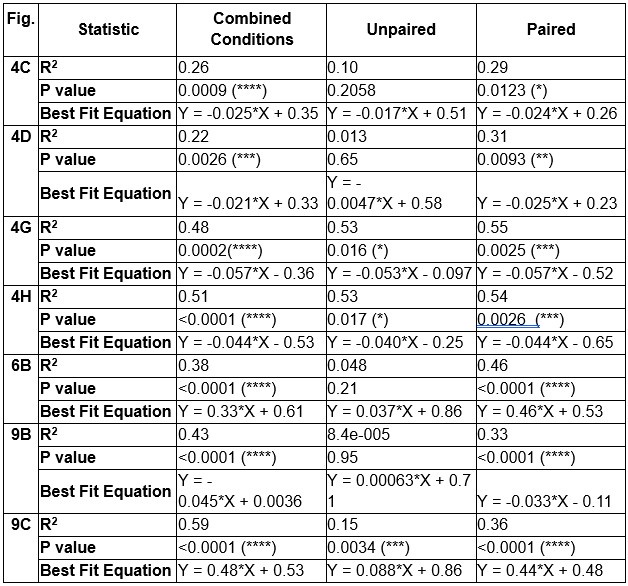
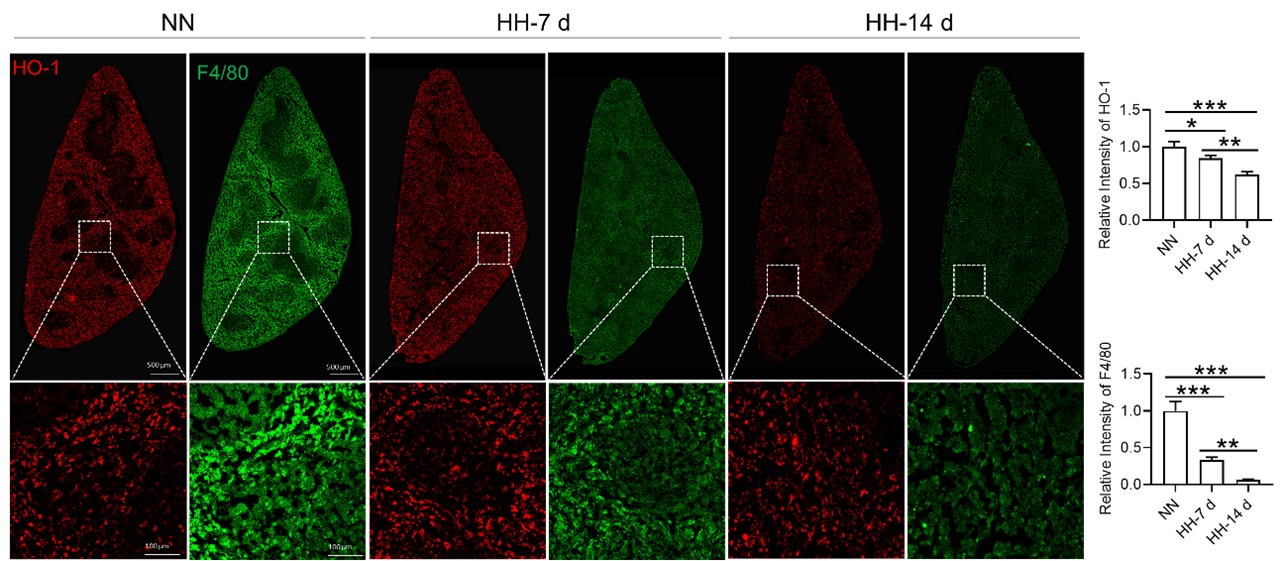
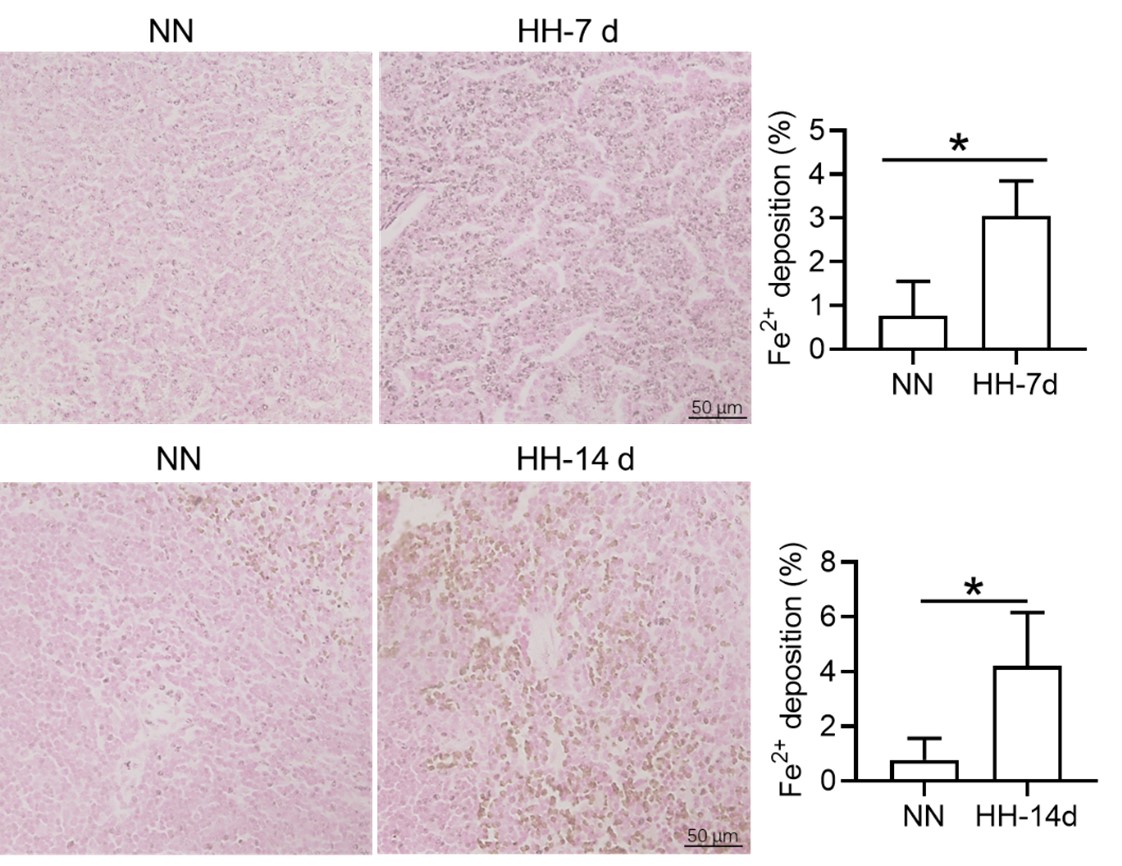
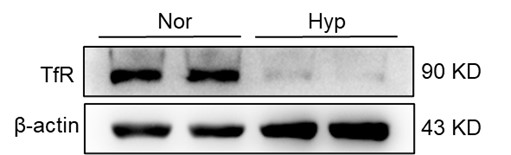
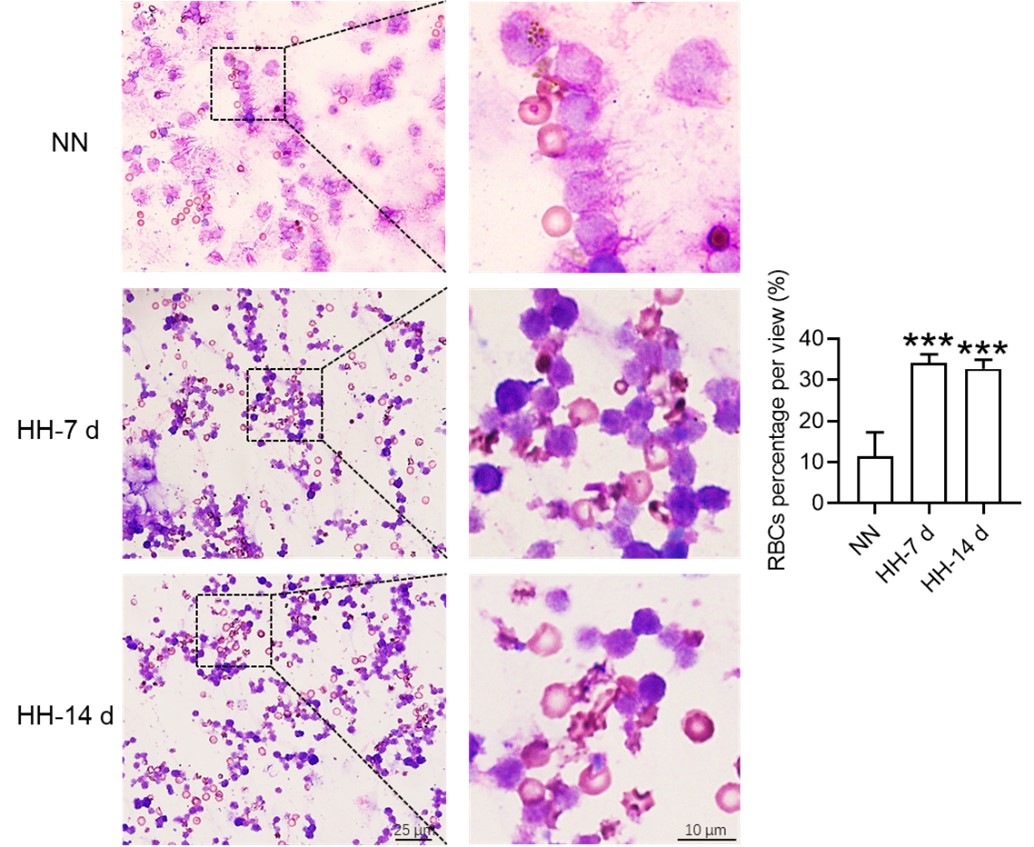
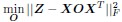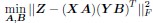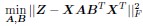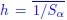Author response:
Notes to Editors
We previously received comments from three reviewers at Biological Psychiatry, which we have addressed in detail below. The following is a summary of the reviewers’ comments along with our responses.
