2013.08.004