Cai et al. [117] interviewed 21 pathologists who used a deep neural network to aid in thediagnosis of prostate cancer. The interviews showed that pathologists needed to learn moreabout the network’s strengths and limitations to use it effectively. They also wanted to knowthe design objective of the network and the kind of data on which it was trained.
10,000 Matching Annotations
- Last 7 days
-
glassmanlab.seas.harvard.edu glassmanlab.seas.harvard.edu
-
-
Roy et al. [715] explored what happens when users doing a task with a simulated crane needto choose between further using automation and manually continuing the task. The authorsshowed that the decision of whether to use automation is affected by the users’ perceptionof its accuracy as well as how easy it is to do the task themselves.
-
The performance of the system must be reliable and controllable. Its behavior should be safe, and the way it is designed and used should be ethical [768]. Users need to trust the system's decisions and ability. It should be made clear to the user what it can and cannot do.
statements that describe assertions of desirable system properties
-
such systems should be designed to take into account the fact that automated results will inevitably be incorrect on occasion.
statements that describe assertions of desirable system properties
-
users can be trained to understand not only the decision-making tasks but also the underpinning capabilities and limitations of the automation solution.
statements that describe assertions of desirable system properties
-
When the system fails, users need to be able to redirect it. To avoid biases and discrimination, some level of transparency and explainability is required.
statements that describe assertions of desirable system properties
-
In other scenarios, the agent may be continuously monitoring and analyzing theuser’s actions and proactively acting to assist the user.
-
They typi-cally do so by using some kind of AI that takes over part of a task from the user.
Tags
- user needs
- concept: agent
- ai-pending
- ai-user-approved
- concept: mixed-initiative interface
- concept: delegation
- possible desirable system properties
- user knowledge desires
- concept: ai-assisted decision making
- factors influencing human-AI team performance
- factors influencing user behavior
Annotators
URL
-
-
social-media-ethics-automation.github.io social-media-ethics-automation.github.io
-
Reflection Questions
This example about storing gender made me realize that data design is always a trade-off between simplicity and inclusiveness. While using fixed categories can make data easier to organize and analyze, it can also exclude people whose identities don’t fit those options. At the same time, allowing completely open input can make the data messy and harder to use. It makes me think that there is no perfect solution, and designers have to balance usability with fairness when deciding how to store information.
-
-
social-media-ethics-automation.github.io social-media-ethics-automation.github.io
-
Julia Evans. Examples of floating point problems. January 2023. URL: https://jvns.ca/blog/2023/01/13/examples-of-floating-point-problems/ (visited on 2023-11-24).
The author talks about the different use cases of floating points as well as explaining what they are. Floating point numbers are numbers that contain decimals, similar to integers with the exception that they can be longer and tend to be more precise forms of keeping track of numerical data; floats are often used in more complex math operations.
-
hannon Bond. Elon Musk wants out of the Twitter deal. It could end up costing at least $1 billion. NPR, July 2022. URL: https://www.npr.org/2022/07/08/1110539504/twitter-elon-musk-deal-jeopardy (visited on 2023-11-24).
The article discusses how Elon Musk wanted to halt his acquisition of Twitter once information regarding real users vs bots had come out. Muskl faced legal challengers but the discourse resulted in lower acquisition costs from his legal team. The article highlights in the business or economic sense how bots are viewed when it comes to platforms and how in a way, they can be a negative feature, which indicates a lack of platform use and therefore lower revenue generation for corporations.
-
Julia Evans. Examples of floating point problems. January 2023. URL: https://jvns.ca/blog/2023/01/13/examples-of-floating-point-problems/ (visited on 2023-11-24).
I was reading the Julia Evans article, and what stuck out to me was specifically the odometer example. This was supposed to be a program that tracked 10,000 km, but it only actually tracked 262 km because floating-point errors kept accumulating over time. I originally thought of floating point as a super technical concept, but this example makes it seem much clearer and less technical to me, and makes me think twice about how much we should trust computers when it comes to accuracy.
-
W3Schools. Introduction to HTML. URL: https://www.w3schools.com/html/html_intro.asp (visited on 2023-11-24).
This source provides a foundational summary of HyperText Markup Language (HTML), explaining that it is the standard markup language for creating web pages by describing the structure of a webpage through a series of elements. A key detail from the source is that HTML elements tell the browser how to display content, using tags like
for headings and
for paragraphs to label pieces of content.
-
Ruta Butkute. The dark side of voluntourism selfies. June 2018. URL: https://kinder.world/articles/you/the-dark-side-of-voluntourism-selfies-18537 (visited on 2023-11-24).
I found this article interesting because it introduced me to points that I have never considered before. Specifically, I feel that I have seen too many of these "voluntourism" selfies on social media platforms, and I considered them normal without considering the real implications. For instance, the fact that these pictures create the generalization that all of Africa is in poverty.
-
My last name is to long, what do I do? June 2019. Section: Get your taxes done using TurboTax. URL: https://ttlc.intuit.com/community/taxes/discussion/my-last-name-is-to-long-what-do-i-do/00/655670 (visited on 2023-11-24).
While reading this post, I noticed the user is dealing with a problem where their last name is too long for the system, and others in the comments are sharing advice or similar situations. I realize how systems like TurboTax simplify real-world information by setting character limits. In reality, names don’t have a fixed length, so this can be frustrating for people with longer names. I can relate to this because my own name is also quite long, and I’ve had similar issues where some characters get cut off when entering my information. So I often have to double check multiple times to make sure everything is correct. It also shows that some users are not fully supported by these systems and I wonder why these limits exist and whether they could be made more flexible.
-
-
social-media-ethics-automation.github.io social-media-ethics-automation.github.io
-
“Design justice is a framework for analysis of how design distributes benefits and burdens between various groups of people. Design justice focuses explicitly on the ways that design reproduces and/or challenges the matrix of domination (white supremacy, heteropatriarchy, capitalism, ableism, settler colonialism, and other forms of structural inequality).” It’s also about which groups get to be part of the design process itself.
Design justice is an interesting concept. I feel we usually forget how the users/those having their data collected are impacted by the data collection. Also it makes me wonder how harmful is the data companies collect on us, and subsequently do we have an expectation of data privacy whenusing companies platforms.
-
-
social-media-ethics-automation.github.io social-media-ethics-automation.github.io
-
In addition to representing data with different data storage methods, computers can also let you add additional constraints on what can be saved. So, for example, you might limit the length of a tweet to 280 characters, even though the computer can store longer strings.
This shows how the constraints provide efficiency. For example if you add more than 280 characters, the system will be slow and not efficient.
-
-
social-media-ethics-automation.github.io social-media-ethics-automation.github.io
-
If we download information about a set of tweets (text, user, time, etc.) to analyze later, we might consider that set of information as the main data, and our metadata might be information about our download process, such as when we collected the tweet information, which search term we used to find it, etc.
I think it's super interesting how data is collected across various apps and also downloadable into a compact format. I recall a friend utilizing this tool after his account on TikTok was compromised in order to retain his original algorithim which was created over the course of 6 years. Data accessibility is particularly useful in enhancing quality of life through information retention in case of account breaches.
-
Metadata is information about some data. So we often think about a dataset as consisting of the main pieces of data (whatever those are in a specific situation), and whatever other information we have about that data (metadata).
This part helped me better understand what metadata actually means. Before, I only thought about the main content like text or photos as “data,” but I didn’t realize that information like time, user, and interactions can also be very important. In real life, I feel like metadata might even reveal more about a person than the content itself. For example, when we use social media, patterns like when we post or who we interact with can say a lot about our habits. This makes me think more about privacy and how much information we are unintentionally sharing.
-
Metadata is information about some data. So we often think about a dataset as consisting of the main pieces of data (whatever those are in a specific situation), and whatever other information we have about that data (metadata).
Learning about metadata made me realize how much information I have that is going out to the public that I do not even know about. Every photo I take shares a location and a timestamp, and most apps track when and how the app is being used, so it sort of bothers me how so much of my information and schedule is going out to these companies, which can potentially sell it to other groups or organizations.
-
-
Local file Local file
-
s testament to the power of community-spiritedness anda willingness to identify with the entire region
Toqueville
-
These localities would retain many of the functions enjoyed to-day, including education, police and fire protection, limited zoningpowers, libraries, and community services. They could elect councilmembers and administrators, but their boundaries would be subject toperiodic reconfiguration
Sort of talking about a council manager type beat
-
is their limited capacity to achieve major publicworks
Maybe just say fuck it and give it all under the purview of the state
-
Although it is unclear whether such places have morecivic capacity than other types of communities (such questions await ex-ploration in future research), some limited anecdotal evidence suggeststhat they might
The cost benefit is something I remain skeptical of
-
The best way torevitalize suburban civic life is to promote greater racial and economicintegration.
I'm liking the "ya diversity" standpoint
-
it is unreason-able to expect that in today’s metropolitan areas, economic and racialdiversity could be maintained in communities under 50,000 in size.
In reality the relocation and political/physical infrastructure this would take is like fucking impossible
-
particularly among whitesand the affluent seeking to protect their racial and economic privileges
And sort of a moral disregard
-
If we want to address the problems of democracy insuburbia, we need to rearrange the configuration of local governments inmetropolitan areas.
This is the arrow chart we say above
-
between the neighboring towns rather thanamong citizens
Which only serves to further nationalize the issue
-
too often shield residents from the conflicts and problemsthat exist within their greater metropolitan community
Insular concerns
-
Municipalities with larger popula-tions lose vital civic capacity as residents tune out local politics; thesmaller the local unit, the more citizens are involved in community af-fairs
But you need diversity which is harder to get w/ small populations
-
ocalities with a weak civic capacity have lesscapability of making local government responsive and fewer options foraddressing social problems; consequently, they will be subject to greatersocial tension.
The suburbs have greater social tension, but on what scale?
-
However,in communities with greater civic capacity, citizens can be more easilyorganized and mobilized.
Important as the building block for government action and accountability
-
Bypreventing municipal institutions from addressing such conflicts, politicalfragmentation undermines the much-lauded role of America’s localities asarenas for democratic governance
Limiting the issues that matter at the local level
-
any issue involving the redistribution of wealth to groupswith less revenue-producing capability will not be advanced by a locality
Because affluent places have disproportionate power
-
lso limits effectivecitizen participation in the decision-making process
The citizens it actually matters to have less of a say
-
using local institutionsto limit the scope of conflict in metropolitan areas.
Not involving all the affected actors
-
Any change in the number of participants,Schattschneider argues, changes the results.
Right so its great when its contained to EG but eventually EG wants to bug providence
-
Civic withdrawal insegregated suburbs may not only lead to a narrow vision of self amongcitizens; it may also preempt the opportunity for learning essential demo-cratic skills and a broader understanding of community.
Which would be bad for dsemocracy if we could actually prove that was the cause
-
find that neitheris antecedent to the other.
Both arose at the same time
-
political participationcorresponds to higher trust, but more trusting people are also more likelyto be politically active, although campaign work seems to increase levelsof trust more than vice versa
Unclear which way the causation flows
-
The social segregation of suburbs is not only harmful todemocratic practice but impoverishing of the human spirit.
Also a breakdown in putmans capital
-
the social resources and links to other people that facilitate the achieve-ment of goals.
Mobilization
-
he in the end acquires a habit and taste forserving them.
So optimistic
-
the civil societyperspective views citizen participation as not simply important for demo-cratic organization but essential for realizing one’s humanity.
In that case the suburbs are defiantly NOT good
-
interdependent
right, the local level might be ok but step it up anymore and we've got a probelm
-
The ordering of these competing preferences depends upon how Irealize myself at any particular moment, an understanding that is pro-foundly shaped by my institutional and social circumstances.
So rational actors who are detached is not a good theory
-
who is fully formedbefore the confrontation begins.
But probably society has some say in how you are formed, I'm following
-
derive theirincome and other services from the remainder of the metropolis
But a different polity reaps the benefits
-
divorce themselves from the greater community with whichthey interact
The urb bruh
-
only to highlight the antidemocratic character of suburban institu-tional arrangements
Points out that there is going to be an inherent inequality between municipalities
-
the multi-faceted and inherently social condition of human existence
I mean not if you believe Hobbes
-
not from democratic actionbut from the potential mobility of revenue-producing citizens.
Proactive gov.
-
they can simply“vote with their feet” and move to another jurisdiction
Self sorting leads to same-ness which is not a problem as we see above, might also be a problem for the causation the author wants to claim as self sorting would be selection bias. Of course ability to sort is not perfect eh.
-
These models startwith the Hobbesian-like premise that individuals in a political system areisolated and autonomous, with the added condition that all individualsare motivated to act in ways that maximize the utility of their actions.
Latter, maybe, former, hell no
-
If all people in a society think alike, then anyone member can speak for the group.
This presupposes that economic and racial homogeneity equates to political homogeneity
-
also varies with the diver-sity of opinion in a polity.
The views of a pop will not be stagnant and neither will the population and so civic action accounts for changes
-
create a protected sphere enabling indi-viduals to pursue their own goals in relative isolation.
Def of the suburbs bruh
-
Similarly, if a person shares a politywith people who have an identical set of preferences, then the necessityof mass participation is quite low, as any one voter can articulate theviews of many.
homogeneity
-
Ifrepresentatives are adequately representing the aggregate of citizen inter-ests, then the level of citizen participation either necessary or possible inthe governing process is fairly low
The problem to be concerned about is the inter-municipal conflicts
-
the content of political life
I mean also they are having a say over residents –> voters
-
who par-ticipates?
The question should be "where participates"
-
Most important, the findings highlight the often over-looked role of social contexts and institutions in civic life
The place you live shapes the politics you participate in
-
suburbanization alsoeliminates many of the incentives that draw citizens into the publicrealm.
But hypothetically their needs are being met
-
the civic benefits of smaller size are undermined.
By a lack of diversity, still fuzzy about the mechanism here
-
The suburb superficially restores thedream of Jeffersonian democracy
Big claim
-
decry the isolating, banal, andoverly privatized quality of suburban areas
The writing in the book is pretty good
-
-
bookshelf.vitalsource.com bookshelf.vitalsource.com
-
Albert Einstein once famously said that God does not play dice with the universe; apparently the same cannot be said about humans and the Earth.
This reminds me of God’s command to humans. When He gave us dominion over the earth, He did not give us the rights to do whatever we want, but to care and maintain(stewarding) the earth.
-
As forests disappear,
Forests disappearing seems like an easy fix for more land, however trees and forests will ultimately cause much damage due to the carbon dioxide that they provide.
-
To deal with these environmental problems
To maintain a healthy/stable economy and society it is vital to keep your environment ready and able for the problems that nature might bring.
-
Economic growth is a rough indicator of our species’ relationship with the environment
A great way to help with economic growth is utilizing the environment and finding what in your society has greatest value that other societies do not have.
-
-
www.biorxiv.org www.biorxiv.org
-
Since scav-5 and scav-6 are paralogs of scav-4, we analysed their functions in lipid accumulation using scav-5(ok1606) deletion mutants and scav-6 knockout alleles generated in this study through CRISPR/Cas9-mediated gene editing (Figure 4B). We found that when fed with JUb74, both scav-5(-) and scav-6(-) mutants had moderately reduced LD sizes, but not to the extent of scav-4(-) mutants (Figure 4E). Previous promoter reporter studies showed that scav-5 and scav-6 were expressed in the intestine.34 We constructed translational reporters for both genes and found weak or no signals for SCAV-5::TagRFP possibly due to low protein levels. The SCAV-6::TagRFP fusion protein was expressed in the intestine and was localized to the apical membrane (Figure 4C). From the fluorescent intensity, the scav-6 expression appeared to be weaker than the scav-4 expression. Moreover, scav-4(-) scav-6(-) double mutants had the same LD diameter as scav-4(-) single mutants (Figure 4F). The above results suggested that SCAV-4 may play a more significant role than the other two paralogs in intestinal lipid uptake.
I'm surprised that the scav-5 and scav-6 paralogs were both able to reduce the large LD phenotype to the same extent as scav-4 (there doesn't appear to be significant difference between the mutants). To me this suggests either they each contribute a third of the BCFA uptake, or that they operate together to internalize BCFAs. The scav-4;scav-6 double mutant suggests the first idea isn't correct as you don't see a stronger effect there. Do you think its possible these transporters are working as a complex? I would be interested to see if you can rescue each of these mutants with scav-4 expression, or if rescue requires all receptors to be present.
-
In a small-scale screen of a few thousand haploid genomes, we isolated an allele unk28, which led to the formation of supersized LDs under the JUb74 diet (Figure 4A).
It's amazing you were able to find a GOF mutation that exacerbated this phenotype! I think it would help accentuate this if you included a picture of wt animals on the JUb74 food, next to the L462F animals on JUB74 food in Fig 4A to contrast that shift in LD size.
-
Conversely, we also supplemented either specific BCFAs or a mixture of them to the OP50 culture but were not able to increase the BCFA levels above 30%, likely because OP50 could not take in large amount of BCFAs (Figure S2C). Feeding those BCFA-treated OP50 to wild-type C. elegans did not induce large LDs (Figure S2D). Thus, we concluded that, in addition to the dosage effect, a threshold existed for the minimal level of BCFAs that could induce large LDs.
Could you preform the experiment in C. elegans defined media supplemented with BCFAs? That would fully remove other variables from the Microbacterium and confirm this is solely due to BCFAs.
-
-
drive.google.com drive.google.comview1
-
In a narrative all writing studies scholarsare familiar with, much of the teaching of writing in late 19 th- andearly- to mid-20th-century America focused on the object producedby writing, not the process of writing a text. This focus on the prod-uct of writing reinforced the idea of writing as a skill some peoplejust had. Essays were usually written once and were done, for goodor ill.
I feel like schools are the reason why i hesitate at times to turn in work on time even though I MAY have gotten the prompt down just fine for points like my zine.... it's that anxiety i get that makes me think what I've written is not enough.