Reviewers 1 and 2 sought clearer justification for studying the cognition-mental health overlap (covariation) and its neuroimaging correlates. In the revised manuscripts, we expanded the Introduction and Discussion to explicitly outline the theoretical implications of investigating this overlap with machine learning. We also added nuance to the interpretation of the observed associations.
Reviewer 1 raised concerns about the accessibility of the machine learning methodology for readers without expertise in this field. We revised the Methods section to provide a clearer, step-by-step explanation of our machine learning approach, particularly the two-level machine learning through stacking. We also enhanced the description of the overall machine learning design, including model training, validation, and testing.
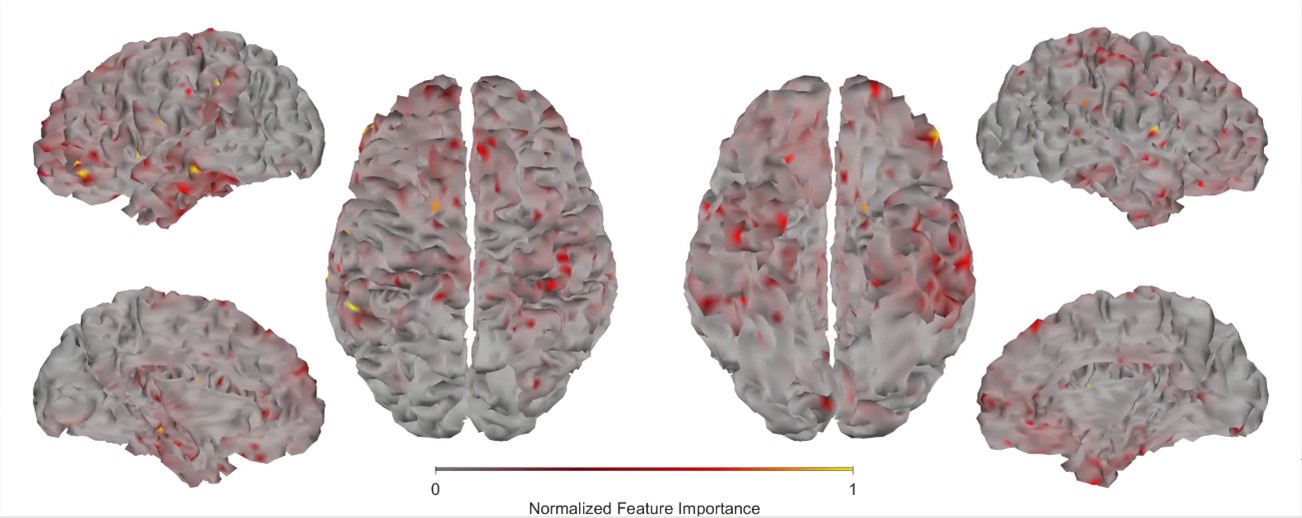
In response to Reviewer 2’s request for deeper interpretation of our findings and stronger theoretical grounding, we have expanded our discussion by incorporating a thorough interpretation of how mental health indices relate to cognition, material that was previously included only in supplementary materials due to word limit constraints. We have further strengthened the theoretical justification for our study design, with particular emphasis on the importance of examining shared variance between cognition and mental health through the derivation of neural markers of cognition. Additionally, to enhance the biological interpretation of our results, we included new analyses of feature importance across neuroimaging modalities, providing clearer insights into which neural features contribute most to the observed relationships.
Notably, Reviewer 3 acknowledged the strength of our study, including multimodal design, robust analytical approach, and clear visualization and interpretation of results. Their comments were exclusively methodological, underscoring the manuscript’s quality.
Reviewer 1:
The authors try to bridge mental health characteristics, global cognition and various MRI-derived (structural, diffusion and resting state fMRI) measures using the large dataset of UK Biobank. Each MRI modality alone explained max 25% of the cognitionmental health covariance, and when combined together 48% of the variance could be explained. As a peer-reviewer not familiar with the used methods (machine learning, although familiar with imaging), the manuscript is hard to read and I wonder what the message for the field might be. In the end of the discussion the authors state '... we provide potential targets for behavioural and physiological interventions that may affect cognition', the real relevance (and impact) of the findings is unclear to me.
Thank you for your thorough review and practical recommendations. We appreciate your constructive comments and suggestions and hope our revisions adequately address your concerns.
Major questions
(1) The methods are hard to follow for people not in this specific subfield, and therefore, I expect that for readers it is hard to understand how valid and how useful the approach is.
Thank you for your comment. To enhance accessibility for readers without a machine learning background, we revised the Methods section to clarify our analyses while retaining important technical details needed to understand our approach. Recognizing that some concepts may require prior knowledge, we provide detailed explanations of each analysis step, including the machine learning pipeline in the Supplementary Methods.
Line 188: “We employed nested cross-validation to predict cognition from mental health indices and 72 neuroimaging phenotypes (Fig. 1). Nested cross-validation is a robust method for evaluating machine-learning models while tuning their hyperparameters, ensuring that performance estimates are both accurate and unbiased. Here, we used a nested cross-validation scheme with five outer folds and ten inner folds.
We started by dividing the entire dataset into five outer folds. Each fold took a turn being held out as the outerfold test set (20% of the data), while the remaining four folds (80% of the data) were used as an outer-fold training set. Within each outer-fold training set, we performed a second layer of cross-validation – this time splitting the data into ten inner folds. These inner folds were used exclusively for hyperparameter tuning: models were trained on nine of the inner folds and validated on the remaining one, cycling through all ten combinations.
We then selected the hyperparameter configuration that performed best across the inner-fold validation sets, as determined by the minimal mean squared error (MSE). The model was then retrained on the full outer-fold training set using this hyperparameter configuration and evaluated on the outer-fold test set, using four performance metrics: Pearson r, the coefficient of determination ( R<sup>2</sup>), the mean absolute error (MAE), and the MSE. This entire process was repeated for each of the five outer folds, ensuring that every data point is used for both training and testing, but never at the same time. We opted for five outer folds instead of ten to reduce computational demands, particularly memory and processing time, given the substantial volume of neuroimaging data involved in model training. Five outer folds led to an outer-fold test set at least n = 4 000, which should be sufficient for model evaluation. In contrast, we retained ten inner folds to ensure robust and stable hyperparameter tuning, maximising the reliability of model selection.
To model the relationship between mental health and cognition, we employed Partial Least Squares Regression (PLSR) to predict the g-factor from 133 mental health variables. To model the relationship between neuroimaging data and cognition, we used a two-step stacking approach [15–17,61] to integrate information from 72 neuroimaging phenotypes across three MRI modalities. In the first step, we trained 72 base (first-level) PLSR models, each predicting the g-factor from a single neuroimaging phenotype. In the second step, we used the predicted values from these base models as input features for stacked models, which again predicted the g-factor. We constructed four stacked models based on the source of the base predictions: one each for dwMRI, rsMRI, sMRI, and a combined model incorporating all modalities (“dwMRI Stacked”, “rsMRI Stacked”, “sMRI Stacked”, and “All MRI Stacked”, respectively). Each stacked model was trained using one of four machine learning algorithms – ElasticNet, Random Forest, XGBoost, or Support Vector Regression – selected individually for each model (see Supplementary Materials, S6).
For rsMRI phenotypes, we treated the choice of functional connectivity quantification method – full correlation, partial correlation, or tangent space parametrization – as a hyperparameter. The method yielding the highest performance on the outer-fold training set was selected for predicting the g-factor (see Supplementary Materials, S5).
To prevent data leakage, we standardized the data using the mean and standard deviation derived from the training set and applied these parameters to the corresponding test set within each outer fold. This standardization was performed at three key stages: before g-factor derivation, before regressing out modality-specific confounds from the MRI data, and before stacking. Similarly, to maintain strict separation between training and testing data, both base and stacked models were trained exclusively on participants from the outer-fold training set and subsequently applied to the corresponding outer-fold test set.
To evaluate model performance and assess statistical significance, we aggregated the predicted and observed g_factor values from each outer-fold test set. We then computed a bootstrap distribution of Pearson’s correlation coefficient (_r) by resampling with replacement 5 000 times, generating 95% confidence intervals (CIs) (Fig. 1). Model performance was considered statistically significant if the 95% CI did not include zero, indicating that the observed associations were unlikely to have occurred by chance.”
(2) If only 40% of the cognition-mental health covariation can be explained by the MRI variables, how to explain the other 60% of the variance? And related to this %: why do the author think that 'this provides us confidence in using MRI to derive quantitative neuromarkers of cognition'?
Thank you for this insightful observation. Using the MRI modalities available in the UK Biobank, we were able to account for 48% of the covariation between cognition and mental health. The remaining 52% of unexplained variance may arise from several sources. One possibility is the absence of certain neuroimaging modalities in the UK Biobank dataset, such as task-based fMRI contrasts, positron emission tomography, arterial spin labeling, and magnetoencephalography/electroencephalography. Prior research from our group and others has consistently demonstrated strong predictive performance from specific task-based fMRI contrasts, particularly those derived from tasks like the n-Back working memory task and the face-name episodic memory task, none of which is available in the UK Biobank.
Moreover, there are inherent limitations in using MRI as a proxy for brain structure and function. Measurement error and intra-individual variability, such as differences in a cognitive state between cognitive assessments and MRI acquisition, may also contribute to the unexplained variance. According to the Research Domain Criteria (RDoC) framework, brain circuits represent only one level of neurobiological analysis relevant to cognition. Other levels, including genes, molecules, cells, and physiological processes, may also play a role in the cognition-mental health relationship.