-
-
www.biorxiv.org www.biorxiv.org
-
eLife Assessment
This study offers valuable insights into how humans detect and adapt to regime shifts, highlighting dissociable contributions of the frontoparietal network and ventromedial prefrontal cortex to sensitivity to signal diagnosticity and transition probabilities. The combination of an innovative instructed-probability task, Bayesian behavioural modelling, and model-based fMRI analyses provides solid support for the main claims. The addition of new model-comparison figures in revision effectively addresses the previously noted potential confound between posterior switch probability and time in the neuroimaging results. At the behavioural level, while the computational model captures the pattern of "system neglect" well, qualitatively distinct mechanisms, such as hyper-prior attraction toward experiment-wise mean parameters, reporting biases, or probability-outlier underweighting, could produce similar behavioural signatures and cannot be fully disambiguated with the current design alone; however, converging evidence from the authors' prior work partially mitigates this concern.
-
Reviewer #1 (Public review):
Summary:
The study examines human biases in a regime-change task, in which participants have to report the probability of a regime change in the face of noisy data. The behavioral results indicate that humans display systematic biases, in particular, overreaction in stable but noisy environments and underreaction in volatile settings with more certain signals. fMRI results suggest that a frontoparietal brain network is selectively involved in representing subjective sensitivity to noise, while the vmPFC selectively represents sensitivity to the rate of change.
Strengths:
- The study relies on a task that measures regime-change detection primarily based on descriptive information about the noisiness and rate of change. This distinguishes the study from prior work using reversal-learning or change-point tasks in which participants are required to learn these parameters from experiences. The authors discuss these differences comprehensively.
- The study uses a simple Bayes-optimal model combined with model fitting, which seems to describe the data well. The model is comprehensively validated.
- The authors apply model-based fMRI analyses that provide a close link to behavioral results, offering an elegant way to examine individual biases.
Weaknesses:
The authors have adequately addressed my prior concerns.
-
Reviewer #3 (Public review):
This study concerns how observers (human participants) detect changes in the statistics of their environment, termed regime shifts. To make this concrete, a series of 10 balls are drawn from an urn that contains mainly red or mainly blue balls. If there is a regime shift, the urn is changed over (from mainly red to mainly blue) at some point in the 10 trials. Participants report their belief that there has been a regime shift as a % probability. Their judgement should (mathematically) depend on the prior probability of a regime shift (which is set at one of three levels) and the strength of evidence (also one of three levels, operationalized as the proportion of red balls in the mostly-blue urn and vice versa). Participants are directly instructed of the prior probability of regime shift and proportion of red balls, which are presented on-screen as numerical probabilities. The task therefore differs from most previous work on this question in that probabilities are instructed rather than learned by observation, and beliefs are reported as numerical probabilities rather than being inferred from participants' choice behaviour (as in many bandit tasks, such as Behrens 2007 Nature Neurosci).
The key behavioural finding is that participants over-estimate the prior probability of regime change when it is low, and under estimate it when it is high; and participants over-estimate the strength of evidence when it is low and under-estimate it when it is high. In other words participants make much less distinction between the different generative environments than an optimal observer would. This is termed 'system neglect'. A neuroeconomic-style mathematical model is presented and fit to data.
Functional MRI results how that strength of evidence for a regime shift (roughly, the surprise associated with a blue ball from an apparently red urn) is associated with activity in the frontal-parietal orienting network. Meanwhile at time-points where the probability of a regime shift is high, there is activity in another network including vmPFC. Both networks show individual differences effects, such that people who were more sensitive to strength of evidence and prior probability show more activity in the frontal-parietal and vmPFC-linked networks respectively.
Strengths
(1) The study provides a different task for looking at change-detection and how this depends on estimates of environmental volatility and sensory evidence strength, in which participants are directly and precisely informed of the environmental volatility and sensory evidence strength rather than inferring them through observation as in most previous studies
(2) Participants directly provide belief estimates as probabilities rather than experimenters inferring them from choice behaviour as in most previous studies
(3) The results are consistent with well-established findings that surprising sensory events activate the frontal-parietal orienting network whilst updating of beliefs about the word ('regime shift') activates vmPFC.
Weaknesses
(1) The use of numerical probabilities (both to describe the environments to participants, and for participants to report their beliefs) may be problematic because people are notoriously bad at interpreting probabilities presented in this way, and show poor ability to reason with this information (see Kahneman's classic work on probabilistic reasoning, and how it can be improved by using natural frequencies). Therefore the fact that, in the present study, people do not fully use this information, or use it inaccurately, may reflect the mode of information delivery.
In the response to this comment the authors have pointed out their own previous work showing that system neglect can occur even when numerical probabilities are not used. This is reassuring but there remains a large body of classic work showing that observers do struggle with conditional probabilities of the type presented in the task,
(2) Although a very precise model of 'system neglect' is presented, many other models could fit the data.
For example, you would get similar effects due to attraction of parameter estimates towards a global mean - essentially application of a hyper-prior in which the parameters applied by each participant in each block are attracted towards the experiment-wise mean values of these parameters. For example, the prior probability of regime shift ground-truth values [0.01, 0.05, 0.10] are mapped to subjective values of [0.037, 0.052, 0.069]; this would occur if observers apply a hyper-prior that the probability of regime shift is about 0.05 (the average value over all blocks). This 'attraction to the mean' is a well-established phenomenon and cannot be ruled out with the current data (I suppose you could rule it out by comparing to another dataset in which the mean ground-truth value was different).
More generally, any model in which participants don't fully use the numerical information they were given would produce apparent 'system neglect'. Four qualitatively different example reasons are: 1. Some individual participants completely ignored the probability values given. 2. Participants did not ignore the probability values given, but combined them with a hyperprior as above. 3. Participants had a reporting bias where their reported beliefs that a regime-change had occurred tend to be shifted towards 50% (rather than reporting 'confident' values such 5% or 95%). 4. Participants underweighted probability outliers resulting in underweighting of evidence in the 'high signal diagnosticity' environment (10.1016/j.neuron.2014.01.020 )
In summary I agree that any model that fits the data would have to capture the idea that participants don't differentiate between the different environments as much as they should, but I think there are a number of qualitatively different reasons why they might do this - of which the above are only examples.
-
Author response:
The following is the authors’ response to the previous reviews
eLife Assessment
This study offers valuable insights into how humans detect and adapt to regime shifts, highlighting dissociable contributions of the frontoparietal network and ventromedial prefrontal cortex to sensitivity to signal diagnosticity and transition probabilities. The combination of an innovative instructed-probability task, Bayesian behavioural modeling, and model-based fMRI analyses provides a solid foundation for the main claims; however, major interpretational limitations remain, particularly a potential confound between posterior switch probability and time in the neuroimaging results. At the behavioural level, reliance on explicitly instructed conditional probabilities leaves open alternative explanations that complicate attribution to a single computational mechanism, such that clearer disambiguation between competing accounts and stronger control of temporal and representational confounds would further strengthen the evidence.
Thank you. In this revision, we addressed Reviewer 3’s remaining concern on the potential confound between posterior probability and time in neuroimaging results. First, as suggested by the reviewer, we provided images of activations for the effect of Pt and delta Pt after controlling for intertemporal prior in GLM-2. Second, we compared the effect of Pt and delta Pt between GLM-1 (without intertemporal prior) and GLM-2 (with intertemporal prior) and showed the results in a new figure (Figure 4).
Regarding issue on reliance on explicitly instructed probabilities, we wish to point out that most of the concerns such as response mode and regression to the mean were addressed in the original behavioral paper by Massey and Wu (2005). Please see our response to this point in detail in Weakness (2) posted by Reviewer 3.
Public Reviews:
Reviewer #1 (Public review):
Summary:
The study examines human biases in a regime-change task, in which participants have to report the probability of a regime change in the face of noisy data. The behavioral results indicate that humans display systematic biases, in particular, overreaction in stable but noisy environments and underreaction in volatile settings with more certain signals. fMRI results suggest that a frontoparietal brain network is selectively involved in representing subjective sensitivity to noise, while the vmPFC selectively represents sensitivity to the rate of change.
Strengths:
- The study relies on a task that measures regime-change detection primarily based on descriptive information about the noisiness and rate of change. This distinguishes the study from prior work using reversal-learning or change-point tasks in which participants are required to learn these parameters from experiences. The authors discuss these differences comprehensively.
- The study uses a simple Bayes-optimal model combined with model fitting, which seems to describe the data well. The model is comprehensively validated.
- The authors apply model-based fMRI analyses that provide a close link to behavioral results, offering an elegant way to examine individual biases.
Weaknesses:
The authors have adequately addressed my prior concerns.
Thank you for reviewing our paper and providing constructive comments that helped us improve our paper.
Reviewer #3 (Public review):
Thank you again for reviewing the manuscript. In this revision, we focused on addressing your concern on the potential confound between posterior probability and time in neuroimaging results. First, we presented whole-brain results of subjects’ probability estimates (Pt, their subjective posterior probability of switch) after controlling for the effect of time on probability of switch (the intertemporal prior). Second, we compared the effect of probability estimates (Pt) on vmPFC and ventral striatum activity—which we found to correlate with Pt—with and without including intertemporal prior in the GLM. These results will be summarized in a new figure (Figure 4) in the revised manuscript.
As suggested by the reviewer, we also added slice-by-slice images of the whole-brain results on Pt and delta Pt in the supplement in addition to the Tables of Activation so that the activated brain regions can be clearly seen through these images.
This study concerns how observers (human participants) detect changes in the statistics of their environment, termed regime shifts. To make this concrete, a series of 10 balls are drawn from an urn that contains mainly red or mainly blue balls. If there is a regime shift, the urn is changed over (from mainly red to mainly blue) at some point in the 10 trials. Participants report their belief that there has been a regime shift as a % probability. Their judgement should (mathematically) depend on the prior probability of a regime shift (which is set at one of three levels) and the strength of evidence (also one of three levels, operationalized as the proportion of red balls in the mostly-blue urn and vice versa). Participants are directly instructed of the prior probability of regime shift and proportion of red balls, which are presented on-screen as numerical probabilities. The task therefore differs from most previous work on this question in that probabilities are instructed rather than learned by observation, and beliefs are reported as numerical probabilities rather than being inferred from participants' choice behaviour (as in many bandit tasks, such as Behrens 2007 Nature Neurosci).
The key behavioural finding is that participants over-estimate the prior probability of regime change when it is low, and under estimate it when it is high; and participants over-estimate the strength of evidence when it is low and under-estimate it when it is high. In other words participants make much less distinction between the different generative environments than an optimal observer would. This is termed 'system neglect'. A neuroeconomic-style mathematical model is presented and fit to data.
Functional MRI results how that strength of evidence for a regime shift (roughly, the surprise associated with a blue ball from an apparently red urn) is associated with activity in the frontal-parietal orienting network. Meanwhile at time-points where the probability of a regime shift is high, there is activity in another network including vmPFC. Both networks show individual differences effects, such that people who were more sensitive to strength of evidence and prior probability show more activity in the frontal-parietal and vmPFC-linked networks respectively.
Strengths
(1) The study provides a different task for looking at change-detection and how this depends on estimates of environmental volatility and sensory evidence strength, in which participants are directly and precisely informed of the environmental volatility and sensory evidence strength rather than inferring them through observation as in most previous studies
(2) Participants directly provide belief estimates as probabilities rather than experimenters inferring them from choice behaviour as in most previous studies
(3) The results are consistent with well-established findings that surprising sensory events activate the frontal-parietal orienting network whilst updating of beliefs about the word ('regime shift') activates vmPFC.
Weaknesses
(1) The use of numerical probabilities (both to describe the environments to participants, and for participants to report their beliefs) may be problematic because people are notoriously bad at interpreting probabilities presented in this way, and show poor ability to reason with this information (see Kahneman's classic work on probabilistic reasoning, and how it can be improved by using natural frequencies). Therefore the fact that, in the present study, people do not fully use this information, or use it inaccurately, may reflect the mode of information delivery.
In the response to this comment the authors have pointed out their own previous work showing that system neglect can occur even when numerical probabilities are not used. This is reassuring but there remains a large body of classic work showing that observers do struggle with conditional probabilities of the type presented in the task.
Thank you. Yes, people do struggle with conditional probabilities in many studies. However, as our previous work suggested (Massey and Wu, 2005), system-neglect was likely not due to response mode (having to enter probability estimates or making binary predictions, and etc.).
(2) Although a very precise model of 'system neglect' is presented, many other models could fit the data.
For example, you would get similar effects due to attraction of parameter estimates towards a global mean - essentially application of a hyper-prior in which the parameters applied by each participant in each block are attracted towards the experiment-wise mean values of these parameters. For example, the prior probability of regime shift ground-truth values [0.01, 0.05, 0.10] are mapped to subjective values of [0.037, 0.052, 0.069]; this would occur if observers apply a hyper-prior that the probability of regime shift is about 0.05 (the average value over all blocks). This 'attraction to the mean' is a well-established phenomenon and cannot be ruled out with the current data (I suppose you could rule it out by comparing to another dataset in which the mean ground-truth value was different).
More generally, any model in which participants don't fully use the numerical information they were given would produce apparent 'system neglect'. Four qualitatively different example reasons are: 1. Some individual participants completely ignored the probability values given. 2. Participants did not ignore the probability values given, but combined them with a hyperprior as above. 3. Participants had a reporting bias where their reported beliefs that a regime-change had occurred tend to be shifted towards 50% (rather than reporting 'confident' values such 5% or 95%). 4. Participants underweighted probability outliers, resulting in underweighting of evidence in the 'high signal diagnosticity' environment (10.1016/j.neuron.2014.01.020 )
In summary I agree that any model that fits the data would have to capture the idea that participants don't differentiate between the different environments as much as they should, but I think there are a number of qualitatively different reasons why they might do this - of which the above are only examples - hence I find it problematic that the authors present the behaviour as evidence for one extremely specific model.
We thank the reviewer for this comment. We thank you for putting out that there are alternative models that can describe the over- and underreaction seen in the dataset. Massey and Wu (2005) dealt with this possibility in their original paper. Their concern was not so much about alternative ways of modeling their results, but in terms of alternative psychological processes. For example, asymmetric noise accounts have been posited in the judgment and decision making literature as possible accounts of phenomena like over-confidence. They addressed what might be crudely called “regression/attraction to the mean” in two ways. First, they looked at median responses as well as mean responses (because medians are less affected by the regressive effect) and found the same patterns of over- and underreactions. Second, they also generated sequences that matched particular posterior probabilities (so that over- and underreaction cannot be explained by regression to the mean) and still found under- and overreactions.
We also wish to point out in the judgment and decision making literature starting from Edwards (1968), there is a long history of using normative Bayesian model as the starting model and subsequently develop quasi-Bayesian models (like the system-neglect model) to describe systematic deviations from the normative Bayesian.
Finally, we want to clarify that our primary goal is not to engage in model fitting exercise that examines different possible models. To us, what is more important is that system neglect is a psychologically motivated hypothesis. It is built on the idea that the lack of sensitivity to the system parameters is due to the fact that people focus primarily on the signals and secondarily on the system parameters that generate the signals. Massey and Wu (2005) dealt with a host of other potential explanations through experimental manipulations and data analysis. In this paper, we built on Massey and Wu to examine the neurocomputational basis that gives rise to over- and underreactions.
(3) Despite efforts to control confounds in the fMRI study, including two control experiments, I think some confounds remain.
For example, a network of regions is presented as correlating with the cumulative probability that there has been a regime shift in this block of 10 samples (Pt). However, regardless of the exact samples shown, Pt always increases with sample number (as by the time of later samples, there have been more opportunities for a regime shift)? To control for this the authors include, in a supplementary analysis, an 'intertemporal prior.' I would have preferred to see the results of this better-controlled analysis presented in the main figure. From the tables in the SI it is very difficult to tell how the results change with the includion of the control regressors.
Thank you. In response, we added a new figure, now Figure 4, showing the results of Pt and delta Pt from GLM-2 where we added the intertemporal prior as a regressor to control for temporal confounds. We compared Pt and delta Pt results in vmPFC and ventral striatum between GLM-1 and GLM-2. We also showed the results on intertemporal prior on vmPFC and ventral striatum from GLM-2.
On the other hand, two additional fMRI experiments are done as control experiments and the effect of Pt in the main study is compared to Pt in these control experiments. Whilst I admire the effort in carrying out control studies, I can't understand how these particular experiment are useful controls. For example, in experiment 3 participants simply type in numbers presented on the screen - how can we even have an estimate of Pt from this task?
We thank the reviewer for this comment. On the one hand, the effect of Pt we see in brain activity can be simply due to motor confounds and the purpose of Experiment 3 was to control for them. Our question was, if subjects saw the similar visual layout and were just instructed to press buttons to indicate two-digit numbers, would we observe the vmPFC, ventral striatum, and the frontoparietal network like what we did in the main experiment (Experiment 1)?
On the other hand, the effect of Pt can simply reflect probability estimates of that the current regime is the blue regime, and therefore not particularly about change detection. In Experiment 2, we tested that idea, namely whether what we found about Pt was unique to change detection. In Experiment 2, subjects estimated the probability that the current regime is the blue regime (just as they did in Experiment 1) except that there were no regime shifts involved. In other words, it is possible that the regions we identified were generally associated with probability estimation and not particularly about probability estimates of change. We used Experiment 2 to examine whether this were true.
To make the purpose of the two control experiments clearer, we updated the paragraph describing the control experiments on page 9:
“To establish the neural representations for regime-shift estimation, we performed three fMRI experiments (n = 30 subjects for each experiment, 90 subjects in total). Experiment 1 was the main experiment, while Experiments 2 to 3 were control experiments that ruled out two important confounds (Fig. 1E). The control experiments were designed to clarify whether any effect of subjects’ probability estimates of a regime shift, P<sub>t</sub>, in brain activity can be uniquely attributed to change detection. Here we considered two major confounds that can contribute to the effect of P<sub>t</sub>. First, since subjects in Experiment 1 made judgments about the probability that the current regime is the blue regime (which corresponded to probability of regime change), the effect of P<sub>t</sub> did not particularly have to do with change detection. To address this issue, in Experiment 2 subjects made exactly the same judgments as in Experiment 1 except that the environments were stationary (no transition from one regime to another was possible), as in Edwards (1968) classic “bookbag-and-poker chip” studies. Subjects in both experiments had to estimate the probability that the current regime is the blue regime, but this estimation corresponded to the estimates of regime change only in Experiment 1. Therefore, activity that correlated with probability estimates in Experiment 1 but not in Experiment 2 can be uniquely attributed to representing regime-shift judgments. Second, the effect of P<sub>t</sub> can be due to motor preparation and/or execution, as subjects in Experiment 1 entered two-digit numbers with button presses to indicate their probability estimates. To address this issue, in Experiment 3 subjects performed a task where they were presented with two-digit numbers and were instructed to enter the numbers with button presses. By comparing the fMRI results of these experiments, we were therefore able to establish the neural representations that can be uniquely attributed to the probability estimates of regime-shift.”
To further make sure that the probability-estimate signals in Experiment 1 were not due to motor confounds, we implemented an action-handedness regressor in the GLM, as we described below on page 19:
“Finally, we note that in GLM-1, we implemented an “action-handedness” regressor to directly address the motor-confound issue, that higher probability estimates preferentially involved right-handed responses for entering higher digits. The action-handedness regressor was parametric, coding -1 if both finger presses involved the left hand (e.g., a subject pressed “23” as her probability estimate when seeing a signal), 0 if using one left finger and one right finger (e.g., “75”), and 1 if both finger presses involved the right hand (e.g., “90”). Taken together, these results ruled out motor confounds and suggested that vmPFC and ventral striatum represent subjects’ probability estimates of change (regime shifts) and belief revision.”
(4) The Discussion is very long, and whilst a lot of related literature is cited, I found it hard to pin down within the discussion, what the key contributions of this study are. In my opinion it would be better to have a short but incisive discussion highlighting the advances in understanding that arise from the current study, rather than reviewing the field so broadly.
Thank you. We thank the reviewer for pushing us to highlight the key contributions. In response, we added a paragraph at the beginning of Discussion to better highlight our contributions:
“In this study, we investigated how humans detect changes in the environments and the neural mechanisms that contribute to how we might under- and overreact in our judgments. Combining a novel behavioral paradigm with computational modeling and fMRI, we discovered that sensitivity to environmental parameters that directly impact change detection is a key mechanism for under- and overreactions. This mechanism is implemented by distinct brain networks in the frontal and parietal cortices and in accordance with the computational roles they played in change detection. By introducing the framework in system neglect and providing evidence for its neural implementations, this study offered both theoretical and empirical insights into how systematic judgment biases arise in dynamic environments.”
Recommendations for the authors:
Reviewer #3 (Recommendations for the authors):
Thank you for pointing out the inclusion of the intertemporal prior in glm2, this seems like an important control that would address my criticism. Why not present this better-controlled analysis in the main figure, rather than the results for glm1 which has no effective control of the increasing posterior probability of a reversal with time?
Thank you for this suggestion. We added a new figure (Figure 4) that showed results of Pt and delta Pt from GLM-2. We also compared the effect of Pt and delta Pt between GLM-1 and GLM-2. We found that the effect of Pt and delta Pt did not differ between GLM-1 and GLM-2. GLM-1 and GLM-2 differed on whether various task-related regressors contributing to Pt, including the intertemporal prior, were included in the model. In GLM-1, those task-related regressors were not included. In GLM-2, the task-related regressors were included in addition to Pt and delta P.
The reason we kept results from GLM-1 (Figure 3) was primarily because we wanted to compare the effect of Pt between experiments under identical GLM. In other words, the regressors in GLM-1 was identical across all 3 experiments. In Experiments 1 and 2, Pt and delta Pt were respectively probability estimates and belief updates that current regime was the Blue regime. In Experiment 3, Pt and delta Pt were simply the number subjects were instructed to press (Pt) and change in number between successive periods (delta Pt).
Here is the section in the main text where we discussed the new Figure 4 on page 19-22:
We further examined the robustness of P<sub>t</sub> and ∆P<sub>t</sub> representations in vmPFC and ventral striatum in three follow-up analyses. In the first analysis, we implemented a GLM (GLM-2 in Methods) that, in addition to P<sub>t</sub> and ∆P<sub>t</sub>, included various task-related variables contributing to P<sub>t</sub> as regressors. Specifically, to account for the fact that the probability of regime change increased over time, we included the intertemporal prior as a regressor in GLM-2. The intertemporal prior is the natural logarithm of the odds in favor of regime shift in the t-th period,
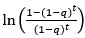 , where q is transition probability and t = 1, …, 10is the period (Eq. 1 in Methods). It describes normatively how the prior probability of change increased over time regardless of the signals (blue and red balls) the subjects saw during a trial. Including it along with P<sub>t</sub> would clarify whether any effect of P<sub>t</sub> can otherwise be attributed to the intertemporal prior. We found that the results of P<sub>t</sub> and ∆P<sub>t</sub> in the vmPFC and ventral striatum in GLM-2 were identical to those in GLM-1 (Fig. 4): Fig. 4A was meant to depict the results in slices identical to those shown in Fig. 3B for results based on GLM-1. For slice-by-slice results, see Fig. S7 in SI for results based on GLM-1 and Fig. S9 for GLM-2. For Tables of activations, see Tables S1-S3 in SI for GLM-1 and Tables S7-S9 for GLM-2. In a separate, independent region-of-interest (ROI) analysis on vmPFC and ventral striatum (Fig. 4BC; see Independent regions-of-interest (ROIs) analysis in Methods for details), we further compared the effect of both P<sub>t</sub> and ∆P<sub>t</sub> between GLM-1 and GLM-2. For P<sub>t</sub>, the difference between GLM-1 and GLM-2 was not significant (paired t-test, t(58) = −0.72, p = 0.47 in vmPFC, t(58) = −0.21, p = 0.83 in ventral striatum), while the effect of P<sub>t</sub> from GLM-1 (one sample t-test, t(29) = −3,82, p <.01 in vmPFC; t(29) = −3.06, p <.01 in ventral striatum) and GLM-2 was significant (one-sample t-test, t(29) = −2.69, p =.01 in vmPFC; t(29) = −2.50, p .02 in ventral striatum). For ∆P<sub>t</sub>, the difference between GLM-1 and GLM-2 was not significant (paired t-test, t(58) = −0.07, p =0.94 in vmPFC; t(58) = −0.14, p =0.88 in ventral striatum), while the effect of from GLM-1 (one-sample t-test, t(29) = −3.12, p <.01 in vmPFC; t(29) = −4.14, p <.01 in ventral striatum) and GLM-2 was significant (one-sample t-test, t(29) = −2.92, p <.01 in vmPFC; t(29) = −3.59, p <.01 in ventral striatum). For the intertemporal prior, activity in both vmPFC and ventral striatum did not correlate significantly with the intertemporal prior (one-sample t-test, t(29) = −0.07, p =0.95 in vmPFC; t(29) = −0.53, p =0.60 in ventral striatum). All the t-tests described above were two-tailed. Taken together, these results suggest that vmPFC and ventral striatum represented P<sub>t</sub> and ∆P<sub>t</sub> regardless of whether the intertemporal prior and other task-related regressors contributing to P<sub>t</sub> were included in the GLM. We also did not find that vmPFC and ventral striatum to represent the intertemporal prior. In the second analysis, we implemented a GLM that replaced P<sub>t</sub> with the log odds of P<sub>t</sub>, 1n (P<sub>t</sub>/(1 - P<sub>t</sub>)) (Fig. S10 in SI). In the third analysis, we implemented a GLM that examined P<sub>t</sub> separately on periods when change-consistent (blue balls) and change-inconsistent (red balls) signals appeared (Fig. S11 in SI). Each of these analyses showed significant correlation with P<sub>t</sub> in vmPFC and ventral striatum, further establishing the robustness of the P<sub>t</sub> findings.
, where q is transition probability and t = 1, …, 10is the period (Eq. 1 in Methods). It describes normatively how the prior probability of change increased over time regardless of the signals (blue and red balls) the subjects saw during a trial. Including it along with P<sub>t</sub> would clarify whether any effect of P<sub>t</sub> can otherwise be attributed to the intertemporal prior. We found that the results of P<sub>t</sub> and ∆P<sub>t</sub> in the vmPFC and ventral striatum in GLM-2 were identical to those in GLM-1 (Fig. 4): Fig. 4A was meant to depict the results in slices identical to those shown in Fig. 3B for results based on GLM-1. For slice-by-slice results, see Fig. S7 in SI for results based on GLM-1 and Fig. S9 for GLM-2. For Tables of activations, see Tables S1-S3 in SI for GLM-1 and Tables S7-S9 for GLM-2. In a separate, independent region-of-interest (ROI) analysis on vmPFC and ventral striatum (Fig. 4BC; see Independent regions-of-interest (ROIs) analysis in Methods for details), we further compared the effect of both P<sub>t</sub> and ∆P<sub>t</sub> between GLM-1 and GLM-2. For P<sub>t</sub>, the difference between GLM-1 and GLM-2 was not significant (paired t-test, t(58) = −0.72, p = 0.47 in vmPFC, t(58) = −0.21, p = 0.83 in ventral striatum), while the effect of P<sub>t</sub> from GLM-1 (one sample t-test, t(29) = −3,82, p <.01 in vmPFC; t(29) = −3.06, p <.01 in ventral striatum) and GLM-2 was significant (one-sample t-test, t(29) = −2.69, p =.01 in vmPFC; t(29) = −2.50, p .02 in ventral striatum). For ∆P<sub>t</sub>, the difference between GLM-1 and GLM-2 was not significant (paired t-test, t(58) = −0.07, p =0.94 in vmPFC; t(58) = −0.14, p =0.88 in ventral striatum), while the effect of from GLM-1 (one-sample t-test, t(29) = −3.12, p <.01 in vmPFC; t(29) = −4.14, p <.01 in ventral striatum) and GLM-2 was significant (one-sample t-test, t(29) = −2.92, p <.01 in vmPFC; t(29) = −3.59, p <.01 in ventral striatum). For the intertemporal prior, activity in both vmPFC and ventral striatum did not correlate significantly with the intertemporal prior (one-sample t-test, t(29) = −0.07, p =0.95 in vmPFC; t(29) = −0.53, p =0.60 in ventral striatum). All the t-tests described above were two-tailed. Taken together, these results suggest that vmPFC and ventral striatum represented P<sub>t</sub> and ∆P<sub>t</sub> regardless of whether the intertemporal prior and other task-related regressors contributing to P<sub>t</sub> were included in the GLM. We also did not find that vmPFC and ventral striatum to represent the intertemporal prior. In the second analysis, we implemented a GLM that replaced P<sub>t</sub> with the log odds of P<sub>t</sub>, 1n (P<sub>t</sub>/(1 - P<sub>t</sub>)) (Fig. S10 in SI). In the third analysis, we implemented a GLM that examined P<sub>t</sub> separately on periods when change-consistent (blue balls) and change-inconsistent (red balls) signals appeared (Fig. S11 in SI). Each of these analyses showed significant correlation with P<sub>t</sub> in vmPFC and ventral striatum, further establishing the robustness of the P<sub>t</sub> findings.As a further point I could not navigate the tables of fMRI activations in SI and recommend replacing or supplementing these with images. For example I cannot actually find a vmPFC or ventral striatum cluster listed for the effect of Pt in GLM1 (version in table S1), which I thought were the main results? Beyond that, comparing how much weaker (or not) those results are when additional confound regressors are included in GLM2 seems impossible.
As suggested by the reviewer, we added slice-by-slice images showing the effect of Pt and delta Pt (Figure S9 in SI for GLM-2 and Figure S7 for GLM-1). The clusters in blue represent Pt effect, the clusters in orange represent delta Pt effect. As can be seen, both Pt and delta Pt are represented in the vmPFC and ventral striatum.
-
-
social-media-ethics-automation.github.io social-media-ethics-automation.github.io
-
Open a social media interface (not the one you’ve been working with) and choose a view (e.g., a list of posts, an individual post, an author page etc.). First identify as many pieces of information you can see the screen (without doing anything). For each piece of information: What data types might be used to represent that data on a computer? How is this data a simplification of reality? That is, what does it not capture? Who does it work best for, and who does it not work well for? Did the user(s) directly provide that data, or was it collected automatically by the social media site?
On Instagram, a single post distills complex human experiences into a structured collection of Strings (usernames and captions), Integers (like counts), DateTimes (relative post age), and Binary/Blob data (the image or video itself). These digital artifacts act as a significant simplification of reality by flattening three-dimensional, multi-sensory moments into a 2D frame that lacks the physical context, emotional depth, or the "unfiltered" events occurring just outside the camera's view. This system works exceptionally well for "influencers" and brands who benefit from highly curated, aesthetic-first storytelling that drives rapid engagement.
-
What pieces of information you think should be immediately visible to users
When considering the design of social media sites, I think the pieces of information that should be immediately visible to users are other users' display names, profile pictures, and mutual connections that they may share. I think that these are good pieces of information to be immediately visible because, it will draw the user in and get them to engage more with others profiles. By only sharing basic information, users have enough factors to judge whether or not they want to engage with this user.
-
Open a social media interface (not the one you’ve been working with) and choose a view (e.g., a list of posts, an individual post, an author page etc.). First identify as many pieces of information you can see the screen (without doing anything). For each piece of information: What data types might be used to represent that data on a computer? How is this data a simplification of reality? That is, what does it not capture? Who does it work best for, and who does it not work well for? Did the user(s) directly provide that data, or was it collected automatically by the social media site?
TikTok only shows the number of likes as an integer data type, meaning it tells me how many people liked a video, but it does not show different emotions like Facebook, where users can react with various feelings. So we cannot really tell whether people truly enjoyed the video or just saved or liked it to share with others. It does not clearly reflect viewers’ real feelings, including mine. Another example is text data such as usernames and profile pictures which are based on users’ personal preferences and do not necessarily reflect who they are in real life. This is why there are many fake accounts on social media, created for different purposes. Sometimes when scrolling on TikTok, I wonder why I see unfamiliar videos that I have never searched for or talked about. I think this happens because the platform collects data from my followers, and if they like certain types of videos, similar content may also appear on my feed.
-
-
Local file Local file
-
The Stuarts ... belonged essentially to the conquering Norman race ... not so theWallaces, whose three Scotch generations could not so utterly have obliterated allsympathy with the Cambrian cradle of their family, but that the savage injusticeand cruelty of the Plantagenet conquest of Wales ... must have struck them withpeculiar horror and indignation ... The Wallaces had found shelter from Englishbondage in Scotland ... when they found [they were liable to come under Englishmasters there as well] they determined to resist for themselves to the uttermost oftheir power
VERY GOOD SLAYYY - CLEARLY, WHILE AN ARISTOCRAT, HE LAYS CLAIM TO THE WELSH AND HOW THE ENGLISH WERE UNJUST!!!
-
here were also echoesof the Old Testament, where a vineyard is the usual image for the people of God, and, perhapseven more poignantly, the Gospel of St. John, where Christ speaks of himself as the true vine,and his disciples as
Very very slay! It was kinda highly a religious project!!!
-
Nothing about Bute suggests he had much taste for personal grandeur; it was the pleasure ofworking on the designing and building of his projects that impelled him. He once famouslyremarked that he had 'comparativly little interest in a thing after it is finished. 163 He wasdeeply and personally involved in all his projects. Burges was more of a collaborator than anemployee, others awaited his visits, ideas and judgements with a mixture of pleasure andtrepidation. 64 Bute was also extremely price-consciou
Very interesting for architecture!!
-
it was a part of his dislike of the wreck of old and beautiful
SLAYYY FOR ARCHITECTUREEE
-
The rest is by no means satisfactory and has been thevictim of every barbarism since the Renaissance
Interestingly describes baroque and classical as 'barbarous' while gothic was usually described this way!
-
considering these three courses there is no doubt at all, that in any age otherthan the present the last mentioned one is that which would most certainly havebeen adopted in as much as it is the most suited to the circumstances of the case;for we must never lose sight of the fact that Cardiff Castle is not an antiquarianruin but the seat of the Marquess of Bute
SLAYYY - IT NEEDED TO BE GRAND, IT WAS A NOBLE PLACE AND NEEDED TO BE NOBLE, IT NEEDED TO BE MODERN AND REFLECT THE INDUSTRIAL MIGHT OF THE BUTES!!!
-
n 1865, Bute had met one of the most original architects of the Victorian period, WilliamBurges.37 Both men were passionately interested in history. Bute had fallen in love with theGothic style before his ninth birthday, the style in which Burges invariably built. It is notclear if they first met because Burges had already been asked to prepare a report on restoringCardiff Castle, or if he was asked to make the report following a chance encounter
slayyyy background to the architecture with burges!!!
-
Cardiff Castlehadlong been an inconveniently crampedhousefor a nobleman,or, indeed, any well-to-do man.It was simply too small for entertaining. The secondMarquesshad found it so himself, andthe first had rarely usedit at all
CARDIFF CASTLE
-
f ... as seems ... likely this conversion or perversion is the result of priestlyinfluences acting upon a weak, ductile and naturally superstitious mind, we mayexpect the continual eclipse of all intellectual vigour; for these influences willnever leave the Marquis but darken and darken around him as long as he lives.The Roman Church knows well how to treat such cases and how to use them forher own advantage
Instead, he engaged with a maginificent architectural wonder?
-
t would be his day of freedom, the day, also,when he would not be able to shelter behind his promises given to delay his choice of a Church
interesting that the day he got his majority was the day he joined the church!
-
It is well illustrated byhis difficulty earlier that year in trying to present a stained glass window to Cumnock parishchurch. The Presbyterians took a strict view of the Second Commandment which forbad
Interesting that he wished to gift this to people!