Nonetheless, neuroimaging provides a valuable window into the biological mechanisms underlying this overlap – insights that cannot be gleaned from behavioural data alone. We have now incorporated these considerations into the Discussion section.
Line 658: “Although recent debates [18] have challenged the predictive utility of MRI for cognition, our multimodal marker integrating 72 neuroimaging phenotypes captures nearly half of the mental health-explained variance in cognition. We demonstrate that neural markers with greater predictive accuracy for cognition also better explain cognition-mental health covariation, showing that multimodal MRI can capture both a substantial cognitive variance and nearly half of its shared variance with mental health. Finally, we show that our neuromarkers explain a substantial portion of the age- and sex-related variance in the cognition-mental health relationship, highlighting their relevance in modeling cognition across demographic strata.
The remaining unexplained variance in the relationship between cognition and mental health likely stems from multiple sources. One possibility is the absence of certain neuroimaging modalities in the UK Biobank dataset, such as task-based fMRI contrasts, positron emission tomography, arterial spin labeling, and magnetoencephalography/electroencephalography. Prior research has consistently demonstrated strong predictive performance from specific task-based fMRI contrasts, particularly those derived from tasks like the n-Back working memory task and the face-name episodic memory task, none of which is available in the UK Biobank [15,17,61,69,114,142,151].
Moreover, there are inherent limitations in using MRI as a proxy for brain structure and function. Measurement error and intra-individual variability, such as differences in a cognitive state between cognitive assessments and MRI acquisition, may also contribute to the unexplained variance. According to the RDoC framework, brain circuits represent only one level of neurobiological analysis relevant to cognition [14]. Other levels, including genes, molecules, cells, and physiological processes, may also play a role in the cognition-mental health relationship.
Nonetheless, neuroimaging provides a valuable window into the biological mechanisms underlying this overlap – insights that cannot be gleaned from behavioural data alone. Ultimately, our findings validate brain-based neural markers as a fundamental neurobiological unit of analysis, advancing our understanding of mental health through the lens of cognition.”
Regarding our confidence in using MRI to derive neural markers for cognition, we base this on the predictive performance of MRI-based models. As we note in the Discussion (Line 554: “Consistent with previous studies, we show that MRI data predict individual differences in cognition with a medium-size performance (r ≈ 0.4) [15–17, 28, 61, 67, 68].”), the medium effect size we observed (r ≈ 0.4) agrees with existing literature on brain-cognition relationships, confirming that machine learning leads to replicable results. This effect size represents a moderate yet meaningful association in neuroimaging studies of aging, consistent with reports linking brain to behaviour in adults (Krämer et al., 2024; Tetereva et al., 2022). For example, a recent meta-analysis by Vieira and colleagues (2022) reported a similar effect size (r = 0.42, 95% CI [0.35;0.50]). Our study includes over 15000 participants, comparable to or more than typical meta-analyses, allowing us to characterise our work as a “mega-analysis”. And on top of this predictive performance, we found our neural markers for cognition to capture half of the cognition-mental health covariation, boosting our confidence in our approach.
Krämer C, Stumme J, da Costa Campos L, Dellani P, Rubbert C, Caspers J, et al. Prediction of cognitive performance differences in older age from multimodal neuroimaging data. GeroScience. 2024;46:283–308.
Tetereva A, Li J, Deng JD, Stringaris A, Pat N. Capturing brain cognition relationship: Integrating task‐based fMRI across tasks markedly boosts prediction and test‐retest reliability. NeuroImage. 2022;263:119588.
(3) Imagine that we can increase the explained variance using multimodal MRI measures, why is it useful? What does it learn us? What might be the implications?
We assume that by variance, Reviewer 1 referred to the cognition-mental health covariation mentioned in point 2) above.
If we can increase the explained cognition-mental health covariation using multimodal MRI measures, it would mean that we have developed a reasonable neuromarker that is close to RDoC’s neurobiological unit of analysis for cognition. RDoC treats cognition as one of the main basic functional domains that transdiagnostically underly mental health. According to RDoC, mental health should be studied in relation to cognition, alongside other domains such as negative and positive valence systems, arousal and regulatory systems, social processes, and sensorimotor functions. RDoC further emphasizes that each domain, including cognition, should be investigated not only at the behavioural level but also through its neurobiological correlates. This means RDoC aims to discover neural markers of cognition that explain the covariation between cognition and mental health. For us, we approach the development of such neural markers using multimodal neuroimaging. We have now explained the motivation of our study in the first paragraph of the Introduction.
Line 43: “Cognition and mental health are closely intertwined [1]. Cognitive dysfunction is present in various mental illnesses, including anxiety [2, 3], depression [4–6], and psychotic disorders [7–12]. National Institute of Mental Health’s Research Domain Criteria (RDoC) [13,14] treats cognition as one of the main basic functional domains that transdiagnostically underly mental health. According to RDoC, mental health should be studied in relation to cognition, alongside other domains such as negative and positive valence systems, arousal and regulatory systems, social processes, and sensorimotor functions. RDoC further emphasizes that each domain, including cognition, should be investigated not only at the behavioural level but also through its neurobiological correlates. In this study, we aim to examine how the covariation between cognition and mental health is reflected in neural markers of cognition, as measured through multimodal neuroimaging.”
More specific issues:
Introduction
(4) In the intro the sentence 'in some cases, altered cognitive functioning is directly related to psychiatric symptom severity' is in contrast to the next sentence '... are often stable and persist upon alleviation of psychiatric symptoms'.
Thank you for pointing this out. The first sentence refers to cases where cognitive deficits fluctuate with symptom severity, while the second emphasizes that core cognitive impairments often remain stable even during symptom remission. To avoid this confusion, we have removed these sentences.
(5) In the intro the text on the methods (various MRI modalities) is not needed for the Biol Psych readers audience.
We appreciate your comment. While some members of our target audience may have backgrounds in neuroimaging, machine learning, or psychiatry, we recognize that not all readers will be familiar with all three areas. To ensure accessibility for those who are not familiar with neuroimaging, we included a brief overview of the MRI modalities and quantification methods used in our study to provide context for the specific neuroimaging phenotypes. Additionally, we provided background information on the machine learning techniques employed, so that readers without a strong background in machine learning can still follow our methodology.
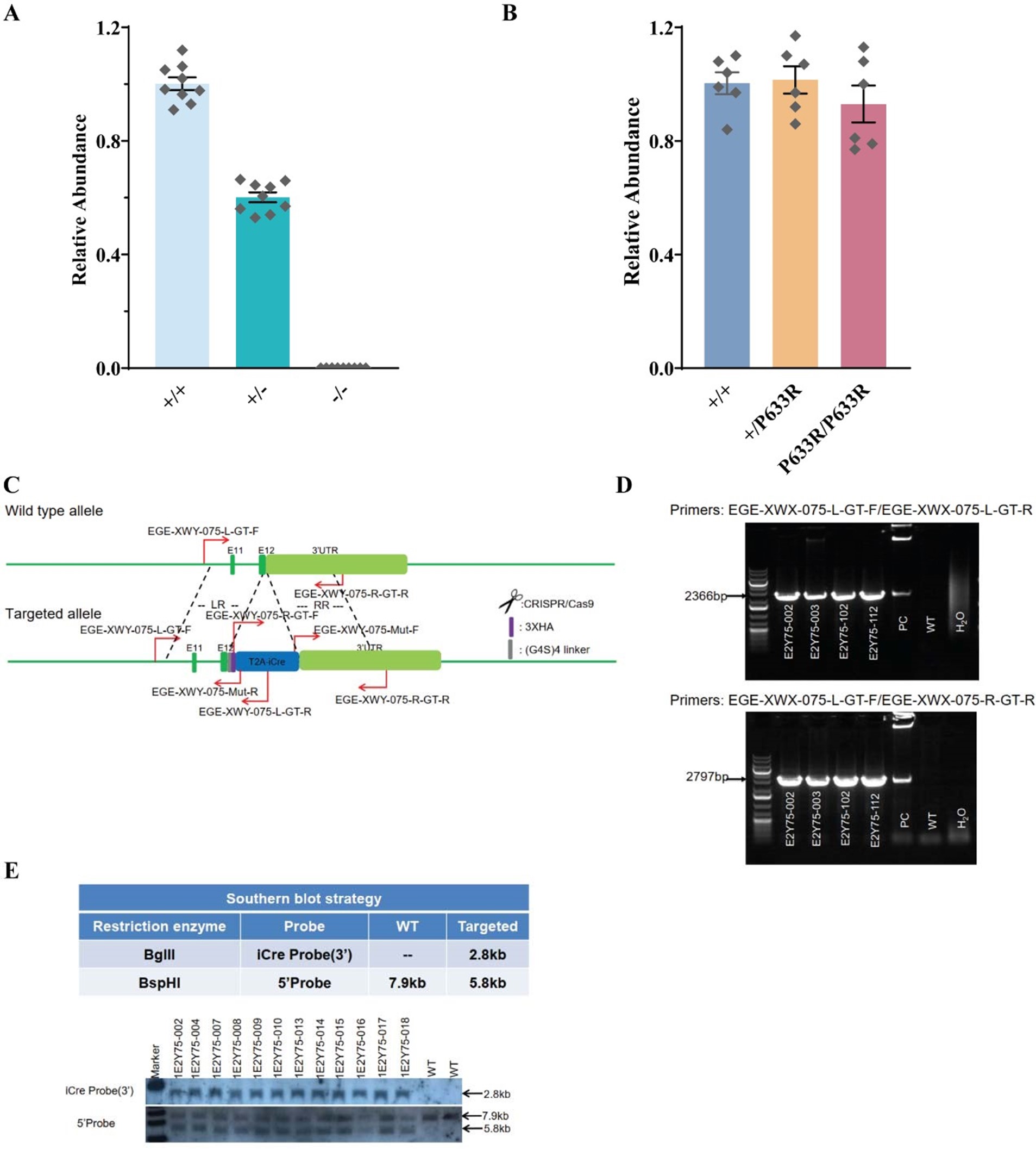
(6) Regarding age of the study sample: I understand that at recruitment the subjects' age ranges from 40 to 69 years. At MRI scanning the age ranges between about 46 to 82. How is that possible? And related to the age of the population: how did the authors deal with age in the analyses, since age is affecting both cognition as the brain measures?
Thank you for noticing this. In the Methods section, we first outline the characteristics of the UK Biobank cohort, including the age at first recruitment (40-69 years). Table 1 then shows the characteristics of participant subsamples included in each analysis. Since our study used data from Instance 2 (the second in-person visit), participants were approximately 5-13 years older at scanning, resulting in the age range of 46 to 82 years. We clarified the Table 1 caption as follows:
Line 113: “Table 1. Demographics for each subsample analysed: number, age, and sex of participants who completed all cognitive tests, mental health questionnaires, and MRI scanning”
We acknowledge that age may influence cognitive and neuroimaging measures. In our analyses, we intentionally preserved age-related variance in brain-cognition relationships across mid and late adulthood, as regressing out age completely would artificially remove biologically meaningful associations. At the same time, we rigorously addressed the effects of age and sex through additional commonality analyses quantifying age and sex contributions to the relationship between cognition and mental health.
As noted by Reviewer 1 and illustrated in Figure 8, age and sex shared substantial overlapping variance with both mental health and neuroimaging phenotypes in explaining cognitive outcomes. For example, in Figure 8i, age and sex together accounted for 43% of the variance in the cognition-mental health relationship:
(2.76 + 1.03) / (2.76 + 1.03 + 3.52 + 1.45) ≈ 0.43
Furthermore, neuromarkers from the all-MRI stacked model explained 72% of this age/sexrelated variance:
2.76 / (2.76 + 1.03) ≈ 0.72
This indicates that our neuromarkers captured a substantial portion of the cognition-mental health covariation that varied with age and sex, highlighting their relevance in age/sex-sensitive cognitive modeling.
In the Methods, Results, and Discussion, we say:
Methods
Line 263: “To understand how demographic factors, including age and sex, contribute to this relationship, we also conducted a separate set of commonality analyses treating age, sex, age2, age×sex, and age2×sex as an additional set of explanatory variables (Fig. 1).”
Results
Line 445: “Age and sex shared substantial overlapping variance with both mental health and neuroimaging in explaining cognition, accounting for 43% of the variance in the cognition-mental health relationship. Multimodal neural marker of cognition based on three MRI modalities (“All MRI Stacked”) explained 72% of this age and sex-related variance (Fig. 8i–l and Table S21).”
Discussion
Line 660: “We demonstrate that neural markers with greater predictive accuracy for cognition also better explain cognition-mental health covariation, showing that multimodal MRI can capture both a substantial cognitive variance and nearly half of its shared variance with mental health. Finally, we show that our neuromarkers explain a substantial portion of the age- and sex-related variance in the cognition-mental health relationship, highlighting their relevance in modeling cognition across demographic strata.”
(7) Regarding the mental health variables: where characteristics with positive value (e.g. happiness and subjective wellbeing) reversely scored (compared to the negative items, such as anxiety, addition, etc)?
We appreciate you noting this. These composite scores primarily represent standard clinical measures such as the GAD-7 anxiety scale and N-12 neuroticism scale. We did not reverse the scores to keep their directionality, therefore making interpretability consistent with the original studies the scores were derived from (e.g., Davis et al., 2020; Dutt et al., 2022). Complete descriptive statistics for all mental health indices and detailed derivation procedures are provided in the Supplementary Materials (S2). On Page 6, Supplementary Methods, we say:
Line 92: “Composite mental health scores included the Generalized Anxiety Disorder (GAD-7), the Posttraumatic Stress Disorder (PTSD) Checklist (PCL-6), the Alcohol Use Disorders Identification Test (AUDIT), the Patient Health Questionnaire (PHQ-9) [12], the Eysenck Neuroticism (N-12), Probable Depression Status (PDS), and the Recent Depressive Symptoms (RDS-4) scores [13, 14]. To calculate the GAD-7, PCL-6, AUDIT, and PHQ-9, we used questions introduced at the online follow-up [12]. To obtain the N-12, PDS, and RDS-4 scores [14], we used data collected during the baseline assessment [13, 14].
We subcategorized depression and GAD based on frequency, current status (ever had depression or anxiety and current status of depression or anxiety), severity, and clinical diagnosis (depression or anxiety confirmed by a healthcare practitioner). Additionally, we differentiated between different depression statuses, such as recurrent depression, depression triggered by loss, etc. Variables related to self-harm were subdivided based on whether a person has ever self-harmed with the intent to die.
To make response scales more intuitive, we recorded responses within the well-being domain such that the lower score corresponded to a lesser extent of satisfaction (“Extremely unhappy”) and the higher score indicated a higher level of happiness (“Extremely happy”). For all questions, we assigned the median values to “Prefer not to answer” (-818 for in-person assessment and -3 for online questionnaire) and “Do not know” (-121 for in-person assessment and -1 for online questionnaire) responses. We excluded the “Work/job satisfaction” question from the mental health derivatives list because it included a “Not employed” response option, which could not be reasonably coded.
To calculate the risk of PTSD, we used questions from the PCL-6 questionnaire. Following Davis and colleagues [12], PCL-6 scores ranged from 6 to 29. A PCL-6 score of 12 or below corresponds to a low risk of meeting the Clinician-Administered PTSD Scale diagnostic criteria. PCL-6 scores between 13 and 16 and between 17 and 25 are indicative of an increased risk and high risk of PTSD, respectively. A score of above 26 is interpreted as a very high risk of PTSD [12, 15]. PTSD status was set to positive if the PCL-6 score exceeded or was equal to 14 and encompassed stressful events instead of catastrophic trauma alone [12].
To assess alcohol consumption, alcohol dependence, and harm associated with drinking, we calculated the sum of the ten questions from the AUDIT questionnaire [16]. We additionally subdivided the AUDIT score into the alcohol consumption score (questions 1-3, AUDIT-C) and the score reflecting problems caused by alcohol (questions 4-10, AUDIT-P) [17]. In questions 2-10 that followed the first trigger question (“Frequency of drinking alcohol”), we replaced missing values with 0 as they would correspond to a “Never” response to the first question.
An AUDIT score cut-off of 8 suggests moderate or low-risk alcohol consumption, and scores of 8 to 15 and above 15 indicate severe/harmful and hazardous (alcohol dependence or moderate-severe alcohol use disorder) drinking, respectively [16, 18]. Subsequently, hazardous alcohol use and alcohol dependence status correspond to AUDIT scores of ≥ 8 and ≥ 15, respectively. The “Alcohol dependence ever” status was set to positive if a participant had ever been physically dependent on alcohol. To reduce skewness, we logx+1-transformed the AUDIT, AUDIT-C, and AUDIT-P scores [17].”
Davis KAS, Coleman JRI, Adams M, Allen N, Breen G, Cullen B, et al. Mental health in UK Biobank – development, implementation and results from an online questionnaire completed by 157 366 participants: a reanalysis. BJPsych Open. 2020;6:e18.
Dutt RK, Hannon K, Easley TO, Griffis JC, Zhang W, Bijsterbosch JD. Mental health in the UK Biobank: A roadmap to selfreport measures and neuroimaging correlates. Hum Brain Mapp. 2022;43:816–832.
(8) In the discussion section (page 23, line 416-421), the authors refer to specific findings that are not described in the results section > I would add these findings to the main manuscript (including the discussion / interpretation).
We appreciate your careful reading. We agree that our original Results section did not explicitly describe the factor loadings for mental health in the PLSR model, despite discussing their implications later in the paper. We needed to include this part of the discussion in the Supplementary Materials to meet the word limit of the original submission. However, in response to your suggestion, we have now added the results regarding factor loadings to the Results section. We also moved the discussion of the association between mental health features and general cognition from the Supplementary Material to the manuscript’s Discussion.
Results
Line 298: “On average, information about mental health predicted the g-factor at R<sup>2</sup><sub>mean</sub> = 0.10 and r<sub>mean</sub> \= 0.31 (95% CI [0.291, 0.315]; Fig. 2b and 2c and Supplementary Materials, S9, Table S12). The magnitude and direction of factor loadings for mental health in the PLSR model allowed us to quantify the contribution of individual mental health indices to cognition. Overall, the scores for mental distress, alcohol and cannabis use, and self-harm behaviours relate positively, and the scores for anxiety, neurological and mental health diagnoses, unusual or psychotic experiences, happiness and subjective well-being, and negative traumatic events relate negatively to cognition.”
Discussion
Line 492: “Factor loadings derived from the PLSR model showed that the scores for mental distress, alcohol and cannabis use, and self-harm behaviours related positively, and the scores for anxiety, neurological and mental health diagnoses, unusual or psychotic experiences, happiness and subjective well-being, and negative traumatic events related negatively to the g-factor. Positive PLSR loadings of features related to mental distress may indicate greater susceptibility to or exaggerated perception of stressful events, psychological overexcitability, and predisposition to rumination in people with higher cognition [72]. On the other hand, these findings may be specific to the UK Biobank cohort and the way the questions for this mental health category were constructed. In particular, to evaluate mental distress, the UK Biobank questionnaire asked whether an individual sought or received medical help for or suffered from mental distress. In this regard, the estimate for mental distress may be more indicative of whether an individual experiencing mental distress had an opportunity or aspiration to visit a doctor and seek professional help [73]. Thus, people with better cognitive abilities and also with a higher socioeconomic status may indeed be more likely to seek professional help.