-
herever he went, Bute found the destruction of irreplaceable remains and ruins, and he wasincensed. One of the six circular Churches known had been pulled down in 1829. He travelledthe greater part of a day to view a broch standing in a manse glebe. It had been reduced fromits original 50 feet high to provide the stone for building walls around the fields.
SLAYYY he felt angered at the ruins - it is thus arguable that, in seeing the state of Castell Coch, he sought to restore it to its glory as a home away from home for the rich
-
n hot climates, Bute slept naked, and he was much amused by Mr Bistani who 'took offnothing except his outer clothes, not even his stockings.176 Perhaps what struck Bute most inthe palace, however, were the courtyards and rooms open to the air either above or througharches. The amazing mixture of room and open air fascinated him. The 'most perfectapartment' was the on
Likely innfluenced the islamic room in blah?
-
e was,however, still dwelling on his wrongs, and especially upon his financial wrongs. Bute wasgenerous with his charities, and surrounded by sons of some of the richer British families. Theold wrong which he felt had been done him by Stuart was still very real t
is this why he rebuilt castell coch - the outside was for the people, and he went onto build schools and other thigs in the gothic style that were for ordinary people
-
This is not surprising, as English establishment attitudes, severely alienated from Scotland ahundred years before during the Jacobite rebellions, and only slowly thawing under the wavesof the picturesque mediaevalism let loose by Sir Walter Scott and fashionable 'Balmorality'promoted by Victoria, still had little sympathy for the independent rights of the Scottishpeople, or understanding of the profoundly different culture which they cherished
mirror in the welsh?
-
Sophia had edited her father's journals and her sister's poems. Now she assisted her son tostart a small newspaper. The Mount Stuart Weekly Journal was begun at the end of 1858 to4convey to our absent friends some knowledge of how we are occupied' in which it succeedsnow as then. Bute was the editor, and he copied the whole out in his own hand, and as a goodeditor should, he solicited contributions from all the talented associates he could find.Occasionally, however, he was force
really really interesting, he found his own later on!
-
From the moonlight drive to the ferry at Folkestone, it was an enchanted time forthe young Bute, so much so that themes from it were to haunt his adulthood.
interesting, did this impact his love for gothic architecture?
-
Lady Bute fought back with vigour. Determined to have her own home, and with the southWales trustees on her side, Lord James was forced to leave Cardiff Castle in the summer of1849.61
interesting, did he wish to restore it to avenge his mum?
-
Lord Bute naturally wanted to take his son to Cardiff, the town he had created. Lady Sophiathought he intended to spend the whole spring there
Cardiff was significant for the bute family, it was a land in which they had built --> like the marcher lords? idk
-
ohn DavieS4 gives afascinating account of how Lord Bute created a thriving industrial complex out of this formeragricultural land. The city of Cardiff was largely of his making, springing up around the dockshe built; and he blazed the way in the creation of new collierie
background for bute and industry by his dad!
-
There was also the question of Bute's Catholicism. There were many aristocratic Catholicconverts in the nineteenth century and they married into the old Catholic families, as Butehimself did. Yet Bute was extraordinary amongst them. His faith spilled over into hispatronage of the arts, and into his scholarship: indeed his faith, his scholarship and his art fedone other
slayyyyyyy
-
-
fhsu.pressbooks.pub fhsu.pressbooks.pub
-
see our chapter on Lateral Reading
Could you link to the other chapter to open in a new tab?
-
-
fhsu.pressbooks.pub fhsu.pressbooks.pub
-
full article
It would be helpful to set the link to open in a new tab
-
-
fhsu.pressbooks.pub fhsu.pressbooks.pub
-
Running Out of Time When you are a student taking many classes simultaneously and facing many deadlines, it may be hard to devote the time needed to doing good scholarship and accurately representing the sources you have used. Research takes time. The sooner you can start and the more time you can devote to it, the better your work will be. From the beginning, be sure to include in your notes where you found information you could quote, paraphrase, and summarize in your final product.
Duplicate paragraph
-
-
is.herbwoman.com is.herbwoman.com
-
Hypericum perforatum L. Hypericaceae — Ofanjarðarhlutar sem eru uppskornir við blómgun
Ljónslappi (Hypericum perforatum) 🔥 mild bólgueyðing ⚡ getur hjálpað við taugatengda verki
Ljónslappi fyrir brjósk: ➕ mjög óbein áhrif ➖ ekki relevant sem liðajurt
-
Valerian officinalis L. Caprifoliaceae (áður Valerianaceae) — Rót og rhizome (þurrkað)
Garðabrúða (Valerian officinalis) 😌 minnkar spennu og verkjatilfinningu 🧘♀️ bætir svefn → betri endurheimt
Valeríana fyrir brjósk: ➕ óbein áhrif (slökun, svefn) ➖ ekki „joint herb“
-
Hjartafró L. Lapiaceae — Lauf og ofanjarðarhlutar (ferskt eða vandlega þurrkað við lágan hita)
Hjartafró (Leonurus cardiaca) Það sem hún gerir: 🧘♀️ róar taugakerfi ❤️ styður hjartastarfsemi 🔥 mild bólgueyðandi
Hjartafró fyrir brjósk: ➕ mjög óbein áhrif ➖ ekki relevant sem „joint herb“
-
-
is.herbwoman.com is.herbwoman.com
-
Ginkgo biloba L. Ginkgoaceae — Lauf (staðlað útdráttur úr þurrkuðum laufblöðum)
Ginkgo (Ginkgo biloba) Ginkgo fyrir brjósk:
🩸 bætir blóðflæði 🧬 ver frumur 🔥 mild bólgueyðing ➡️ óbein áhrif
-
Withania somnifera (L.) Dunal Solanaceae — Rót (þurrkuð rót og rótarþykkni)
Ashwagandha (Withania somnifera) Ashwagandha fyrir brjósk:
🔥 bólgueyðandi 🛡️ ónæmisstillandi 🧘♀️ adaptogen ➡️ mjög óbein en mikilvæg áhrif
-
-
web.archive.org web.archive.orgPreface1
-
The skills acquired from learning to design programs
Básicamente para aprender y adquirir naturalmente una habilidad hay que practicar, mantenerse en ese estado para que nuestra memoria y nuestro cuerpo se configuren para este tipo de acciones realizadas y se transfiera a nuestra memoria como una habilidad adquirida. Todo este proceso se normaliza con el tiempo y se vuelva algo casi mecánico.
-
-
www.biorxiv.org www.biorxiv.org
-
eLife Assessment
This study represents an important advance in our understanding of how certain inhibitors affect the behavior of voltage gated potassium channels. Robust molecular dynamics simulation and analysis methods lead to a new proposed inhibition mechanism with convincing strength of support. This study has considerable significance for the fields of ion channel physiology and pharmacology and could aid in development of selective inhibitors for protein targets.
-
Reviewer #3 (Public review):
Summary
In this manuscript, Zhang et al. investigate the conduction and inhibition mechanisms of the Kv2.1 channel, with a particular focus on the distinct effects of TEA and RY785 on Kv2 potassium channels. Using microsecond-scale molecular dynamics simulations, the authors characterize K⁺ ion permeation and RY785-mediated inhibition within the central pore. Their results reveal an inhibition mechanism that differs from those described for other Kv channel inhibitors.
Strengths
The study identifies a distinctive inhibitory mode for RY785, which binds along the channel walls in the open-state structure while still permitting a reduced level of K⁺ conduction. In addition, the authors propose a long-range allosteric coupling between RY785 binding in the central pore and changes in the structural dynamics of Kv2.1. Overall, this is a well-organized and carefully executed study, employing robust simulation and analysis methodologies. The work provides novel mechanistic insights into voltage-gated potassium channel inhibition and may offer useful guidance for future structure-based drug design efforts.
Weaknesses:
As noted in the Discussion, this study focuses primarily on the major binding site within the central pore and was not designed to systematically assess other potential allosteric binding sites for RY785. A more comprehensive structural and biophysical evaluation of possible additional binding sites would be a valuable direction for future investigations.
Comments on revisions:
The authors have addressed my comments.
-
Author response:
The following is the authors’ response to the previous reviews
Public Reviews:
Reviewer #1 (Public review):
Summary:
The authors were seeking to identify a molecular mechanism whereby the small molecule RY785 selectively inhibits Kv2.1 channels. Specifically, the authors sought to explain some of the functional differences that RY785 exhibits in experimental electrophysiology experiments as compared to other Kv inhibitors, namely the charged and non-specific inhibitor tetraethylammonium (TEA). The authors used a recently published cryo-EM Kv2.1 channel structure in the open activated state and performed a series of multi-microsecond-long all-atom molecular dynamics simulations to study Kv2.1 channel conduction under the applied membrane voltage with and without RY785 or TEA present. They observed that while TEA directly blocks K+ permeation by occluding ion permeation pathway, RY785 binds to multiple non-polar residues near the hydrophobic gate of the channel driving it to a semi-closed non-conductive state. They confirmed this mechanism using an additional set of simulations and used it to explain experimental electrophysiology data,
Strengths:
The total length of simulation time is impressive, totaling many tens of microseconds. The authors develop their own forcefield parameters for the RY785 molecule based on extensive QM based parameterization. The computed permeation rate of K+ ions through the channel observed under applied voltage conditions is in reasonable agreement with experimental estimates of the single channel conductance. The authors have performed extensive simulations with the apo channel as well as both TEA and RY785. The simulations with TEA reasonably demonstrate that TEA directly blocks K+ permeation by binding in the center of the Kv2.1 channel cavity, preventing K+ ions from reaching the SCav site. The authors conclude that RY785 likely stabilizes a partially closed conformation of the Kv2.1 channel and thereby inhibits K+ current. This conclusion is plausible given that RY785 makes stable contacts with multiple hydrophobic residues in the S6 helix, which they can also validate using a recently published closed-state Kv2.1 channel cryo-EM structure. This further provides a possible mechanism for the experimental observations that RY785 speeds up the deactivation kinetics of Kv2 channels from a previous experimental electrophysiology study.
Weaknesses:
The authors, however, did not directly observe this semi-closed channel conformation and in fact acknowledge that more direct simulation evidence would require extensive enhanced-sampling simulations beyond the scope of this study. They have not estimated the effect of RY785 binding on the protein-based hydrophobic pore constriction, which may further substantiate their proposed mechanism. And while the authors quantified K+ permeation, they have not made any estimates of the ligand binding affinities or rates, which could have been potentially compared to experiment and used to validate their models.
However, despite those relatively minor weaknesses, the conclusions of the study are convincing, and overall this is a solid study helping us to understand two distinct molecular mechanisms of the voltage-gated potassium channel Kv2.1 inhibition by TEA and RY785, respectively.
Reviewer #2 (Public review):
Summary
In this manuscript, Zhang et al. investigate the conduction and inhibition mechanisms of the Kv2.1 channel, with a particular focus on the distinct effects of TEA and RY785 on Kv2 potassium channels. Using microsecond-scale molecular dynamics simulations, the authors characterize K⁺ ion permeation and RY785-mediated inhibition within the central pore. Their results reveal an inhibition mechanism that differs from those described for other Kv channel inhibitors.
Strengths
The study identifies a distinctive inhibitory mode for RY785, which binds along the channel walls in the open-state structure while still permitting a reduced level of K⁺ conduction. In addition, the authors propose a long-range allosteric coupling between RY785 binding in the central pore and changes in the structural dynamics of Kv2.1. Overall, this is a well-organized and carefully executed study, employing robust simulation and analysis methodologies. The work provides novel mechanistic insights into voltage-gated potassium channel inhibition and may offer useful guidance for future structure-based drug design efforts.
Weaknesses:
The study needs to consider the possibility of multiple binding sites for PY785, particularly given its impact on voltage sensors and gating currents. Specifically, the potential for allosteric binding sites in the voltage-sensing domain (VSD) should be assessed, as some allosteric modulators with thiazole moieties are known to bind VSD domains in multiple voltage-gated sodium channels (Ahuja et al., 2015; Li et al., 2022; McCormack et al., 2013; Mulcahy et al., 2019). Increasing structural and functional evidence supports the existence of multiple ligand-binding modes in voltage-gated ion channels. For example, polyunsaturated fatty acids have been shown to bind to KCNQ1 at both the voltage sensor domain and the pore domain (https://doi.org/10.1085/jgp.202012850). Similarly, cannabidiol has been structurally resolved in Nav1.7 at two distinct sites, one in a fenestration and another near the IFM-binding pocket (https://doi.org/10.1038/s41467-023-39307-6). These advances illustrate that ligand effects cannot always be interpreted based solely on a single binding site identified previously.
Reviewing Editor:
The comments of the reviewers seem thoughtful and constructive. The weaknesses noted in reviews mainly concern mismatch between expectations, created by reading the Abstract, and data in the manuscript. The mismatch could be reconciled by either new simulations examining a semi-open state of the gate and additional RY785 binding sites, or by adjusting wording of the Abstract and Discussion to make it more clear that such simulations were not done.
The Abstract and Discussion have been revised to make clear the computer-simulations presented in our study were designed to specifically validate or refute the hypothesis that RY785 is recognized by the pore domain, not the voltage sensors.
Recommendations for the authors:
Reviewer #1 (Recommendations for the authors):
The authors addressed all the major issues in the original submission identified by the reviewers. I noticed a few minor issues, listed below, which can potentially fix small errors and further improve the readability of the manuscript.
p.3 tetramethyl-ammonium -> tetraethylammonium
p.7 "Snapshot of the final snapshot" -> "Snapshot of the final simulation coordinates"
p. 8 "sigma value" - please spell out what it is.
p. 9 "one or other subunit of the tetramer" -> "one or another subunit of the tetramer" or "one or more subunits of the tetramer"
p 15 "(the net charge of these constructs is thus zero)." -> ""(the net charge of these constructs is zero for these systems)." Please note that using ionizable amino acid residues in their default protonation state does not guarantee net zero charge of the system since the number of cationic and anionic residues is generally not the same.
p. 15 "Two K+ ions were initially positioned in the selectivity filter, one coordinated by residues 373..." Please indicate at which ion binding sites S_1, S_2, e.g. K+ were located and what the residue names are .
SI Figs. S3-S20. Please indicate in the figure captions that all those data are for RY785
SI Fig. S22 and SI Table S1 captions "shown in Fig. S20" -> "shown in Fig. S21"
We thank the Reviewer for this thorough proofreading. We have made the necessary corrections.
Reviewer #2 (Recommendations for the authors):
The authors have addressed most of my comments satisfactorily, with the exception of the first point. Below, I provide further clarification regarding my concern.
First, it appears that the authors may have misunderstood what is meant by the possibility of multiple binding sites for RY785. This does not imply that the central pore is excluded as a binding site. Rather, it refers to the possibility that, in addition to a pore-domain site, the ligand may interact with additional binding sites, either simultaneously or in a statedependent manner. Increasing structural and functional evidence supports the existence of multiple ligand-binding modes in voltage-gated ion channels. For example, polyunsaturated fatty acids have been shown to bind to KCNQ1 at both the voltage sensor domain and the pore domain (https://doi.org/10.1085/jgp.202012850). Similarly, cannabidiol has been structurally resolved in Nav1.7 at two distinct sites, one in a fenestration and another near the IFM-binding pocket (https://doi.org/10.1038/s41467-02339307-6). These advances illustrate that ligand ecects cannot always be interpreted based solely on a single binding site identified previously. Therefore, even if one assumes that there is no precedent for a small-molecule inhibitor that simultaneously acts on both the voltage sensor and pore domain, this does not exclude the possibility that a ligand may bind to both regions in dicerent functional states.
The Reviewer’s opinion came across clearly in the previous version. We however disagree that a computational investigation of the possibility that RY785 binds to the voltagesensors is well-advised at this point, given that the model we propose seemingly ocers a rationale for the inhibitory ecects observed experimentally. Our opinion is also that there is no compelling precedent for the mechanism of inhibition envisaged by the Reviewer – and would argue that neither of the two studies referenced above are compelling examples. As we stated in our previous response to the Reviewer, we believe that the logical next step in this research will be to validate or refute the computational prediction we have put forward, experimentally.
In addition, the present computational study does not provide direct mechanistic evidence to explain the statement that RY785 accelerates voltage-sensor deactivation. Specifically, no simulations were performed to model pore-domain closure or voltage-sensor motion upon RY785 binding. Moreover, alternative binding sites were neither explored nor explicitly excluded, as the simulations only involved placing a single molecule of TEA or RY785 approximately 10 Å below the cytoplasmic gate. Under these conditions, conclusions regarding ecects on voltage-sensor dynamics remain speculative.
That is a fair characterization.
These concerns do not detract from the overall quality of this otherwise strong computational study. There are several straightforward ways to address this issue. For example:
(1) Perform molecular docking or related screening approaches to evaluate potential ligand-binding sites beyond the central pore, particularly in regions proximal to the voltage sensor. This should not impose a substantial additional computational burden for a computational chemistry group.
(2) Revise the abstract and discussion to clarify that the current work focuses exclusively on pore-domain binding and does not explore possible additional binding sites near the voltage sensor. Explicitly stating this limitation would help prevent potential overinterpretation by readers.
We have opted for (2), as noted above.
-
-
powo.science.kew.org powo.science.kew.org
-
Solanaceae
same family as the mandrake
-
-
ccrc.tc.columbia.edu ccrc.tc.columbia.edu
-
Our results were consistent with the idea that each community has a limited amount of experience and resources to help build up student capital in their college-going population.
a very important issue that does need more addressing
-
This approach also has the benefit of addressing systemic inequities built into society.
incredibily important.
-
gathered the resources, traits, and skills they need to navigate complex social and academic environments.
everyone has different ways of learning and teaching but there's really only one way that is taught and it's outdated. most students and teachers have to conform to that outdated standard
-
sense of control over the world.
def what most people crave and make assumptions for a sense of safety
-
-
garden-plan-lto-b.vercel.app garden-plan-lto-b.vercel.app
-
Garden Plan customer
These people haven't purchased the plan - so should be "Garden Plan User"
-
If you're reading this, you probably know the feeling. You want a real garden, the kind that actually feeds your family, but every year you end up with patchy results, wasted seeds, and a half-empty bed by July. I know that feeling because I lived it for a long time before I figured this out.In 2006, my dad Edwin joined his brother managing Bountiful Blessings Farm in middle Tennessee. I went full-time there in 2008, and we ran a Winter CSA from October through March. Keeping fresh produce on people's tables through the dead of winter is the hardest test of a gardener I can think of. We leaned hard on Eliot Coleman's books, on what my dad had learned over the years, and on a lot of trial and error in the field.Around the same time, my dad and I launched an online garden training business together. We'd been working alongside each other for years, and we wanted to teach what we knew to people growing food in their own backyards. That's where I learned the real problem isn't growing food. It's planning. Most people don't fail because they're bad gardeners. They fail because nobody ever gave them a real plan that maps out their season from start to finish.So we started building one. First it was spreadsheets and custom checklists for our students. Then it became "Click 'N Drop Gardening Calendars." Then it became Seedtime, the app, the plans, the cheat sheets, all of it, because what people really needed was one place where the layout, the timing, the rotation, and the techniques all lived together.The $2,000 Plug & Play Garden Plan you're looking at right now is the foundation of all of it. It's the same plan I use on my own garden. It's the plan we use to teach beginners and experienced gardeners alike how to actually feed themselves from a backyard space.If you're frustrated, if you've tried to garden and it just hasn't clicked, this is what I'd hand you if you walked up my driveway and asked me where to start. No fluff, no theory. Just the plan that works.
If you're reading this, you probably know the feeling. You want a real garden, the kind that actually feeds your family, but every year you end up with patchy results, wasted seeds, and a half-empty bed by July. I know that feeling and it's not fun.
In 2006, my dad Edwin joined his brother managing Bountiful Blessings Farm in middle Tennessee. I went full-time there in 2008, and we ran a Winter CSA from October through March. Keeping fresh produce on people's tables through the dead of winter is the hardest test of a gardener I can think of. We leaned hard on Eliot Coleman's books, on what my dad and uncle had learned over the years, and on a lot of trial and error in the field.
A few years later, I launched an online garden training business and eventually pulled in my dad in to work with me. We'd been working alongside each other for years, and we wanted to teach what we knew to people growing food in their own backyards. That's where I learned the real struggle for a lot of people isn't just growing food. It's planning. Most people don't fail at growing a continual harvest of fresh food because they're bad gardeners. They fail because they don't know what to do and when to do it. They get overwhelmed, stressed, and nobody ever gave them a real plan that maps out their season from start to finish.
So we started solving this problem. First it was spreadsheets that we planned to turn into custom checklists for our students. Then it became "Click 'N Drop Gardening Calendars." Then it became Seedtime, the app, our plug and play garden plan, the cheat sheets, all of it, because what people really needed was one place where the layout, the timing, the rotation, and the techniques all lived together.
The $2,000 Plug & Play Garden Plan you're looking at right now is the result of all of it. It's the same plan I used on my own garden. It's the plan we use to teach beginners and experienced gardeners alike how to actually feed themselves and make the most from a backyard space.
If you're frustrated, if you've tried to garden and it just hasn't clicked, this is what I'd hand you if you walked up my driveway and asked me where to start. No fluff, no theory. Just the plan that works.
-
-
marianbull.substack.com marianbull.substack.com
-
Asking yourself, What am I doing? will help you overcome the habit of wanting to complete things quickly
How to be present.
-
Recently, I’ve been trying to do the dishes in silence. The easiest way to get them done, of course, is to blast some music, or put a podcast into my ears, to separate my mind from my body and distract myself from the task at hand. This will always be the post-dinner-party strategy, the only way to get it done. But in an effort to repair my relationship with time and attention and wean myself off of the devices that drain me, I have been trying to become more comfortable alone in my own head. Some people call dish-washing meditative; I don’t think I’ll ever get there. I am a subsistence dish-washer, not a fanatic. The act does not bring me delight, but I know that abstaining from it will only bring me stress. So I do it, and quietly. Sometimes I’ll even ask myself those words from Miller’s epigraph: what am I doing? The question feels like pause, not doubt.
This is a well written summation, honest and encouraging.
-
Sometimes when I don’t feel like doing the dishes, I’ll set a timer for 8 minutes, and tell myself that when it goes off, I can stop. But I never do. I’m either done or I keep going, it always works.
This actually seems like it could be very effective.
-
Cleaning, that is: before refrigeration was commonplace, cleaning after cooking meant keeping your family safe.
The nobility of protecting one's family.
-
The end point of cooking is not eating; it is cleaning.We dream, we plan, we crave, we shop, we chop, we fry, we simmer, we garnish, we serve. And then we eat, sometimes alone and sometimes not, and when the eating is done, a mess remains, record of our pleasure.
This reminds me of C.S. Lewis' Hross take on the full experience.
-
-
dsl.richmond.edu dsl.richmond.edu
-
By the late 1960s, an estimated 10,249 families had been displaced by urban renewal projects in Boston, 32% of which were families of color.
This is one of the cities were people of color weren't in majority. It is also important that the context of Boston building highways and expressways and that Boston is a very old city in the U.S gives a little more insight.
-
"culture of clearance"
This is a interesting quote when put with the context of how during this era over 330,000 families , not people families where displaced. Add that they didn't even include certain years and demographics it paints a smudge on this "golden era" of the U.S.
-
By the late 1960s, an estimated 4,077 families had been displaced by urban renewal projects in Atlanta, 89% of which were families of color.
This is a much higher number between the one in Chicago 89% compared to 64%. Granted Chicago is larger but, the density of the color disparity is alarming.
-
-
www.biorxiv.org www.biorxiv.org
-
eLife Assessment
In this manuscript, based on electron microscopy observations of C. elegans embryos, the authors make the bold claim that the plasma membrane ruptures during cell division and that closure of this opening by membrane extension contributes to cytokinesis. Although the findings are potentially valuable, the evidence in support of the authors' claims is inadequate.
-
-
web.hypothes.is web.hypothes.is
-
Human Centered Learning
Human-centered learning aligns well to the social learning concept. Social annotation delivers this.
-
-
www.sciencedirect.com www.sciencedirect.com
-
在路径 I 中,磺胺甲噁唑的异噁唑环受到 CUF/PMS 体系中活性氧物种的攻击,生成 P1、P2 和 P3。这些分解产物 P1、P2 和 P3 分别氧化为 P4 和 P5,这被视为磺胺甲噁唑的经典氧化路径[64]。根据路径 II,P6 的形成源于异噁唑环上甲基和苯环上–NH 2 的氧化,随后因 S-N 键断裂转化为 P7 和 P8[65]。在路径 III 中,磺胺甲噁唑的 S-N 键可直接被活性氧物种断裂,矿化为 P5 和 P9。接着,TP 9 的异噁唑环因羟基化被活性氧物种攻击形成 P10,随后由于·HO 与烯烃双键之间的高反应活性进一步生成 P11[66]。 此外,生成的 P9 分别通过偶联反应和异恶唑环的亲电取代进一步转化为 P12。P13 的形成可能涉及源自 P10 的–NH 2 基团的氮中心自由基与中间产物的偶联。随后,异恶唑环发生开环反应,生成 P14,后者被活性氧物种氧化为 P15。最终,这些中间产物将进一步矿化为小分子(P16、P17 和 P18)。
Ce-UiO-66-F催化PMS降解SAs的中间产物 The intermediates of CUF/PMS system were identified by the HPLC-MS (in Figure S17), which was used to investigate the possible degradation pathway of sulfamethoxazole. As shown in Figure 13d, the degradation pathway of sulfamethoxazole could be summarized as hydroxylation, deamination, sulfonamide (S-N) cleavage and desulfonation. In pathway I, the isoxazole ring of sulfamethoxazole was attacked by ROSs in CUF/PMS to generate P1, P2 and P3. The breakdown products P1, P2 and P3 oxidized into P4 and P5, respectively, which was viewed as a classic oxidation pathway of sulfamethoxazole [64]. According to pathway II, the formation of P6 resulted from the oxidation of methyl group on the isoxazole ring and –NH2 on the benzene ring and then converted into P7 and P8 due to the broken S-N bond [65]. For pathway III, the S-N bond of sulfamethoxazole could be directly broken by ROSs to be mineralized into P5 and P9. Then, the isoxazole ring of TP 9 was attacked by ROSs to form P10 due to hydroxylation, and then further generated P11, caused by high reactivity between ·HO and olefinic double bonds [66]. In addition, the generated P9 was further converted into P12 via coupling reaction and electrophilic replacement of the isoxazole ring, respectively [67]. The formation of P13 might be involved in the coupling of N-centered radical derived from by –NH2 group of P10 and intermediate products [68]. Then, the isoxazole ring opening reaction occurred resulting in the generation of P14, which was oxidized by ROSs into P15. Finally, these intermediates would be further mineralized into small molecules (P16, P17 and P18). W. Peng, J. Liao, Y. Yan, L. Chen, C. Ge, S. Lin Enriched nitrogen-doped carbon derived from expired drug with dual active sites as effective peroxymonosulfate activator: Ultra-fast sulfamethoxazole degradation and mechanism insight Chem. Eng. J., 446 (2022), Article 137407, 10.1016/j.cej.2022.137407 View PDF View articleView in ScopusGoogle Scholar [65] Y. Chen, D. Chen, X. Bai Binary MOFs-derived Mn-Co3O4 for efficient peroxymonosulfate activation to remove sulfamethoxazole: Oxygen vacancy-assisted high-valent cobalt-oxo species generation Chem. Eng. J., 479 (2024), Article 147886, 10.1016/j.cej.2023.147886 View PDF View articleView in ScopusGoogle Scholar [66] Y. Bao, W.J. Lee, T.-T. Lim, R. Wang, X. Hu Pore-functionalized ceramic membrane with isotropically impregnated cobalt oxide for sulfamethoxazole degradation and membrane fouling elimination: Synergistic effect between catalytic oxidation and membrane separation Appl. Catal. B-Environ., 254 (2019), pp. 37-46, 10.1016/j.apcatb.2019.04.081 View PDF View articleView in ScopusGoogle Scholar [67] M. Xu, H. Zhou, Z. Wu, N. Li, Z. Xiong, G. Yao, B. Lai Efficient degradation of sulfamethoxazole by NiCo2O4 modified expanded graphite activated peroxymonosulfate: Characterization, mechanism and degradation intermediates J. Hazard. Mater., 399 (2020), Article 123103, 10.1016/j.jhazmat.2020.123103 View PDF View articleView in ScopusGoogle Scholar [68] R. Guo, Y. Wang, J. Li, X. Cheng, D.D. Dionysiou Sulfamethoxazole degradation by visible light assisted peroxymonosulfate process based on nanohybrid manganese dioxide incorporating ferric oxide Appl. Catal. B-Environ., 278 (2020), Article 119297, 10.1016/j.apcatb.2020.119297 View PDF View articleView in ScopusGoogle Scholar
-
-
bafybeie6mvbv4iu6ohvg72edpp7n2cgwp455whdktehy2fkanyqsnk26ui.ipfs.dweb.link bafybeie6mvbv4iu6ohvg72edpp7n2cgwp455whdktehy2fkanyqsnk26ui.ipfs.dweb.link
-
https://bafybeie6mvbv4iu6ohvg72edpp7n2cgwp455whdktehy2fkanyqsnk26ui.ipfs.dweb.link/?💻/asus/🧊/me/📓/2026/04
Here we have an IndyWeb Applet that provides a permanent link to launching IPFS Desktop WbUI in an iframe in a page so that the local IPFS resource can be seen and annotated
-
-
arxiv.org arxiv.org
-
We characterize errors as a forest-structured Forest of Errors (FoE) and conclude that FoE makes the First the Best
主流观点认为推理错误是随机的、孤立的,可以通过更多探索来避免。但作者提出错误实际上具有森林结构特性,会相互影响和放大,这种系统性错误的观点挑战了人们对模型错误本质的传统理解。
-
This observation challenges widely accepted test-time scaling laws, leading us to hypothesize that errors within the reasoning path scale concurrently with test time.
大多数AI研究者认为推理时间越长,模型探索越充分,结果应该越好。作者却挑战这一共识,认为推理过程中的错误会随着时间同步增长,导致长时间推理反而会降低质量,这是一个颠覆性的观点。
-
The First is The Best, where alternative solutions are not merely suboptimal but potentially detrimental.
大多数人认为在大型推理模型中探索多种解决方案可以提高最终结果的质量,因为这种方法类似于人类的多角度思考。但作者认为第一个解决方案实际上是最好的,后续的替代方案不仅更差,甚至可能是有害的,这与主流的推理模型设计理念相悖。
-
-
www.technologyreview.com www.technologyreview.com
-
For small entrepreneurs in the US, deciding what to sell and where to make it has traditionally been a slow, labor-intensive process that can take months. Now that work is increasingly being done by AI tools like Accio, which help connect businesses with manufacturers in countries including China and India.
大多数人认为全球化会削弱小型企业的竞争力,但作者认为AI正在赋予小企业前所未有的全球供应链接入能力。AI工具如Accio正在消除地理障碍,使小型企业家能够以前所未有的速度和效率连接国际制造商,这挑战了关于规模经济的传统认知。
-
Sellers say that while AI tools have made it easier to come up with ideas and get a business off the ground, they do not replace the core skills that make someone good at e-commerce.
在AI热潮中,大多数人认为AI将使电子商务创业变得更容易,使技能变得不那么重要。但作者认为AI实际上放大了已有技能的价值,优秀的企业家仍然需要决策能力、执行速度和订单交付能力,这些是AI无法替代的核心竞争力。
-
McClary took the process from there, contacting the supplier himself to discuss the revised design. Within a month, the new version of the Guardian flashlight was back up for sale on Amazon and on his brand's website.
大多数人认为AI会完全取代人类在产品开发中的角色,但作者认为AI实际上增强了人类决策者的能力。Mike McClary使用AI工具缩短了产品开发周期,但仍需要亲自与供应商沟通并做出最终决策,这表明AI是辅助工具而非替代品。
-
-
huggingface.co huggingface.co
-
SOTA models of different architectures and parameter scales exhibit highly consistent failure patterns on the same set of hard samples, suggesting that the performance bottleneck stems from shared deficiencies in training data rather than architecture itself.
大多数人认为不同架构的模型会有不同的失败模式和弱点,但作者发现无论架构和参数规模如何,SOTA模型在相同困难样本上表现出高度一致的失败模式,这表明性能瓶颈源于训练数据的共同缺陷,而非架构差异,这一发现挑战了模型多样化的传统观点。
-
Without any architectural modification, MinerU2.5-Pro achieves 95.69 on OmniDocBench v1.6, improving over the same-architecture baseline by 2.71 points and surpassing all existing methods including models with over 200× more parameters.
大多数人认为更大的模型架构必然带来性能提升,但作者仅通过数据工程和训练策略优化,在保持1.2B参数架构不变的情况下,超越了参数量超过200倍的现有模型,这挑战了'越大越好'的行业共识,证明了数据质量的重要性。
-
-
huggingface.co huggingface.co
-
the design of the retrieval and cache policy, especially how they decide what to keep, reuse, or drop across scenes, seems to be what actually drives the latency and throughput gains
大多数研究者可能关注模型架构或算法创新来提升性能,但评论者指出检索和缓存策略的设计才是延迟和吞吐量提升的关键。这一观点挑战了AI研究中过度关注模型本身的倾向,暗示系统优化和资源管理策略可能比模型架构创新对性能影响更大,这是一个反直觉的系统设计见解。
-
they fuse streaming data construction with a unified model so the memory supports both real-time q&a and long-horizon interaction, which is nontrivial under strict latency constraints
大多数系统设计者可能认为实时问答和长时程交互需要不同的处理架构,但作者通过融合流式数据构建和统一模型,使内存同时支持这两种功能。这一设计挑战了实时系统处理复杂性的常规认知,表明在严格的延迟约束下实现多功能整合是可行的,这为实时AI助手的设计提供了新思路。
-
We release the AURA model together with a real-time inference framework to facilitate future research
大多数人认为先进的视频理解模型通常会被商业公司保留作为专有技术,但作者选择开源模型和实时推理框架。这一反直觉的决策挑战了AI研究中常见的封闭做法,表明作者更注重推动领域发展而非商业利益,这可能加速整个视频理解领域的技术进步。
-
It achieves state-of-the-art performance on streaming benchmarks and supports a real-time demo system with ASR and TTS running at 2 FPS on two 80G accelerators
大多数人认为实时视频处理需要极高的计算资源和帧率才能有效,但作者仅用两块80G加速器就实现了2 FPS的实时系统,并达到了最先进的性能。这一结果挑战了高性能视频处理需要大量计算资源的共识,暗示通过优化算法和架构可以显著降低实时视频处理的计算门槛。
-
-
huggingface.co huggingface.co
-
leading baselines achieve only about half the accuracy at the same efficiency
作者暗示当前主流的KV缓存压缩方法在相同效率水平下只能达到约一半的准确率,这表明现有方法存在根本性缺陷。这一尖锐的批评挑战了当前领域内的技术路线,暗示大多数同行可能一直在错误的方向上优化KV压缩。
-
-
hackernoon.com hackernoon.com
-
amplifies the false narrative that technology and creativity are at odds, and that existing rights holders must be compensated by AI companies for changing industry dynamics.
大多数人认为技术创新与创意保护之间存在根本冲突,但作者认为这种观点是错误的叙事。这一挑战性论点打破了技术进步必然损害创作者权益的二元对立思维,暗示两者可以共存共赢。
-
The government has so far favoured a pro-innovation, sector-led approach, prioritising voluntary principles over hard regulation.
大多数人认为政府会迅速采取立法行动保护创作者权益,但作者指出英国政府实际上倾向于自愿原则而非硬性监管。这一观点挑战了公众对政府会在AI版权问题上采取强硬措施的预期,揭示了政策制定的实际倾向。
-
introducing a commercial text and data mining exception for AI training would expand the AI sector in the country.