Limited evidence supports a positive association between self-harm behaviours and cognitive abilities, with some studies indicating higher cognitive performance as a risk factor for non-suicidal self-harm. Research shows an inverse relationship between cognitive control of emotion and suicidal behaviours that weakens over the life course [73,74]. Some studies have found a positive correlation between cognitive abilities and the risk of nonsuicidal self-harm, suicidal thoughts, and suicidal plans that may be independent of or, conversely, affected by socioeconomic status [75,76]. In our study, the magnitude of the association between self-harm behaviours and cognition was low (Fig. 2), indicating a weak relationship.
Positive PLSR loadings of features related to alcohol and cannabis may also indicate the influence of other factors. Overall, this relationship is believed to be largely affected by age, income, education, social status, social equality, social norms, and quality of life [79–80]. For example, education level and income correlate with cognitive ability and alcohol consumption [79,81–83]. Research also links a higher probability of having tried alcohol or recreational drugs, including cannabis, to a tendency of more intelligent individuals to approach evolutionary novel stimuli [84,85]. This hypothesis is supported by studies showing that cannabis users perform better on some cognitive tasks [86]. Alternatively, frequent drinking can indicate higher social engagement, which is positively associated with cognition [87]. Young adults often drink alcohol as a social ritual in university settings to build connections with peers [88]. In older adults, drinking may accompany friends or family visits [89,90]. Mixed evidence on the link between alcohol and drug use and cognition makes it difficult to draw definite conclusions, leaving an open question about the nature of this relationship.
Consistent with previous studies, we showed that anxiety and negative traumatic experiences were inversely associated with cognitive abilities [90–93]. Anxiety may be linked to poorer cognitive performance via reduced working memory capacity, increased focus on negative thoughts, and attentional bias to threatening stimuli that hinder the allocation of cognitive resources to a current task [94–96]. Individuals with PTSD consistently showed impaired verbal and working memory, visual attention, inhibitory function, task switching, cognitive flexibility, and cognitive control [97–100]. Exposure to traumatic events that did not reach the PTSD threshold was also linked to impaired cognition. For example, childhood trauma is associated with worse performance in processing speed, attention, and executive function tasks in adulthood, and age at a first traumatic event is predictive of the rate of executive function decline in midlife [101,102]. In the UK Biobank cohort, adverse life events have been linked to lower cognitive flexibility, partially via depression level [103].
In agreement with our findings, cognitive deficits are often found in psychotic disorders [104,105]. We treated neurological and mental health symptoms as predictor variables and did not stratify or exclude people based on psychiatric status or symptom severity. Since no prior studies have examined isolated psychotic symptoms (e.g., recent unusual experiences, hearing unreal voices, or seeing unreal visions), we avoid speculating on how these symptoms relate to cognition in our sample.
Finally, negative PLSR loadings of the features related to happiness and subjective well-being may be specific to the study cohort, as these findings do not agree with some previous research [107–109]. On the other hand, our results agree with the study linking excessive optimism or optimistic thinking to lower cognitive performance in memory, verbal fluency, fluid intelligence, and numerical reasoning tasks, and suggesting that pessimism or realism indicates better cognition [110]. The concept of realism/optimism as indicators of cognition is a plausible explanation for a negative association between the g-factor and friendship satisfaction, as well as a negative PLSR loading of feelings that life is meaningful, especially in older adults who tend to reflect more on the meaning of life [111]. The latter is supported by the study showing a negative association between cognitive function and the search for the meaning of life and a change in the pattern of this relationship after the age of 60 [112]. Finally, a UK Biobank study found a positive association of happiness with speed and visuospatial memory but a negative relationship with reasoning ability [113].”
(9) In the discussion section (page 24, line 440-449), the authors give an explanation on why the diffusion measure have limited utility, but the arguments put forward also concern structural and rsfMRI measures.
Thank you for this important observation. Indeed, the argument about voxel-averaged diffusion components (“… these metrics are less specific to the properties of individual white matter axons or bundles, and instead represent a composite of multiple diffusion components averaged within a voxel and across major fibre pathways”) could theoretically apply across other MRI modalities. We have therefore removed this point from the discussion to avoid overgeneralization. However, we maintain our central argument about the biological specificity of conventional tractography-derived diffusion metrics as their particular sensitivity to white matter microstructure (e.g., axonal integrity, myelin content) may make them better suited for detecting neuropathological changes than dynamic cognitive processes. This interpretation aligns with the mixed evidence linking these metrics to cognitive performance, despite their established utility in detecting white matter abnormalities in clinical populations (e.g., Bergamino et al., 2021; Silk et al., 2009). We clarify this distinction in the manuscript.
Line 572: “The somewhat limited utility of diffusion metrics derived specifically from probabilistic tractography in serving as robust quantitative neuromarkers of cognition and its shared variance with mental health may stem from their greater sensitivity and specificity to neuronal integrity and white matter microstructure rather than to dynamic cognitive processes. Critically, probabilistic tractography may be less effective at capturing relationships between white matter microstructure and behavioural scores cross-sectionally, as this method is more sensitive to pathological changes or dynamic microstructural alterations like those occurring during maturation. While these indices can capture abnormal white matter microstructure in clinical populations such as Alzheimer’s disease, schizophrenia, or attention deficit hyperactivity disorder (ADHD) [117–119], the empirical evidence on their associations with cognitive performance is controversial [114, 120–126].”
Bergamino M, Walsh RR, Stokes AM. Free-water diffusion tensor imaging improves the accuracy and sensitivity of white matter analysis in Alzheimer’s disease. Sci Rep. 2021;11:6990.
Silk TJ, Vance A, Rinehart N, Bradshaw JL, Cunnington R. White-matter abnormalities in attention deficit hyperactivity disorder: a diffusion tensor imaging study. Hum Brain Mapp. 2009;30:2757–2765.
Reviewer 2:
This is an interesting study combining a lot of data to investigate the link between cognition and mental health. The description of the study is very clear, it's easy to read for someone like me who does not have a lot of expertise in machine learning.
We thank you for your thorough review and constructive feedback. Your insightful comments have helped us identify conceptual and methodological aspects that required improvement in the manuscript. We have incorporated relevant changes throughout the paper, and below, we address each of your points in detail.
Comment 1: My main concern with this manuscript is that it is not yet clear to me what it exactly means to look at the overlap between cognition and mental health. This relation is r=0.3 which is not that high, so why is it then necessary to explain this overlap with neuroimaging measures? And, could it be that the relation between cognition and mental health is explained by third variables (environment? opportunities?). In the introduction I miss an explanation of why it is important to study this and what it will tell us, and in the discussion I would like to read some kind of 'answer' to these questions.
Thank you. It’s important to clarify why we investigated the relationship between cognition and mental health, and what we found using data from the UK Biobank.
Conceptually, our work is grounded in the Research Domain Criteria (RDoC; Insel et al., 2010) framework. RDoC conceptualizes mental health not through traditional diagnostic categories, but through core functional domains that span the full spectrum from normal to abnormal functioning. These domains include cognition, negative and positive valence systems, arousal and regulatory systems, social processes, and sensorimotor functions. Within this framework, cognition is considered a fundamental domain that contributes to mental health across diagnostic boundaries. Meta-analytic evidence supports a link between cognitive functioning and mental health (Abramovitch, et al., 2021; East-Richard, et al., 2020). In the context of a large, population-based dataset like the UK Biobank, this implies that cognitive performance – as measured by various cognitive tasks – should be meaningfully associated with available mental health indicators.
However, because cognition is only one of several functional domains implicated in mental health, we do not expect the covariation between cognition and mental health to be very high. Other domains, such as negative and positive valence systems, arousal and regulatory systems, or social processing, may also play significant roles. Theoretically, this places an upper bound on the strength of the cognition-mental health relationship, especially in normative, nonclinical samples.
Our current findings from the UK Biobank reflect this. Most of the 133 mental health variables showed relatively weak individual correlations with cognition (mean r \= 0.01, SD = 0.05, min r \= –0.08, max r \= 0.17; see Figure 2). However, using a PLS-based machine learning approach, we were able to integrate information across all mental-health variables to predict cognition, yielding an out-of-sample correlation of r = 0.31 [95% CI: 0.29, 0.32].
We believe this estimate approximates the true strength of the cognition-mental health relationship in normative samples, consistent with both theoretical expectations and prior empirical findings. Theoretically, this aligns with the RDoC view that cognition is one of several contributing domains. Empirically, our results are consistent with findings from our previous mega-analysis in children (Wang et al., 2025). Moreover, in the field of gerontology, an effect size of r = 0.31 is not considered small. According to Brydges (2019), it falls around the 70th percentile of effect sizes reported in gerontological studies and approaches the threshold for a large effect (r \= 0.32). Given that most studies report within-sample associations, our out-of-sample results are likely more robust and generalizable (Yarkoni & Westfall, 2017).
To answer, “why is it then necessary to explain this overlap with neuroimaging measures”, we again draw on the conceptual foundation of the RDoC framework. RDoC emphasizes that each functional domain, such as cognition, should be studied not only at the behavioural level but also across multiple neurobiological units of analysis, including genes, molecules, cells, circuits, physiology, and behaviour.
MRI-based neural markers represent one such level of analysis. While other biological systems (e.g., genetic, molecular, or physiological) also contribute to the cognition-mental health relationship, neuroimaging provides unique insights into the brain mechanisms underlying this association – insights that cannot be obtained from behavioural data alone.
In response to the related question, “Could the relationship between cognition and mental health be explained by third variables (e.g., environment, opportunities)?”, we note that developing a neural marker of cognition capable of capturing its relationship with mental health is the central aim of this study. Using the MRI modalities available in the UK Biobank, we were able to account for 48% of the covariation between cognition and mental health.