大多数人认为放宽数据挖掘限制会促进AI创新和增长,但作者认为这种例外实际上不会扩大AI产业。这一观点与科技行业普遍倡导的'更多数据等于更好AI'的信念相悖,挑战了数据自由流动的主流叙事。
-
-
yaseenghanem.com yaseenghanem.com
-
when setting up a new Macbook it presents FileVault as an optional checkbox and I can certainly tell you that there are many people (including my younger self and my family member with the Intel Mac) who do not know what it is and choose to disable it instead.
大多数人认为Apple会默认启用关键安全功能如FileVault,以保护用户数据。但作者指出,FileVault实际上是一个可选功能,许多用户(包括他自己和家人)在设置新Mac时会选择禁用它,这挑战了人们对Apple默认安全策略的认知,揭示了系统安全依赖于用户知识而非厂商默认设置的实际情况。
-
using "Open File..." dialog (`⌘+O`) you could still open and view any file on the system and could preview any file that safari could preview (e.g. `.html`, `.htm`, `.txt`, `.pdf`, and image files)
大多数人认为Apple在更新后会修复安全漏洞,恢复模式的浏览器会被严格限制。但作者发现,即使在更新后的版本中,通过使用"打开文件"对话框,仍然可以访问和预览系统上的任何文件,这表明Apple的修复措施并不彻底,违背了人们对安全补效的预期。
-
by "saving" the webpage (`file->save as`) instead of downloading it (which Safari automatically adds an extension for) I could force it to save it as `malicious_file` (with no extension).
大多数人认为浏览器的保存功能是安全的,会自动处理文件扩展名以确保文件类型正确。但作者发现,通过使用非标准的Content-Type和保存网页功能,可以绕过Safari的安全检查,保存任意扩展名的文件,这打破了人们对浏览器文件处理安全机制的普遍认知。
-
macOS decides to boot the `Volumes` partition which includes `Data`, `Macintosh HD`, `macOS Base System`, and `Preboot` systems, and when you choose the `Macintosh HD` it allows you to save the file to the Mac's permanent disk.
大多数人认为macOS恢复模式是只读环境,用于系统修复和恢复,不应该允许对系统分区的写入操作。但作者发现,在恢复模式下,Safari浏览器竟然允许用户将文件直接保存到Mac的永久磁盘上,包括系统分区,这是一个严重的安全漏洞,违背了人们对恢复模式安全性的基本认知。
-
-
-
Because Deep Extract is doing more work, it takes longer than a standard extraction call. That said, measured against the real alternative of someone manually reviewing a 500-page fund statement field by field, it's faster, cheaper, and consistent at scale.
大多数人认为更复杂的处理流程必然意味着更高的成本和更慢的速度。但作者提出Deep Extract虽然执行更多工作且比标准提取调用更耗时,但在大规模应用中仍然比人工审查更快、更便宜、更一致,这一观点挑战了人们对于复杂性与效率之间关系的传统理解。
-
The issue isn't that models are bad at reading documents. It's that single-pass extraction has no mechanism to catch its own mistakes, and models get lazy.
大多数人认为AI模型在文档提取中的低准确率主要是因为模型能力不足或理解能力有限。但作者提出了一个反直觉的观点:问题不在于模型本身,而在于单次提取缺乏自我纠错的机制,导致模型'变懒'。这挑战了对AI能力局限性的传统认知。
-
For the documents that matter most, it gets to 99–100% field accuracy, even out-performing expert human labelers on extraction tasks.
大多数人认为人工智能系统在文档提取任务上总会落后于人类专家,尤其是对于复杂文档。但作者声称Deep Extract可以达到甚至超过人类专家的准确率(99-100%),这是一个相当大胆的断言,挑战了AI在文档处理领域无法超越人类能力的共识。
-
-
-
The demand for these medications has been the most ferocious thing I have witnessed in my working life, and the hardest parts of running a telehealth company, like finding doctors and fulfilling prescriptions, can be entirely outsourced to platforms like CareValidate and OpenLoop.
大多数人认为医疗行业监管严格且难以突破,但作者指出GLP-1药物的需求如此之大以至于一个人可以在短短两个月内创建价值数十亿美元的公司,并将医疗服务的核心功能外包。这一观点挑战了传统医疗行业的复杂性认知,展示了AI如何颠覆传统受监管行业。
-
His affiliates, armed with AI, built fake doctor profiles in Meta ads and made unscrupulous claims about weight loss using fake testimonials.
大多数人认为AI主要提高生产力和创造力,但作者展示了AI如何被用于大规模欺骗和剥削,创建虚假医生档案和虚假宣传。这一反直觉观点揭示了AI技术黑暗面,挑战了人们对AI价值的乐观假设,提醒我们技术中立性背后的伦理问题。
-
Software, he argues, should be approached the same way. It's a new medium, and it deserves a native design language instead of hand-me-down forms from the physical world.
大多数人认为数字界面应该模仿物理世界的设计元素以提高用户熟悉度,但作者认为软件应该有自己独特的设计语言,不应简单复制物理世界的形式。这一观点挑战了 skeuomorphism(拟物化设计)的传统理念,主张数字媒介应有原生表达方式。
-
The cost of understanding what happens in a video has dropped by a factor of roughly 40, while the quality of that understanding has improved dramatically.
大多数人认为AI视频分析仍处于早期阶段且成本高昂,但作者指出AI视频分析成本已大幅下降40倍,质量反而提升。这一反直觉观点暗示视频分析可能已经跨越了实用性的门槛,将催生全新的应用类别,挑战了人们对AI视频处理能力的传统认知。
-
The consistent argument across the Every Slack was that if cache-breaking usage costs more to serve, make those users pay more: Meter the consumption rather than ban the interface.
大多数人认为公司应该通过限制特定工具使用来保护自己的利益,但作者认为Anthropic应该按实际使用量收费而非直接禁止OpenClaw,因为这更符合公平原则和平台发展。这种观点挑战了科技公司常见的封闭生态策略,主张更开放的计量模式。
-
-
research.google research.google
-
Historically, AI evaluation has leaned toward the forest approach. Most researchers settle for 1 to 5 raters per item, assuming this is enough to find a single 'correct' truth.
大多数人认为AI评估领域的现状是合理的,因为1-5名评估者足以找到单一'正确'真相,但作者指出这种假设忽视了人类评估中的自然分歧。这一批判挑战了AI评估领域的现状,暗示当前许多研究结论可能基于不充分的数据收集方法,需要重新审视评估方法的可靠性。
-
The most encouraging finding is that one doesn't need an infinite budget. We found that by optimizing the ratings-per-item ratio correctly... one can achieve highly reproducible results with a modest budget of around 1,000 total annotations.
大多数人认为高质量的AI评估需要大量预算和大量数据,但作者证明通过优化评估者与项目的比例,即使使用适度的总标注量(约1000个)也能实现高度可复现的结果。这一发现挑战了'越多越好'的普遍观念,为资源有限的研究团队提供了实用的评估路径。
-
-
openai.com openai.com
-
the standard communications playbook just doesn't apply to us
大多数企业会遵循标准的公关和沟通策略。但作者认为OpenAI完全不需要遵循这些传统规则,这暗示了OpenAI认为自己已经达到了一个独特的地位,可以打破常规的商业沟通模式,这与其作为行业领导者的自我定位相符,但也可能引发对其沟通透明度的质疑。
-
-
huggingface.co huggingface.co
-
we aim to cover as many methods as possible, the environment is relatively complex. This codebase primarily supports inference for different world model tasks
大多数人可能认为统一框架应该简化复杂性以提高可用性,但作者认为为了覆盖更多方法,复杂环境是必要的,这挑战了'简单即是好'的普遍设计理念,因为作者认为复杂性能提供更全面的功能覆盖。
-
the memory module is where this design finally hits a sweet spot, separating persistence from real-time reasoning
大多数人认为记忆和推理应该是紧密结合的,但作者认为将持久性记忆与实时推理分离是设计的关键创新点,这挑战了传统认知中记忆与推理必须紧密结合的观点,因为作者认为这种分离能更好地管理长期记忆。
-
a world model is a model or framework centered on perception, equipped with interaction and long-term memory capabilities, for understanding and predicting the complex world
大多数人认为世界模型主要是关于预测和模拟物理世界的系统,但作者认为世界模型必须同时具备感知、交互和长期记忆三种核心能力,这挑战了传统上认为世界模型主要是预测系统的观点,因为作者强调理解与预测同样重要。
-
we have kept the memory modules separate for each pipeline — precisely so that memory can be better isolated and iteratively improved during early development.
大多数人可能认为统一架构应该共享内存模块以提高效率,但作者选择为每个管道保持独立的内存模块,这挑战了系统设计的常规优化思路。这种分离方法虽然可能牺牲一些效率,但为早期开发提供了更大的灵活性和迭代空间。
-
our framework is still being refined, and the design of the memory module primarily draws from the description in Cambrian-S, implementing core memory expansion and management functions.
大多数人可能认为世界模型的记忆模块应该是全新设计的创新组件,但作者承认他们的记忆模块主要借鉴了现有工作(Cambrian-S),这挑战了学术界对完全创新方法的期待。这种务实的方法表明,世界模型的发展可能更多依赖于现有技术的整合而非革命性创新。
-
we propose a clear definition: a world model is a model or framework centered on perception, equipped with interaction and long-term memory capabilities, for understanding and predicting the complex world.
大多数人认为世界模型主要关注预测和生成能力,但作者提出世界模型必须同时具备感知、交互和长期记忆能力,这是一个更广泛的定义,挑战了当前AI领域对世界模型的狭隘理解。这种定义扩展了传统预测模型的边界,将交互性和记忆能力作为核心要素。
-
-
hackernoon.com hackernoon.com
-
Reconstructing raw inputs forces models to model irrelevant low-level detail. Predicting in a learned embedding space allows the model to focus on semantically meaningful, causally relevant features.
大多数人认为AI模型需要重建完整的输入数据才能理解世界,但作者认为这种方法迫使模型关注无关的低级细节。相反,在嵌入空间中进行预测可以让模型专注于语义上有意义、因果相关的特征,这是一个反直觉的见解。
-
Whether or not this specific bet pays off, the underlying argument that the next meaningful leap in AI capability requires moving beyond language modeling is increasingly hard to dismiss.
尽管当前AI领域由语言模型主导,但作者认为语言模型范式已经达到其极限,真正的AI进步需要超越这一范式。这与行业主流观点相悖,暗示我们可能正处于AI范式的转折点。
-
AMI Labs is not building a product for immediate deployment. This is a fundamental research effort, likely measured in years before commercial applications emerge.
在当今AI创业公司追求快速变现的环境中,作者认为AMI Labs正在进行的是基础研究,而非产品开发。这与大多数AI初创公司的商业模式背道而驰,暗示真正的AI突破需要长期投入而非短期商业考量。
-
LLMs have no grounded understanding of the physical world. They model the statistical distribution of language about reality, not reality itself.
大多数人认为大型语言模型通过学习物理世界的知识来理解现实,但作者认为它们实际上只是在学习关于现实的文本描述的统计分布,而非理解现实本身。这是一个反直觉的观点,因为它挑战了我们对AI理解能力的普遍认知。
-
-
hackernoon.com hackernoon.com
-
You have to have people that have the ability to rethink the workflow at a scale that AI can execute, versus at a scale that humans can execute.
大多数人认为AI应该适应现有工作流程,但作者提出相反观点:人类需要重新设计工作流程以适应AI的能力范围。这一反直觉观点强调,AI的成功实施不仅需要技术,更需要组织思维方式的根本转变,从人类执行规模转向AI执行规模。
-
-
mistral.ai mistral.ai
-
You don't need a separate agent API. You need to look at every `input()` call, every CWD assumption, every pretty-printed-only output, and ask: what if the user on the other end is a process, not a person?
大多数人认为需要为AI代理创建专门的API或接口,但作者提出反直觉的观点:不需要单独的代理API,而应该重新设计现有的CLI工具,使其同时支持人类和代理。这种统一的方法更加高效,避免了维护两套接口的复杂性。
-
Implicit state is the Enemy
大多数开发者认为当前工作目录(CWD)和环境变量等隐式状态是理所当然的,是提高开发效率的捷径。但作者认为这些隐式状态是敌人,因为它们会给AI代理带来困难。通过使所有状态显式化,不仅解决了代理的问题,也使工具对人类更可预测和可脚本化。
-
The funny part is that none of this made the CLI worse for humans. The TUI picker still works and looks fancy, progress spinners still spin, confirmation dialogs still confirm. We just added a second door.
大多数人认为增加对AI代理的支持会使工具变得复杂,降低人类用户体验。但作者认为,为AI代理添加的功能实际上没有损害人类用户体验,反而通过增加'第二扇门'(非交互式接口)同时改善了两种用户群体的体验。
-
-
arxiv.org arxiv.org
-
Simultaneously, the parallelism in worker layer accelerates the speed of overall task execution, mitigating the significant latency
虽然并行处理在计算领域常见,但将其应用于LLM代理系统中的信息搜索任务可能出人意料,因为大多数LLM应用仍采用顺序处理模式,作者的观点挑战了这一现状。
-
The parallelism in worker layer accelerates the speed of overall task execution, mitigating the significant latency
虽然并行处理在计算领域常见,但作者将其应用于LLM代理系统中的信息搜索任务,挑战了传统顺序处理方法,暗示并行化可能是解决LLM延迟问题的关键。
-
By leveraging aggregation and reflection mechanisms at the Manager layer, our framework enforces strict context isolation to prevent saturation and error propagation
传统观点认为更多的上下文信息总是有益的,但作者提出严格上下文隔离可以防止饱和和错误传播,这与常规的'更多上下文更好'的直觉相悖。
-
Recent agentic search systems have made substantial progress by emphasising deep, multi-step reasoning. However, this focus often overlooks the challenges of wide-scale information synthesis
大多数人认为深度、多步推理是提升代理搜索系统性能的关键,但作者认为这种方法忽视了大规模信息合成的挑战,暗示过度强调推理深度可能不是最优路径。
-
-
arxiv.org arxiv.org
-
Each task includes a unified evaluation framework supporting sandboxed code and APIs, alongside a human reference trajectory annotated with stepwise checkpoints along dual-axis: S-axis and V-axis.
大多数人认为AI评估可以通过简单的自动化测试完成。但作者提出需要复杂的双轴(S-axis和V-axis)人工参考轨迹和沙箱环境支持,这暗示了评估AI代理能力的极端复杂性远超当前行业的普遍认知。这一观点挑战了AI评估的简化主义倾向,强调了人类参与在评估中的不可替代性。
-
Experimental results show the best model, Gemini3-pro, achieves 56.3% overall accuracy, which falls significantly to 23.0% on Level-3 tasks
大多数人认为当前最先进的多模态大模型已经接近或超越人类在复杂任务上的表现。然而,作者的数据表明,即使是最好的模型在复杂现实任务上的表现也远低于预期,准确率从整体56.3%骤降至23.0%。这一发现挑战了AI领域对当前技术能力的乐观评估,揭示了现实世界多模态代理任务的极端复杂性。
-
-
arxiv.org arxiv.org
-
during the inference phase, the framework invokes both memory mechanisms synchronously
作者主张在推理阶段同时调用两种不同的内存机制,这与当前大多数AI系统中采用单一推理路径的做法相悖。这种同步调用机制挑战了人们对AI推理过程应该线性或层次化的普遍认知。
-
the inherent limitations of such a single-paradigm approach pose a fundamental challenge for existing models
作者暗示当前主流LLM代理模型存在根本性架构缺陷,因为它们试图用单一范式解决本质上不同的问题。这一论点挑战了AI社区对现有方法的信心,暗示需要更根本性的架构变革而非渐进式改进。
-
these two challenges are fundamentally distinct: the former relies on fuzzy semantic planning, while the latter demands strict logical constraints
主流AI研究通常将语义规划和逻辑验证视为可以统一处理的问题,但作者明确指出它们是根本不同的挑战。这一观点与当前大多数LLM代理方法相悖,暗示了单一神经网络架构的局限性。
-
-
arxiv.org arxiv.org
-
our GTPO hybrid advantage formulation eliminates the advantage misalignment problem
大多数人认为在强化学习中,优势函数的计算和优化是一个相对直接的过程,但作者指出存在'优势不匹配问题',并提出了GTPO混合优势公式来解决它。这挑战了强化学习中的基本假设,表明即使是优势函数这样的核心概念也需要仔细设计才能在多轮任务中有效工作。
-
the trained 4B model exceeding GPT-4.1 (49.4 percent) and GPT-4o (42.8 percent) despite being 50 times smaller
大多数人认为在复杂任务中,大型语言模型由于其参数量和训练数据的优势,总是能显著超越小型模型。然而,作者展示了他们的方法能让一个小型4B模型在Tau-Bench基准测试中超越GPT-4.1和GPT-4o,这挑战了AI社区对模型规模的普遍信仰。
-
the trained 4B model exceeding GPT-4.1 (49.4 percent) and GPT-4o (42.8 percent) despite being 50 times smaller
大多数人认为GPT-4级别的性能需要同等规模或更大的模型才能实现,但作者展示了他们的4B模型不仅超过了GPT-4.1和GPT-4o,而且模型规模只有后者的1/50。这一发现挑战了AI领域中对模型规模的依赖,暗示了算法创新可能比单纯扩大模型规模更有效。
-
-
arxiv.org arxiv.org
-
model alignment alone does not reliably guarantee the safety of autonomous agents.
大多数人认为模型对齐(alignment)是确保AI系统安全的关键因素,但作者通过实验证明,即使是对齐良好的模型(如Claude Code)在计算机使用代理中也表现出高达73.63%的攻击成功率。这挑战了当前AI安全领域的核心假设,表明仅依赖模型对齐无法解决自主代理的安全问题。
Tags
Annotators
URL
-
-
www.biorxiv.org www.biorxiv.org
-
Note: This response was posted by the corresponding author to Review Commons. The content has not been altered except for formatting.
Learn more at Review Commons
Reply to the reviewers
1. Point-by-point description of the revisions
Reviewer #1 (Evidence, reproducibility and clarity (Required)):
In this study, the authors investigated the effect of nutritional stress (HSD and HFD) on cardiac function by assessing multiple parameters on adult flies. They next identified the adaptive transcriptomic changes in the heart in response to these nutritional stresses and screened for their roles under ND, HSD and HFD. They identified fit gene, encoding a satiety gene, expressed by cardiomyocytes and pericardial cells.
I think the characterisation is thorough; however, the conclusion is not well supported by the evidence. My main concern is that in many graphs, the difference between control and experiment is subtle, and, secondly, the authors showed some conflicting results (e.g. one RNAi showed a reduction of one parameter, however, the other independent RNAi did not. In this case, I believe the authors shouldn't conclude that the RNAi is functionally required, since the RNAis are meant to confirm each other.
First, we thank the reviewer for her/his constructive comments and suggestions. We obtained new results presented in the last version of the manuscript, which consistently support our conclusions and improve the study.
High-Sugar and High-Fat Diets modified cardiac performance
They assessed how HSD and HFD affect Adult fly heart performance. Instead of performing 3 weeks of dietary manipulation as has been done before by other groups, they put adult flies on HSD for 7 days and HFD for only 3 days.
We would like to clarify the nutritional challenge used. Cardiac function of flies was assessed at 10 days after emergence. Flies were put either in ND or HSD during these 10 days (ND and HSD conditions), or in ND for 7 days then transferred on HFD for 3days (HFD condition). Finally, all the females spent 10 days in a diet before being imaged or before hearts/brains dissection.
They found: HSD increases HP and SI, and reduces AI. The difference is too small and not consistent between different control lines. Also, when the difference is this small, p value does not tell much!
They probably intentionally induced a milder effect so that they could assess adaptive transcriptomic changes to this nutritional stress. In Fig. 1D SI is increased under HSD with control-KK, In Fig. S1C, SI is not changed under HSD with control-GD and control-GFP. Instead, DI is increased, which is also opposite to what they showed in Fig. 1 C. HFD increased ESD, EDD, SV, FS and CO.(Hypertrophy). This is not true with control-GD and control-GFP lines though! Comments: They have assessed many parameters in live animals with many different control lines, which is thorough. However, it is hard to draw any conclusions based on these conflicting results. Are these effect KK line specific?
Globally, we agree with the reviewer that the results, presented in the first version of the manuscript, for the control lines were difficult to understand due to the inconsistency of the phenotypes. In this revised version, we performed new results in Figure 1 and __S1 __regarding the effect of 10 days HSD and 3 days HFD exposure vs ND.
105 to 187 flies were imaged for the 3 control conditions, in the 3 diets concomitantly, to increase the power of our analysis. As mentioned in the main text (page 3, line 30-35; page 4, line 1-5), both diets deteriorate cardiac function with HFD leading to consistent phenotypes on heart diameters and rhythm and HSD milder effects. Indeed, the 3 control lines were uniformly affected by HFD after 3 days exposure, whereas 10 days in HSD was not sufficient to quantify a significant effect despite consistent the trends on several phenotypes (EDD, ESD, DI, AI and CO. These results revealed a different sensitivity of the cardiac performance when exposed to sugar and fat.
As described in the text, we were nevertheless confident that our approach would be good to investigate the early molecular dysregulations induced by sugar. This was the purpose of our analysis, presented in the follow-up of the manuscript.
Regarding the small differences measured in the phenotypes in HSD and HFD compared to ND, we would like to outline that the values presented are normalized values to control. Normalization is done for every independent experiment, performed at different dates, and permits the graphical representation of pooled values. Statistical analysis is performed using non-parametric Kruskal-Wallis test accordingly. Values are presented with the X axis cutting the Y axis at 0, this graphical representation also contributes to flattening the differences and p-values indicate their significance.
Analysis of the fly cardiac transcriptome upon nutritional stress
RNA seq to detect differentially expressed genes under HSD and HFD vs ND. Most DE genes are downregulated, which prompts them to assess how the downregulation of these genes adapts the animals to this nutritional stress.
High Sugar Diet downregulated 1c-metabolism and Leloir galactose pathways.
In this revised manuscript, we first present RT-qPCR validating the downregulation of Gnmt, Sardh and Galk expressions in the heart of 10days old HSD-fed females compared to ND-fed ones (Figure S3A).
We apologize for the confused explanations in the first version of the manuscript. We show new results in Figure 3 and __S3 __on the cardiac function of both Gnmt and Sardh, where following reviewer’s suggestion, both genes were knocked down in the heart in ND and Gnmt overexpressed in HSD. No available tools allowed us to test Sardh overexpression in HSD and we could not get some for Galk.
GNMT is downregulated under HSD and HFD.
In ND, GNMT knockdown increased ESD, EDD and CO. Sardh knockdown did the same? However, Sardh knockdown did not affect ESD significantly.
We reanalyze our first data and added new ones, comparing only knockdown or overexpression to the corresponding controls performed in concomitant experiments. Results are now shown in Figure 3C-E; S3C-H. Knocking down Gnmt in the heart increased HP, EDD, ESD and CO, Sardh knockdown in ND resulted in milder phenotypes but inducing significant hypertrophy in ND as Gnmt does. In both cases, FS was not impacted.
Both genes have been previously shown as beneficial to muscular function in time-restricted feeding context (Livelo et al., 2023, Nat.Comm.), illustrating that, even if both enzymes are involved in opposite reaction, their function has the same effect on organ/tissue function, as they did for heart diameters. The text corresponding to results and discussion were updated accordingly (pages 5, 11).
The conclusion here is: GNMT knockdown induces hypertrophy, similar to the effect of HFD.
In HSD, further knockdown of GNMT reduced (rescued) HP, suggesting downregulation of GNMT under HSD is adaptive. Should overexpress GNMT under HSD to see if this manipulation further increases HP, to claim GNMT downregulation is an adaptive change to high sugar stress.
We thank the reviewer for her/his suggestion. We now used UAS-GnmtWT (from FlyORF) to assess the role of Gnmt on cardiac function in HSD.
As shown in (Figure 3C-E; S3C,F), overexpressing Gnmt in the heart in HSD was sufficient to rescue some sugar induced phenotypes or to induce other dysfunctions, when compared to corresponding controls evaluated in the same experiments in ND and HSD. Notably, HP increase and CO decrease are rescued by Gnmt cardiac overexpression in HSD. Interestingly, the cardiac diastolic constriction induced by HSD is associated to increased FS and CO in this genotype in sugar diet. These new results strengthen the positive effect of Gnmt on cardiac function, improving it in HSD and preventing its deterioration in this diet.
Of note, Gnmt overexpression in ND did not trigger cardiac dysfunctions (data not shown).
The results and conclusions have been corrected.
Interestingly, HSD itself tends to decrease AI, a further knockdown of GNMT further decreases AI. This indicates GNMT downregulation under HSD contributes to AI reduction. Together, GNMT downregulation under HSD prevents HP from going higher, while its downregulation causes AI going down.
In the manscript, the authors claim that " Gnmt KD led reduced HP and AI, suggesting that it is able to counteract the effect of HSD observed in control flies on these phenotypes". This is not true according to the logic in Results section 1. As in section 1, the effect of HSD on AI is not significant, so the authors shouldn't say" HS tended to reduce AI".
Our reanalyzes and new results showed no Gnmt impact on AI, so these Figure panels were removed.
Why GNMT knockdown reduced FS under ND (Fig. S3C), while increasing FS under HSD (Fig. 3F)? If GNMT knockdown induces hypertrophy, I would expect it to increase FS.
Gnmt overexpression did not affect cardiac diameters in HSD, but it nevertheless led to an increased contractile efficacy compared to HSD controls (Figure S3F).
These new results strengthen the positive effect of Gnmt on cardiac function, preventing its deterioration in sugar diet. The text was modified accordingly.
High Fat Diet modulated CD36-scavenger receptor and Glut8 orthologues
In this revised manuscript, we present RT-qPCR validating the downregulation of Snmp1 expression and the slight upregulation of nebu’s in the heart of 10days old HFD-fed females compared to ND-fed ones (Figure S3B).
HFD: Snmp1 gene is downregulated, however, both overexpression and knockdown of Snmp1 in ND induced some phenotypes.
Indeed, as mentioned in the revised manuscript (page 6, lines 21-24), in heart of females fed ND, both Snmp1 knockdown (Snmp1KK) and overexpression (Snmp1WT) showed a reduction of EDD and ESD (Figure 3J; S3J) but FS is increased accordingly only in Snmp1KK.
As notified in the text, both downregulation and overexpression of Snmp1 led to side-phenotypes (page 6, lines 24-28): Snmp1KK exhibited abdominal fat increase (Figure S3K) and ostial cells seemed clearly malformed in Snmp1WT (Figure 3M). This may explain why the heart shows the same type of functional impairment in both genotypes.
We now discussed the hypothesis that these similar cardiac dysfunctions may result from Snmp1 being a regulator of organismal or cardiac lipid homeostasis. Indeed, increasing body fat content is deleterious as is increasing the import of fat in the cardiomyocytes. Finally, both affects cardiac cells’ health and functioning.
HFD: nebu has a role in regulating cardiac function under ND.
HSD and HFD revealed the secretory function of the heart
They identified diet-regulated secreted proteins that are required for cardiac dysfunction.
Cardiac Fit expression impacted Cardiac performance.
The author used Hand-G4 to knock down Fit using KK and GD lines, KK line showed a reduction in HP (Fig. 5A), but not GD line (Fig. S5D). How did the author conclude that Fit is required for cardiac function? Also, with the positive data, the difference is too subtle.
We apologize and agree that the contradictory or inconsistent results obtained with the two RNAi lines were confusing.
For this revised version, we first assess the effect of the two RNAi lines (KK and GD) on fit expression in the dissected hearts. RT-qPCR for KK line is presented in Figure S5A. GD line did not show a significant reduction of fit expression when expressed in the heart with Hand>, which can explain the former results presented (not shown but data are available). So, we removed all results obtained with the GD line in this revised version.
To confirm the KK effects, we used fit KO allele (fit81) and truncated version of fit, without its signal peptide (fitDeltaSP), which has a dominant negative effect, both previously published and validated (Sun et al. 2017, Nat. Comm.). These two mutants were used to investigate the cardiac function of fit in our analysis. Results presented in Figure 5 and S5 confirm the phenotypes already observed with the KK line when expressed with Hand> in the heart and with Lsp2> in the fat body.
Our results validate the effect of fit decrease on rhythmicity and contractility, the reverse effects being consistently observed in fit overexpression. In conclusion, we are confident in the requirement of Fit in the regulation of cardiac performance.
These new data are now included in the results section “Cardiac Fit expression impacted Cardiac performance” (pages 8-9)
**Referee cross-commenting**
i agree with the experiments proposed by reviewer 2.
Reviewer #1 (Significance (Required)):
The study aims to examine the effect of diet on cardiac function.
The strength is that a lot of characterisations were done.
the weakness is the functional data regarding fit could not be validated in two different RNAis, thus the evidence is not strong to support the conclusions.
We again would like to thank the reviewer for her/his remarks and suggestions. She/He highlights the weakness of the first analysis and this was an important and constructive feedbacks for us. We strengthened our results by increasing samples, reanalyzing data and performing mandatory new experiments that are now included in this revised version.
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
In this manuscript, Khamvongsa-Charbonnier et al. reported a RNA-seq analysis and RNA interference screening on high-fat and high-sugar-induced cardiomyopathy in Drosophila. The authors uncovered novel genes in 1C-metabolism, galactose metabolism, CD36-scavenger receptor and glucose transporter, as adaptative factors of cardiac function under high-fat and high-sugar treatment. The authors also identified a satiety hormone, Fit, as a cardiokine to control food intake and , expressed by dilp5 secretion. In summary, this study leverages the powerful genetic model Drosophila to uncover a number of new factors in regulating cardiac function under nutritional stresses and potentially offers new insights into molecular mechanisms underlying diet-related cardiac diseases. I have a few concerns, as listed below.
First, we would like to thank the reviewer for her/his comments and suggestions that deeply help us to improve the take-home messages of our manuscript. Following her/his recommendations and suggestions, we can now present a revised and stronger version of our manuscript.
- Quantitative RT-PCR is required to validate the expression patterns of candidate genes identified from the RNAseq analysis.
RT-qPCR have been performed on hearts dissected from 10 days old females fed ND, HSD or HFD. Gnmt, Sardh and Galk validated downregulation are presented in Figure S3A, Snmp1 downregulation and nebu upregulation (trend but non-significant) in Figure S3B, fit downregulation in Figure S5A.
The authors state that the dysregulated gene expression patterns reflect acute adaptation to HSD and HFD stresses. Most of the candidate genes in this study were downregulated upon HSD and HFD. However, it is recommended that overexpression of these genes, rather than knockdown, is needed to confirm whether the downregulation of these candidate genes upon stresses is an adaptative response.
We agree with the reviewer and followed her/his recommendation when tools were accessible for our analysis.
For example, HSD feeding induces the heart period. Knocking down Gnmt, specifically in the heart, under the HSD feeding changes can reduce the heart period. This evidence is insufficient to suggest the protective role of Gnmt under the HSD diet. Gnmt has already been downregulated under the HSD. Further knockdown of Gnmt, instead of returning the Gnmt expression to normal levels, to protect cardiac contractile performance complicates the model.
We thank the reviewer for her/his suggestion. We used UAS-*GnmtWT * (from FlyORF) to perform these experiments.
As shown in (Figure 3C-E; S3C,F), knocking down Gnmt in the heart increased HP, EDD, ESD and CO. In the same Figure panels and in Figure S3F, we showed that overexpressing Gnmt with Hand> in HSD was sufficient to rescue some sugar induced phenotypes or to induce some, when compared to corresponding controls evaluated in the same experiments in ND and HSD. Gnmt overexpression in ND did not trigger cardiac dysfunctions (data not shown).
HP increase and CO decrease are rescued by Gnmt cardiac overexpression in HSD. Interestingly, the cardiac constriction induced by HSD is not rescued by Gnmt overexpression, but it is enough to increase FS and CO in sugar diet. These new results strengthen the positive effect of Gnmt on cardiac function, improving it in HSD and preventing its deterioration in this diet.
Sardh knockdown in ND, resulted in milder phenotypes but induced significant hypertrophy in ND as Gnmt does. No available tools allowed us to test its overexpression in HSD.
Nevertheless, as mentioned and discussed in the manuscript (page 5, line 27-30; page 11, lines 11-14), such protective role of muscular function and integrity has already been characterized in fly IFM in time-restricted feeding experiments for Gnmt and Sardh (Livelo et al., 2023, Nat.Comm.). Our experiments show that both genes encounter the same role in cardiac function upon nutritional stresses. The text was modified accordingly.
The authors suggest that the effect of nebu on heart contractility is not dependent on diet. However, based on the result from Figure 3O-P, the HFD treatment blocks the effect of nebu knockdown on heart contractility. The authors need to further explain these results and modify their conclusions accordingly.
We completely agree with the reviewer. We did not correctly analyze these results. We reanalyze our data, taking into account only the experiments of nebu knockdown that were performed in ND and in HFD concomitantly. Results are shown in Figure 3O-P; S3L-N.
As mentioned in the manuscript (page 7, lines 3-8), nebu knockdown led to identical HP decrease in both diets but its constrictive effect (reduction of heart diameters) in ND is abrogated by fat diet.
We modified the text accordingly in the results and discussion (page 7, lines 8-11; page 12, lines 7-12).
It is a bit confusing that knockdown of fit using Hand-Gal4 induced food intake, but knockdown of fit using tin-Gal4 or Dot-Gal4 did not significantly induce food intake (Fig 6A). The author did not provide any explanation of these results. What is even more confusing is that overexpressing fit using Dot-Gal4 decreased food intake, but overexpressing fit using Hand-Gal4 or tin-Gal4 did not significantly decrease food intake (Fig 6B). Why was the strong food intake phenotype not observed using Hand-Gal4 in both experiments? These confusing results lead to a question, which cell type is responsible for the production of cardiokine, Fit?
We apologize for the misleading results presented in the initial manuscript. We hope that our revised version will clarify Fit function regarding its remote impact.
Concerning the requirement of Fit function and the cell types that produces Fit, the results we obtained when evaluating cardiac performance strongly suggest that both cardiomyocytes and pericardial cells are important and recapitulate the effect of Hand> (Figure 5A-C; S5G-H).
In the case of food intake measurements, we now present results with newly performed food intake experiments for the Hand>fitWT (Figure 6D). They show a significant reduction of food intake in this condition, corroborating the results obtained with Dot>. We add a clarification in the manuscript for this point (page 10, lines 11-16).
When testing the role of cardiac Fit in Dilp5 secretion, the authors subjected flies to starvation stress. However, the main focus of the present study is on HSD and HFD. The RNAseq analysis showed that Fit expression was downregulated by both HSD and HFD. Can the authors show that Dilp5 secretion is reduced by both HSD and HFD? Most importantly, can the authors test whether overexpression of cardiac Fit blocks HSD- or HFD-reduced Dilp5 secretion?
We understand the point raised by the reviewer. First of all, we wanted to correlate the measured impact on food intake, when manipulating fit expression in the heart, to the level of Dilp release, as it has been used and validated in (Sun et al. 2017, Nat. Comm.). In this purpose, we used the same approach and protocol and results are shown in Figure 6 E-F.
As mentioned by the reviewer, fit expression is downregulated in both HSD and HFD (which we confirmed by RT-qPCR in Figure S5A). As suggested by the reviewer, we performed Dilp5 immunostaining on CNS from females that were fed HSD of HFD for 10 days. Our results, in Figure 6B (left panels) and corresponding quantifications in Figure 6C, show that both diets strongly induce a decrease in Dilp5 amount in the IPCs and that it was not due to an altered Dilp2 or Dilp5 expression in the CNS (Figure S6A). In this condition, overexpressing fit, which has a promoting effect on Dilp secretion (Figure 6B, right panels ND), may only have an additive effect. This is shown in Figure 6B-C.