The remaining 52% of unexplained variance may stem from several sources. According to the RDoC framework, neuromarkers could be further refined by incorporating additional neuroimaging modalities (e.g., task-based fMRI, PET, ASL, MEG/EEG, fNIRS) and integrating other units of analysis such as genetic, molecular, cellular, and physiological data.
Once more comprehensive neuromarkers are developed, capturing a greater proportion of the cognition-mental health covariation, they may also lead to new research direction – to investigate how environmental factors and life opportunities influence these markers. However, exploring those environmental contributions lies beyond the scope of the current study.
We discuss these considerations and explain the motivation of our study in the revised Introduction and Discussion.
Line 481: “Our analysis confirmed the validity of the g-factor [31] as a quantitative measure of cognition [31], demonstrating that it captures nearly half (39%) of the variance across twelve cognitive performance scores, consistent with prior studies [63–68]. Furthermore, we were able to predict cognition from 133 mental health indices, showing a medium-sized relationship that aligns with existing literature [69,70]. Although the observed mental health-cognition association is lower than within-sample estimates in conventional regression models, it aligns with our prior mega-analysis in children [69]. Notably, this effect size is not considered small in gerontology. In fact, it falls around the 70th percentile of reported effects and approaches the threshold for a large effect at r = 0.32 [71]. While we focused specifically on cognition as an RDoC core domain, the strength of its relationship with mental health may be bounded by the influence of other functional domains, particularly in normative, non-clinical samples – a promising direction for future research.”
Line 658: “Although recent debates [18] have challenged the predictive utility of MRI for cognition, our multimodal marker integrating 72 neuroimaging phenotypes captures nearly half of the mental health-explained variance in cognition. We demonstrate that neural markers with greater predictive accuracy for cognition also better explain cognition-mental health covariation, showing that multimodal MRI can capture both a substantial cognitive variance and nearly half of its shared variance with mental health. Finally, we show that our neuromarkers explain a substantial portion of the age- and sex-related variance in the cognition-mental health relationship, highlighting their relevance in modeling cognition across demographic strata.
The remaining unexplained variance in the relationship between cognition and mental health likely stems from multiple sources. One possibility is the absence of certain neuroimaging modalities in the UK Biobank dataset, such as task-based fMRI contrasts, positron emission tomography, arterial spin labeling, and magnetoencephalography/electroencephalography. Prior research has consistently demonstrated strong predictive performance from specific task-based fMRI contrasts, particularly those derived from tasks like the n-Back working memory task and the face-name episodic memory task, none of which is available in the UK Biobank [15,17,61,69,114,142,151].
Moreover, there are inherent limitations in using MRI as a proxy for brain structure and function. Measurement error and intra-individual variability, such as differences in a cognitive state between cognitive assessments and MRI acquisition, may also contribute to the unexplained variance. According to the RDoC framework, brain circuits represent only one level of neurobiological analysis relevant to cognition [14]. Other levels, including genes, molecules, cells, and physiological processes, may also play a role in the cognition-mental health relationship.
Nonetheless, neuroimaging provides a valuable window into the biological mechanisms underlying this overlap – insights that cannot be gleaned from behavioural data alone. Ultimately, our findings validate brain-based neural markers as a fundamental neurobiological unit of analysis, advancing our understanding of mental health through the lens of cognition.”
Introduction
Line 43: “Cognition and mental health are closely intertwined [1]. Cognitive dysfunction is present in various mental illnesses, including anxiety [2, 3], depression [4–6], and psychotic disorders [7–12]. National Institute of Mental Health’s Research Domain Criteria (RDoC) [13,14] treats cognition as one of the main basic functional domains that transdiagnostically underly mental health. According to RDoC, mental health should be studied in relation to cognition, alongside other domains such as negative and positive valence systems, arousal and regulatory systems, social processes, and sensorimotor functions. RDoC further emphasizes that each domain, including cognition, should be investigated not only at the behavioural level but also through its neurobiological correlates. In this study, we aim to examine how the covariation between cognition and mental health is reflected in neural markers of cognition, as measured through multimodal neuroimaging.”
Discussion
Line 481: “Our analysis confirmed the validity of the g-factor [31] as a quantitative measure of cognition [31], demonstrating that it captures nearly half (39%) of the variance across twelve cognitive performance scores, consistent with prior studies [63–68]. Furthermore, we were able to predict cognition from 133 mental health indices, showing a medium-sized relationship that aligns with existing literature [69,70]. Although the observed mental health-cognition association is lower than within-sample estimates in conventional regression models, it aligns with our prior mega-analysis in children [69]. Notably, this effect size is not considered small in gerontology. In fact, it falls around the 70th percentile of reported effects and approaches the threshold for a large effect at r = 0.32 [71]. While we focused specifically on cognition as an RDoC core domain, the strength of its relationship with mental health may be bounded by the influence of other functional domains, particularly in normative, non-clinical samples – a promising direction for future research.”
Line 658: “Although recent debates [18] have challenged the predictive utility of MRI for cognition, our multimodal marker integrating 72 neuroimaging phenotypes captures nearly half of the mental health-explained variance in cognition. We demonstrate that neural markers with greater predictive accuracy for cognition also better explain cognition-mental health covariation, showing that multimodal MRI can capture both a substantial cognitive variance and nearly half of its shared variance with mental health. Finally, we show that our neuromarkers explain a substantial portion of the age- and sex-related variance in the cognition-mental health relationship, highlighting their relevance in modeling cognition across demographic strata.
The remaining unexplained variance in the relationship between cognition and mental health likely stems from multiple sources. One possibility is the absence of certain neuroimaging modalities in the UK Biobank dataset, such as task-based fMRI contrasts, positron emission tomography, arterial spin labeling, and magnetoencephalography/electroencephalography. Prior research has consistently demonstrated strong predictive performance from specific task-based fMRI contrasts, particularly those derived from tasks like the n-Back working memory task and the face-name episodic memory task, none of which is available in the UK Biobank [15,17,61,69,114,142,151].
Moreover, there are inherent limitations in using MRI as a proxy for brain structure and function. Measurement error and intra-individual variability, such as differences in a cognitive state between cognitive assessments and MRI acquisition, may also contribute to the unexplained variance. According to the RDoC framework, brain circuits represent only one level of neurobiological analysis relevant to cognition [14]. Other levels, including genes, molecules, cells, and physiological processes, may also play a role in the cognition-mental health relationship.
Nonetheless, neuroimaging provides a valuable window into the biological mechanisms underlying this overlap – insights that cannot be gleaned from behavioural data alone. Ultimately, our findings validate brain-based neural markers as a fundamental neurobiological unit of analysis, advancing our understanding of mental health through the lens of cognition.”
Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research Domain Criteria (RDoC): Toward a New Classification Framework for Research on Mental Disorders. AJP. 2010;167:748–751.
Abramovitch, A., Short, T., & Schweiger, A. (2021). The C Factor: Cognitive dysfunction as a transdiagnostic dimension in psychopathology. Clinical Psychology Review, 86, 102007.
East-Richard, C., R. -Mercier, A., Nadeau, D., & Cellard, C. (2020). Transdiagnostic neurocognitive deficits in psychiatry: A review of meta-analyses. Canadian Psychology / Psychologie Canadienne, 61(3), 190–214.
Wang Y, Anney R, Pat N. The relationship between cognitive abilities and mental health as represented by cognitive abilities at the neural and genetic levels of analysis. eLife. 2025.14:RP105537.
Brydges CR. Effect Size Guidelines, Sample Size Calculations, and Statistical Power in Gerontology. Innovation in Aging. 2019;3(4):igz036.
Yarkoni T, Westfall J. Choosing Prediction Over Explanation in Psychology: Lessons From Machine Learning. Perspect Psychol Sci. 2017;12(6):1100-1122.
Comment 2 Title: - Shouldn't it be "MRI markers" (plural)?
We used the singular form (“marker”) intentionally, as it refers to the composite neuroimaging marker derived from all three MRI modalities in our stacked model. This multimodal marker represents the combined predictive power of all modalities and captures the highest proportion of the mental health-cognition relationship in our analyses.
Comment 3: Introduction - I miss an explanation of why it is useful to look at cognition-mental health covariation
We believe we have sufficiently addressed this comment in our response to Reviewer 2, comment 1 above.
Comment 4: - "Demonstrating that MRI-based neural indicators of cognition capture the covariation between cognition and mental health will thereby support the utility of such indicators for understanding the etiology of mental health" (page 4, line 56-58) - how/why?
Previous research has largely focused on developing MRI-based neural indicators that accurately predict cognitive performance (Marek et al., 2022; Vieira et al., 2020). Building on this foundation, our findings further demonstrate that the predictive performance of a neural indicator for cognition is closely tied to its ability to explain the covariation between cognition and mental health. In other words, the robustness of a neural indicator – its capacity to capture individual differences in cognition – is strongly associated with how well it reflects the shared variance between cognition and mental health.
This insight is particularly important within the context of the RDoC framework, which seeks to understand the etiology of mental health through functional domains (such as cognition) and their underlying neurobiological units of analysis (Insel et al., 2010). According to RDoC, for a neural indicator of cognition to be informative for mental health research, it must not only predict cognitive performance but also capture its relationship with mental health.
Furthermore, RDoC emphasizes the integration of neurobiological measures to investigate the influence of environmental and developmental factors on mental health. In line with this, our neural indicators of cognition may serve as valuable tools in future research aimed at understanding how environmental exposures and developmental trajectories shape mental health outcomes. We discuss this in more detail in the revised Discussion.