Reviewer #2 (Significance (Required)):
In summary, this study leverages the powerful genetic model Drosophila to uncover a number of new factors in regulating cardiac function under nutritional stresses and potentially offers new insights into molecular mechanisms underlying diet-related cardiac diseases.
We again would like to thank the reviewer for her/his remarks and suggestions. Her/His important and constructive feedbacks helped us to improve and strengthen our study. Despite the weak points of the first version, she/he had supportive feedback and we deeply thank her/him. This revised version had improved results and analysis, thanks to the use of new genetic tools that strengthen this analysis.
-
Note: This preprint has been reviewed by subject experts for Review Commons. Content has not been altered except for formatting.
Learn more at Review Commons
Referee #2
Evidence, reproducibility and clarity
In this manuscript, Khamvongsa-Charbonnier et al. reported a RNA-seq analysis and RNA interference screening on high-fat and high-sugar-induced cardiomyopathy in Drosophila. The authors uncovered novel genes in 1C-metabolism, galactose metabolism, CD36-scavenger receptor and glucose transporter, as adaptative factors of cardiac function under high-fat and high-sugar treatment. The authors also identified a satiety hormone, Fit, as a cardiokine to control food intake and , expressed by dilp5 secretion. In summary, this study leverages the powerful genetic model Drosophila to uncover a number of new factors in regulating cardiac function under nutritional stresses and potentially offers new insights into molecular mechanisms underlying diet-related cardiac diseases. I have a few concerns, as listed below.
- Quantitative RT-PCR is required to validate the expression patterns of candidate genes identified from the RNAseq analysis.
- The authors state that the dysregulated gene expression patterns reflect acute adaptation to HSD and HFD stresses. Most of the candidate genes in this study were downregulated upon HSD and HFD. However, it is recommended that overexpression of these genes, rather than knockdown, is needed to confirm whether the downregulation of these candidate genes upon stresses is an adaptative response. For example, HSD feeding induces the heart period. Knocking down Gnmt, specifically in the heart, under the HSD feeding changes can reduce the heart period. This evidence is insufficient to suggest the protective role of Gnmt under the HSD diet. Gnmt has already been downregulated under the HSD. Further knockdown of Gnmt, instead of returning the Gnmt expression to normal levels, to protect cardiac contractile performance complicates the model.
- The authors suggest that the effect of nebu on heart contractility is not dependent on diet. However, based on the result from Figure 3O-P, the HFD treatment blocks the effect of nebu knockdown on heart contractility. The authors need to further explain these results and modify their conclusions accordingly.
- It is a bit confusing that knockdown of fit using Hand-Gal4 induced food intake, but knockdown of fit using tin-Gal4 or Dot-Gal4 did not significantly induce food intake (Fig 6A). The author did not provide any explanation of these results.
What is even more confusing is that overexpressing fit using Dot-Gal4 decreased food intake, but overexpressing fit using Hand-Gal4 or tin-Gal4 did not significantly decrease food intake (Fig 6B). Why was the strong food intake phenotype not observed using Hand-Gal4 in both experiments?
These confusing results lead to a question, which cell type is responsible for the production of cardiokine, Fit? 5. When testing the role of cardiac Fit in Dilp5 secretion, the authors subjected flies to starvation stress. However, the main focus of the present study is on HSD and HFD. The RNAseq analysis showed that Fit expression was downregulated by both HSD and HFD. Can the authors show that Dilp5 secretion is reduced by both HSD and HFD? Most importantly, can the authors test whether overexpression of cardiac Fit blocks HSD- or HFD-reduced Dilp5 secretion?
Significance
In summary, this study leverages the powerful genetic model Drosophila to uncover a number of new factors in regulating cardiac function under nutritional stresses and potentially offers new insights into molecular mechanisms underlying diet-related cardiac diseases.
-
Note: This preprint has been reviewed by subject experts for Review Commons. Content has not been altered except for formatting.
Learn more at Review Commons
Referee #1
Evidence, reproducibility and clarity
In this study, the authors investigated the effect of nutritional stress (HSD and HFD) on cardiac function by assessing multiple parameters on adult flies. They next identified the adaptive transcriptomic changes in the heart in response to these nutritional stresses and screened for their roles under ND, HSD and HFD. They identified fit gene, encoding a satiety gene, expressed by cardiomyocytes and pericardial cells.
I think the characterisation is thorough; however, the conclusion is not well supported by the evidence. My main concern is that in many graphs, the difference between control and experiment is subtle, and, secondly, the authors showed some conflicting results (e.g. one RNAi showed a reduction of one parameter, however, the other independent RNAi did not. In this case, I believe the authors shouldn't conclude that the RNAi is functionally required, since the RNAis are meant to confirm each other.
High-Sugar and High-Fat Diets modified cardiac performance
They assessed how HSD and HFD affect Adult fly heart performance. Instead of performing 3 weeks of dietary manipulation as has been done before by other groups, they put adult flies on HSD for 7 days and HFD for only 3 days. They found: HSD increases HP and SI, and reduces AI. The difference is too small and not consistent between different control lines. Also, when the difference is this small, p value does not tell much! They probably intentionally induced a milder effect so that they could assess adaptive transcriptomic changes to this nutritional stress. In Fig. 1D SI is increased under HSD with control-KK, In Fig. S1C, SI is not changed under HSD with control-GD and control-GFP. Instead, DI is increased, which is also opposite to what they showed in Fig. 1 C.
HFD increased ESD, EDD, SV, FS and CO.(Hypertrophy). This is not true with control-GD and control-GFP lines though!
Comments: They have assessed many parameters in live animals with many different control lines, which is thorough. However, it is hard to draw any conclusions based on these conflicting results. Are these effect KK line specific?
Analysis of the fly cardiac transcriptome upon nutritional stress
RNA seq to detect differentially expressed genes under HSD and HFD vs ND. Most DE genes are downregulated, which prompts them to assess how the downregulation of these genes adapts the animals to this nutritional stress.
High Sugar Diet downregulated 1c-metabolism and Leloir galactose pathways.
GNMT is downregulated under HSD and HFD. In ND, GNMT knockdown increased ESD, EDD and CO. Sardh knockdown did the same? However, Sardh knockdown did not affect ESD significantly.
The conclusion here is: GNMT knockdown induces hypertrophy, similar to the effect of HFD.
In HSD, further knockdown of GNMT reduced (rescued) HP, suggesting downregulation of GNMT under HSD is adaptive. Should overexpress GNMT under HSD to see if this manipulation further increases HP, to claim GNMT downregulation is an adaptive change to high sugar stress. Interestingly, HSD itself tends to decrease AI, a further knockdown of GNMT further decreases AI. This indicates GNMT downregulation under HSD contributes to AI reduction. Together, GNMT downregulation under HSD prevents HP from going higher, while its downregulation causes AI going down.
In the manscript, the authors claim that " Gnmt KD led reduced HP and AI, suggesting that it is able to counteract the effect of HSD observed in control flies on these phenotypes". This is not true according to the logic in Results section 1. As in section 1, the effect of HSD on AI is not significant, so the authors shouldn't say" HS tended to reduce AI".
Why GNMT knockdown reduced FS under ND (Fig. S3C), while increasing FS under HSD (Fig. 3F)? If GNMT knockdown induces hypertrophy, I would expect it to increase FS.
High Fat Diet modulated CD36-scavenger receptor and Glut8 orthologues
HFD: Snmp1 gene is downregulated, however, both overexpression and knockdown of Snmp1 in ND induced some phenotypes.
HFD: nebu has a role in regulating cardiac function under ND.
HSD and HFD revealed the secretory function of the heart
They identified diet-regulated secreted proteins that are required for cardiac dysfunction.
Cardiac Fit expression impacted Cardiac performance.
The author used Hand-G4 to knock down Fit using KK and GD lines, KK line showed a reduction in HP (Fig. 5A), but not GD line (Fig. S5D). How did the author conclude that Fit is required for cardiac function? Also, with the positive data, the difference is too subtle.
Referee cross-commenting
i agree with the experiments proposed by reviewer 2.
Significance
The study aims to examine the effect of diet on cardiac function.
The strength is that a lot of characterisations were done.
the weakness is the functional data regarding fit could not be validated in two different RNAis, thus the evidence is not strong to support the conclusions.
-
-
www.biorxiv.org www.biorxiv.org
-
eLife Assessment
This manuscript uncovers the importance of Vinculin in the maintenance of junctional integrity during neural tube closure in regions of increased mechanical stress, by using sophisticated methods such as laser ablation and live imaging. The manuscript also reports a novel application of an established embryonic stem cell protocol to efficiently generate mutant and transgenic embryos for analysis. The findings are fundamental in nature, significantly improve our understanding of a major research question, and are backed by compelling evidence. Whilst there is much to appreciate in this work, exactly how Vinculin mediates neural fold elevation remains unclear, and addressing this lacuna will significantly improve the strength of the manuscript; in addition, some rewriting for better clarity (including technical/methodological details) and inclusion of possible consequences of the increased number of tight junction gaps in the vinculin mutant would be pertinent.
-
Reviewer #1 (Public review):
Summary:
In many vertebrates, the neural tube closes by folding, elevation, and fusion of bilateral neural folds. Loss of the actin-binding protein Vinculin causes failed cranial neural tube closure in mice and is associated with neural tube defects in human patients, but it was not known how Vinculin contributes to neural tube closure. Here, Prudhomme and colleagues find that neural fold elevation and the apical constriction that drives it initiate normally in Vinculin-deficient mouse embryos, but both arrest before the neural folds fuse. The time of failure coincides with increased mechanical tension within the cranial neural plate. They find that Vinculin localizes to areas of high mechanical stress in the WT neural plate, including multi-cellular junctions and dividing cells, and in the absence of Vinculin, recruitment of Myosin and Apical junction proteins is reduced at these sites. These data support a model in which Vinculin recruits junctional proteins to high-stress areas to maintain junctional integrity during neural tube closure.
Strengths:
The data presented are thorough, rigorous, and convincing. The combination of live imaging and transgenic fluorescent reporters enables direct observation of junctional behaviors within the mouse cranial neural plate and detailed analysis of how these behaviors are disrupted upon loss of Vinculin. The authors make good use of an ESC transplant approach to efficiently generate mutant and transgenic embryos for analysis.
Weaknesses:
Although the loss of junctional integrity, especially at multi-cellular junctions, is clearly and convincingly demonstrated in Vinculin-deficient embryos, it is not clear precisely how this disrupts the elevation of the neural folds to cause exencephaly.
-
Reviewer #2 (Public review):
Summary
Using mouse embryos early in development, this excellent paper from Prudhomme et al. shows that Vinculin's recruitment to adherens junctions during mammalian cranial neural tube closure is essential for maintaining junctional integrity in response to increased tension during this process. Previous work had shown that during neural tube elevation, planar polarity of Myosin II and mechanical forces in the tissue are increased. Additionally, mouse embryos lacking Vinculin were known to display neural tube closure failure, and mutations in human Vinculin had been associated with increased risk of neural tube defects, but the mechanism remained unclear. Here, the authors utilize a high-throughput embryonic stem cell (ESC)-based pipeline to generate Vinculin-depleted embryos, complemented by a conditional mutant lacking Vinculin in the embryonic lineages, to investigate this question. The authors show that Vinculin is not required for force generation, but Vinculin is recruited to cell-cell junctions in a tension-dependent manner and is needed to transmit actomyosin-mediated tension to junctions - particularly tricellular and higher-order multicellular junctions - so that apical constriction can happen during neural fold elevation. Furthermore, they find that Vinculin is required to maintain adhesion during high force events (e.g., rosette resolution and cell division) during neural tube closure. The research builds on previous studies about Vinculin's role in mechanotransduction at cell-cell junctions carried out in cultured epithelial cells, zebrafish cardiomyocytes, or early Xenopus embryos, and investigates how physiological forces required for mouse neural tube closure challenge junction integrity and the important role that Vinculin plays in maintenance of junction integrity and translation of mechanical forces into changes in tissue structure during this process.
Strengths:
This study stands out for its sophisticated use of laser ablation and live imaging in neurulating mouse embryos, enabling quantification of junctional tension, Vinculin recruitment to multicellular junctions, and assessment of junction integrity during neural tube elevation. The authors' use of both ESC-derived Vinculin mutant embryos complemented by a second conditional mutant of Vinculin convincingly demonstrates that their findings are specific to the loss of Vinculin. Additionally, the authors demonstrated proof-of-principle for their ESC-based pipeline with a Shroom3 mutant known to be important for neural tube closure. The Zallen lab's application of the genetically engineered ESC-derived mouse embryo pipeline to efficiently generate larger numbers of mutant mouse embryos exhibiting neural tube closure defects (compared with traditional genetic crossing strategies) that can be utilized for live imaging and mechanical perturbations like laser ablation will be valuable for future work in the field. The authors show that Vinculin depletion disrupts tricellular and multicellular junctions. Notably, over 75% of higher-order (5+) vertices in Vinculin mutant embryos display gaps, but interestingly, about one third of 5+ cell junctions in Control embryos also display gaps, indicating that transient vertex remodeling events are needed for normal neural tube closure. Overall, this is a well-written paper that places the authors' findings within the context of prior literature; their beautiful data that is robustly analyzed and clear figure presentation will make the authors' exciting findings accessible to readers.
Weaknesses:
The criteria for selection of junctions targeted by laser ablation, including specifics of location, Myosin II intensity, and initial junction length, should be more clearly described in the Methods, especially given the use of different reporter strains (MyoIIB-GFP vs. GFP-Plekha7) across figures, which may influence junction selection for laser ablation. Analysis of Myosin II in Vinculin mutant embryos would benefit from staining for active Myosin II (pMRLC), and further examination of actomyosin organization at different stages of neural fold elevation in controls vs. Vinculin mutants would be informative. Although the authors note that ZO-1 gaps are limited to a subset of vertices where adherens junction gaps are detected, the increased frequency of tight junction gaps in Vinculin mutants could have functional significance that should be noted. Finally, inclusion of schematics to detail how the adherens and tight junction gaps were defined and measured at cell vertices, as well as how cell division completion was defined, would improve transparency and strengthen readers' understanding of how the data were quantified.
-
Reviewer #3 (Public review):
Summary:
Prudhomme et al report a detailed analysis of the role of vinculin in maintaining neuroepithelial integrity during cranial neurulation.
Strengths:
The authors use complementary experiments involving super-resolution microscopy, laser ablation, and live imaging of conditional knockout and ESC-derived embryos to demonstrate that loss of vinculin produces wide gaps between the adherens junctions of neuroepithelial cells at later stages of cranial neural fold elevation. The data presented are of extremely high quality, logically presented in a compelling story, and represent a very substantial contribution.
Weaknesses:
The authors are invited to consider the largely minor questions recommended below.
(1) The laser ablations reported are a correlate of cell border, or 'junctional' tension. Please avoid broad statements such as 'mechanical forces are upregulated' (abstract), which invoke gene-like regulation of tissue-level forces (in Newtons). Changes in junctional tension are likely to relate to changes in force generated, but their relationship is not simple: higher tensile stress withstood by the shorter length of junctions in cells with smaller apical surfaces does not necessarily translate into greater force being produced by that cell. The junctional tension readout measured is perfectly relevant to the paper, more so than tissue-level forces would have been.
(2) What is the mechanical mechanism by which loss of vinculin prevents neural fold elevation? The authors present exciting findings about the cellular consequences of losing Vcl at the late elevation stages when the tissue is quantifiably dysmorphic. A clear argument of how Vcl loss could lead to this dysmorphology would strengthen the paper, particularly given that junctional tension defects are excluded and apical non-constriction at the late stage is only mild.
(3) Can the authors comment on the likely impacts of Vcl deletion on the basal domain of the cell? For example, they could cite live-imaging of distinct behaviours in Williams et al Dev Cell 2014, and the NTD phenotypes of some integrin/focal adhesion mutant mice.
(4) The apparent uncoupling of apical area (larger in Vcl KO) from junctional tension (equivalent) in this model is noteworthy. Can the authors speculate on its potential basis?
(5) Live imaging in Figure 7C appears to show a marked reduction in apical area before cleavage furrow formation (T0-18min), suggesting a large apical constriction event (post-mitotic?), as previously reported (e.g., Ampartzidis et al Dev Biol 2023). Do junctional gaps appear during these constrictions?
(6) The live imaging setup used is clearly sufficient to identify differences between genotypes, so this is only a minor point. The gassing conditions listed in the methods specify 5% CO2, but E8.5 embryos also need low O2 to complete cranial closure. Was the O2 level controlled? Was tissue-level shape change observed to be consistent with ongoing neurulation during live-imaging?
(7) Neither the multi-cell laser ablations in the pre-print by De La O cited here, nor the narrower junctional ablations in Bocanegra-Moreno et al., Nat Phys, (2023), identified differences in recoil between developmental stages. Why might those results be different from the findings reported here (e.g., analysis region - not specified in the latter paper)? Limitations to interpreting junctional ablations between cells with different junction lengths include more of the recoil being dissipated by retraction of the longer ablated border.
(8) Is a truncated Vcl expressed in the ESC model, which could bind catenin without an F-actin anchor? The very high-contrast western shown is cropped so it is not clear whether the catenin-binding N-terminus is present. Does the antibody used recognise the head domain (this reviewer could not readily find the information)?
-
-
mhcc.pressbooks.pub mhcc.pressbooks.pub
-
“Medusa” by Alice Pike Barney, Smithsonian American Art Museum and its Renwick Gallery is licensed under CC BY 4.0
Barney's interpretation of Medusa has the signature snakes for hair we see a lot, but for some reason Medusa's expression looks possibly fearful? Possibly bewildered? I also notice that only her head, neck, and left hand are visible while the rest of her body is hidden by what appears to be rock? Could there be some underlying meaning as to why this is?
-