Line 481: “Our analysis confirmed the validity of the g-factor [31] as a quantitative measure of cognition [31], demonstrating that it captures nearly half (39%) of the variance across twelve cognitive performance scores, consistent with prior studies [63–68]. Furthermore, we were able to predict cognition from 133 mental health indices, showing a medium-sized relationship that aligns with existing literature [69,70]. Although the observed mental health-cognition association is lower than within-sample estimates in conventional regression models, it aligns with our prior mega-analysis in children [69]. Notably, this effect size is not considered small in gerontology. In fact, it falls around the 70th percentile of reported effects and approaches the threshold for a large effect at r = 0.32 [71]. While we focused specifically on cognition as an RDoC core domain, the strength of its relationship with mental health may be bounded by the influence of other functional domains, particularly in normative, non-clinical samples – a promising direction for future research.”
Line 658: “Although recent debates [18] have challenged the predictive utility of MRI for cognition, our multimodal marker integrating 72 neuroimaging phenotypes captures nearly half of the mental health-explained variance in cognition. We demonstrate that neural markers with greater predictive accuracy for cognition also better explain cognition-mental health covariation, showing that multimodal MRI can capture both a substantial cognitive variance and nearly half of its shared variance with mental health. Finally, we show that our neuromarkers explain a substantial portion of the age- and sex-related variance in the cognition-mental health relationship, highlighting their relevance in modeling cognition across demographic strata.
The remaining unexplained variance in the relationship between cognition and mental health likely stems from multiple sources. One possibility is the absence of certain neuroimaging modalities in the UK Biobank dataset, such as task-based fMRI contrasts, positron emission tomography, arterial spin labeling, and magnetoencephalography/electroencephalography. Prior research has consistently demonstrated strong predictive performance from specific task-based fMRI contrasts, particularly those derived from tasks like the n-Back working memory task and the face-name episodic memory task, none of which is available in the UK Biobank [15,17,61,69,114,142,151].
Moreover, there are inherent limitations in using MRI as a proxy for brain structure and function. Measurement error and intra-individual variability, such as differences in a cognitive state between cognitive assessments and MRI acquisition, may also contribute to the unexplained variance. According to the RDoC framework, brain circuits represent only one level of neurobiological analysis relevant to cognition [14]. Other levels, including genes, molecules, cells, and physiological processes, may also play a role in the cognition-mental health relationship.
Nonetheless, neuroimaging provides a valuable window into the biological mechanisms underlying this overlap – insights that cannot be gleaned from behavioural data alone. Ultimately, our findings validate brain-based neural markers as a fundamental neurobiological unit of analysis, advancing our understanding of mental health through the lens of cognition.”
Marek S, Tervo-Clemmens B, Calabro FJ, Montez DF, Kay BP, Hatoum AS, et al. Reproducible brain-wide association studies require thousands of individuals. Nature. 2022;603:654–660.
Vieira S, Gong QY, Pinaya WHL, et al. Using Machine Learning and Structural Neuroimaging to Detect First Episode Psychosis: Reconsidering the Evidence. Schizophr Bull. 2020;46(1):17-26.
Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research Domain Criteria (RDoC): Toward a New Classification Framework for Research on Mental Disorders. AJP. 2010;167:748–751.
Comment 5: - The explanation about the stacking approach is not yet completely clear to me. I don't understand how the target variable can be the dependent variable in both step one and two. Or are those different variables? It would be helpful to also give an example of the target variable in line 88 on page 5
Thank you for this excellent question. In our stacking approach, the same target variable, the g-factor, is indeed used across both modeling stages, but with a key distinction in how predictions are generated and integrated.
In the first-level models, we trained separate Partial Least Squares Regression (PLSR) models for each of the 72 neuroimaging phenotypes, each predicting the g-factor independently. The predicted values from these 72 models were then used as input features for the second-level stacked model, which combined them to generate a final prediction of the g-factor. This twostage framework enables us to integrate information across multiple imaging modalities while maintaining a consistent prediction target.
To avoid data leakage, both modeling stages were conducted entirely within the training set for each cross-validation fold. Only after the second-level model was trained was it applied to the outer-fold test participants who were not involved in any part of the model training process.
To improve accessibility, we have revised the Methods section (see Page 10) to clarify this approach, ensuring that the description remains technically accurate while being easier to follow.
Line 188: “We employed nested cross-validation to predict cognition from mental health indices and 72 neuroimaging phenotypes (Fig. 1). Nested cross-validation is a robust method for evaluating machine-learning models while tuning their hyperparameters, ensuring that performance estimates are both accurate and unbiased. Here, we used a nested cross-validation scheme with five outer folds and ten inner folds.
We started by dividing the entire dataset into five outer folds. Each fold took a turn being held out as the outerfold test set (20% of the data), while the remaining four folds (80% of the data) were used as an outer-fold training set. Within each outer-fold training set, we performed a second layer of cross-validation – this time splitting the data into ten inner folds. These inner folds were used exclusively for hyperparameter tuning: models were trained on nine of the inner folds and validated on the remaining one, cycling through all ten combinations.
We then selected the hyperparameter configuration that performed best across the inner-fold validation sets, as determined by the minimal mean squared error (MSE). The model was then retrained on the full outer-fold training set using this hyperparameter configuration and evaluated on the outer-fold test set, using four performance metrics: Pearson r, the coefficient of determination ( R<sup>2</sup>), the mean absolute error (MAE), and the MSE. This entire process was repeated for each of the five outer folds, ensuring that every data point is used for both training and testing, but never at the same time. We opted for five outer folds instead of ten to reduce computational demands, particularly memory and processing time, given the substantial volume of neuroimaging data involved in model training. Five outer folds led to an outer-fold test set at least n = 4 000, which should be sufficient for model evaluation. In contrast, we retained ten inner folds to ensure robust and stable hyperparameter tuning, maximising the reliability of model selection.
To model the relationship between mental health and cognition, we employed Partial Least Squares Regression (PLSR) to predict the g-factor from 133 mental health variables. To model the relationship between neuroimaging data and cognition, we used a two-step stacking approach [15–17,61] to integrate information from 72 neuroimaging phenotypes across three MRI modalities. In the first step, we trained 72 base (first-level) PLSR models, each predicting the g-factor from a single neuroimaging phenotype. In the second step, we used the predicted values from these base models as input features for stacked models, which again predicted the g-factor. We constructed four stacked models based on the source of the base predictions: one each for dwMRI, rsMRI, sMRI, and a combined model incorporating all modalities (“dwMRI Stacked”, “rsMRI Stacked”, “sMRI Stacked”, and “All MRI Stacked”, respectively). Each stacked model was trained using one of four machine learning algorithms – ElasticNet, Random Forest, XGBoost, or Support Vector Regression – selected individually for each model (see Supplementary Materials, S6).
For rsMRI phenotypes, we treated the choice of functional connectivity quantification method – full correlation, partial correlation, or tangent space parametrization – as a hyperparameter. The method yielding the highest performance on the outer-fold training set was selected for predicting the g-factor (see Supplementary Materials, S5).
To prevent data leakage, we standardized the data using the mean and standard deviation derived from the training set and applied these parameters to the corresponding test set within each outer fold. This standardization was performed at three key stages: before g-factor derivation, before regressing out modality-specific confounds from the MRI data, and before stacking. Similarly, to maintain strict separation between training and testing data, both base and stacked models were trained exclusively on participants from the outer-fold training set and subsequently applied to the corresponding outer-fold test set.
To evaluate model performance and assess statistical significance, we aggregated the predicted and observed gfactor values from each outer-fold test set. We then computed a bootstrap distribution of Pearson’s correlation coefficient (r) by resampling with replacement 5 000 times, generating 95% confidence intervals (CIs) (Fig. 1). Model performance was considered statistically significant if the 95% CI did not include zero, indicating that the observed associations were unlikely to have occurred by chance.”
Comment 6: Methods - It's not clear from the text and Figure 1 which 12 scores from 11 tests are being used to derive the g-factor. Figure 1 shows only 8 bullet points with 10 scores in A and 13 tests under 'Cognitive tests' in B. Moreover, Supplement S1 describes 12 tests and 14 measures (Prospective Memory test is in the text but not in Supplementary Table 1).
Thank you for identifying this discrepancy. In the original Figure 1b and in the Supplementary Methods (S1), the “Prospective Memory” test was accidentally duplicated, while it was present in the Supplementary Table 1 (Line 53, Supplementary Table 1). We have now corrected both figures for consistency. To clarify: Figure 1a presents the global mental health and cognitive domains studied, while Figure 1b now accurately lists 1) the 12 cognitive scores from 11 tests used to derive the g-factor (with the Trail Making Test contributing two measures – numeric and alphabetic trails) and 2) the three main categories of mental health indices used as machine learning features.
We also corrected the Supplementary Materials to remove the duplicate test from the first paragraph. In Supplementary Table 1, there were 11 tests listed, and for the Trail Making test, we specified in the “Core measures” column that this test had 2 derivative scores: duration to complete the numeric path (Trail 1) and duration to complete the alphabetic path (Trail 2).
Supplementary Materials, Line 46: “We used twelve scores from the eleven cognitive tests that represented the following cognitive domains: reaction time and processing speed (Reaction Time test), working memory (Numeric Memory test), verbal and numerical reasoning (Fluid Intelligence test), executive function (Trail Making Test), non-verbal fluid reasoning (Matrix Pattern Completion test), processing speed (Symbol Digit Substitution test), vocabulary (Picture Vocabulary test), planning abilities (Tower Rearranging test), verbal declarative memory (Paired Associate Learning test), prospective memory (Prospective Memory test), and visual memory (Pairs Matching test) [1].”
Comment 7: - For the mental health measures: If I understand correctly, the questionnaire items were used individually, but also to create composite scores. This seems counterintuitive, because I would assume that if the raw data is used, the composite scores would not add additional information to that. When reading the Supplement, it seems like I'm not correct… It would be helpful to clarify the text on page 7 in the main text.
You raise an excellent observation regarding the use of both individual questionnaire items and composite scores. This dual approach was methodologically justified by the properties of Partial Least Squares Regression (PLSR), our chosen first-level machine learning algorithm, which benefits from rich feature sets and can handle multicollinearity through dimensionality reduction. PLSR transforms correlated features into latent variables, meaning both individual items and composite scores can contribute unique information to the model. We elaborate on PLSR's mathematical principles in Supplementary Materials (S5).
To directly address this concern, we conducted comparative analyses showing that the PLSR model (a single 80/20% training/test split), incorporating all 133 mental health features (both items and composites), outperformed models using either type alone. The full model achieved superior performance (MSE = 0.458, MAE = 0.537, R² \= 0.112, Pearson r = 0.336, p-value = 6.936e-112) compared to using only composite scores (93 features; MSE = 0.461, MAE = 0.538, R<sup>2</sup> = 0.107, Pearson r = 0.328, p-value = 5.8e-106) or only questionnaire items (40 features; MSE = 0.499, MAE = 0.561, R<sup>2</sup> = 0.033, Pearson r = 0.184, p-value = 2.53e-33). These results confirm that including both data types provide complementary predictive value. We expand on these considerations in the revised Methods section.
Line 123: “Mental health measures encompassed 133 variables from twelve groups: mental distress, depression, clinical diagnoses related to the nervous system and mental health, mania (including bipolar disorder), neuroticism, anxiety, addictions, alcohol and cannabis use, unusual/psychotic experiences, traumatic events, selfharm behaviours, and happiness and subjective well-being (Fig. 1 and Tables S4 and S5). We included both selfreport questionnaire items from all participants and composite diagnostic scores computed following Davis et al. and Dutt et al. [35,36] as features in our first-level (for explanation, see Data analysis section) Partial Least Squares Regression (PLSR) model. This approach leverages PLSR’s ability to handle multicollinearity through dimensionality reduction, enabling simultaneous use of granular symptom-level information and robust composite measures (for mental health scoring details, see Supplementary Materials, S2). We assess the contribution of each mental health index to general cognition by examining the direction and magnitude of its PLSR-derived loadings on the identified latent variables”
Comment 8: - Results - The colors in Figure 4 B are a bit hard to differentiate.
We have updated Figure 4 to enhance colour differentiation by adjusting saturation and brightness levels, improving visual distinction. For further clarity, we split the original figure into two separate figures.
Comment 9: - Discussion - "Overall, the scores for mental distress, alcohol and cannabis use, and self-harm behaviours relate positively, and the scores for anxiety, neurological and mental health diagnoses, unusual or psychotic experiences, happiness and subjective well-being, and negative traumatic events relate negatively to cognition," - this seems counterintuitive, that some symptoms relate to better cognition and others relate to worse cognition. Could you elaborate on this finding and what it could mean?
We appreciate you highlighting this important observation. While some associations between mental health indices and cognition may appear counterintuitive at first glance, these patterns are robust (emerging consistently across both univariate correlations and PLSR loadings) and align with previous literature (e.g., Karpinski et al., 2018; Ogueji et al., 2022). For instance, the positive relationship between cognitive ability and certain mental health indicators like help-seeking behaviour has been documented in other population studies (Karpinski et al., 2018; Ogueji et al., 2022), potentially reflecting greater health literacy and access to care among cognitively advantaged individuals. Conversely, the negative associations with conditions like psychotic experiences mirror established neurocognitive deficits in these domains.
As was initially detailed in Supplementary Materials (S12) and now expanded in our Discussion, these findings likely reflect complex multidimensional interactions. The positive loadings for mental distress indicators may capture: (1) greater help-seeking behaviour among those with higher cognition and socioeconomic resources, and/or (2) psychological overexcitability and rumination tendencies in high-functioning individuals. These interpretations are particularly relevant to the UK Biobank's assessment methods, where mental distress items focused on medical help-seeking rather than symptom severity per se (e.g., as a measure of mental distress, the UK Biobank questionnaire asked whether an individual sought or received medical help for or suffered from mental distress).
Line 492: “Factor loadings derived from the PLSR model showed that the scores for mental distress, alcohol and cannabis use, and self-harm behaviours related positively, and the scores for anxiety, neurological and mental health diagnoses, unusual or psychotic experiences, happiness and subjective well-being, and negative traumatic events related negatively to the g-factor. Positive PLSR loadings of features related to mental distress may indicate greater susceptibility to or exaggerated perception of stressful events, psychological overexcitability, and predisposition to rumination in people with higher cognition [72]. On the other hand, these findings may be specific to the UK Biobank cohort and the way the questions for this mental health category were constructed. In particular, to evaluate mental distress, the UK Biobank questionnaire asked whether an individual sought or received medical help for or suffered from mental distress. In this regard, the estimate for mental distress may be more indicative of whether an individual experiencing mental distress had an opportunity or aspiration to visit a doctor and seek professional help [73]. Thus, people with better cognitive abilities and also with a higher socioeconomic status may indeed be more likely to seek professional help.
Limited evidence supports a positive association between self-harm behaviours and cognitive abilities, with some studies indicating higher cognitive performance as a risk factor for non-suicidal self-harm. Research shows an inverse relationship between cognitive control of emotion and suicidal behaviours that weakens over the life course [73,74]. Some studies have found a positive correlation between cognitive abilities and the risk of nonsuicidal self-harm, suicidal thoughts, and suicidal plans that may be independent of or, conversely, affected by socioeconomic status [75,76]. In our study, the magnitude of the association between self-harm behaviours and cognition was low (Fig. 2), indicating a weak relationship.
Positive PLSR loadings of features related to alcohol and cannabis may also indicate the influence of other factors. Overall, this relationship is believed to be largely affected by age, income, education, social status, social equality, social norms, and quality of life [79–80]. For example, education level and income correlate with cognitive ability and alcohol consumption [79,81–83]. Research also links a higher probability of having tried alcohol or recreational drugs, including cannabis, to a tendency of more intelligent individuals to approach evolutionary novel stimuli [84,85]. This hypothesis is supported by studies showing that cannabis users perform better on some cognitive tasks [86]. Alternatively, frequent drinking can indicate higher social engagement, which is positively associated with cognition [87]. Young adults often drink alcohol as a social ritual in university settings to build connections with peers [88]. In older adults, drinking may accompany friends or family visits [89,90]. Mixed evidence on the link between alcohol and drug use and cognition makes it difficult to draw definite conclusions, leaving an open question about the nature of this relationship.
Consistent with previous studies, we showed that anxiety and negative traumatic experiences were inversely associated with cognitive abilities [90–93]. Anxiety may be linked to poorer cognitive performance via reduced working memory capacity, increased focus on negative thoughts, and attentional bias to threatening stimuli that hinder the allocation of cognitive resources to a current task [94–96]. Individuals with PTSD consistently showed impaired verbal and working memory, visual attention, inhibitory function, task switching, cognitive flexibility, and cognitive control [97–100]. Exposure to traumatic events that did not reach the PTSD threshold was also linked to impaired cognition. For example, childhood trauma is associated with worse performance in processing speed, attention, and executive function tasks in adulthood, and age at a first traumatic event is predictive of the rate of executive function decline in midlife [101,102]. In the UK Biobank cohort, adverse life events have been linked to lower cognitive flexibility, partially via depression level [103].
In agreement with our findings, cognitive deficits are often found in psychotic disorders [104,105]. We treated neurological and mental health symptoms as predictor variables and did not stratify or exclude people based on psychiatric status or symptom severity. Since no prior studies have examined isolated psychotic symptoms (e.g., recent unusual experiences, hearing unreal voices, or seeing unreal visions), we avoid speculating on how these symptoms relate to cognition in our sample.
Finally, negative PLSR loadings of the features related to happiness and subjective well-being may be specific to the study cohort, as these findings do not agree with some previous research [107–109]. On the other hand, our results agree with the study linking excessive optimism or optimistic thinking to lower cognitive performance in memory, verbal fluency, fluid intelligence, and numerical reasoning tasks, and suggesting that pessimism or realism indicates better cognition [110]. The concept of realism/optimism as indicators of cognition is a plausible explanation for a negative association between the g-factor and friendship satisfaction, as well as a negative PLSR loading of feelings that life is meaningful, especially in older adults who tend to reflect more on the meaning of life [111]. The latter is supported by the study showing a negative association between cognitive function and the search for the meaning of life and a change in the pattern of this relationship after the age of 60 [112]. Finally, a UK Biobank study found a positive association of happiness with speed and visuospatial memory but a negative relationship with reasoning ability [113].”
Karpinski RI, Kinase Kolb AM, Tetreault NA, Borowski TB. High intelligence: A risk factor for psychological and physiological overexcitabilities. Intelligence. 2018;66:8–23.
Ogueji IA, Okoloba MM. Seeking Professional Help for Mental Illness: A Mixed-Methods Study of Black Family Members in the UK and Nigeria. Psychol Stud. 2022;67:164–177.
Comment 10: - All neuroimaging factors together explain 48% of the variance in the cognition-mental health relationship. However, this relationship is only r=0.3 - so then the effect of neuroimaging factors seems a lot smaller… What does it mean?
Thank you for raising this critical point. We have addressed this point in our response to Reviewer 1, comment 2, Reviewer 1, comment 3 and Reviewer 2, comment 1.
Briefly, cognition is related to mental health at around r = 0.3 and to neuroimaging phenotypes at around r = 0.4. These levels of relationship strength are consistent to what has been shown in the literature (e.g., Wang et al., 2025 and Vieira et al., 2020). We discussed the relationship between cognition and mental health in our response to Reviewer 2, comment 1 above. In short, this relationship reflects just one functional domain – mental health may also be associated with other domains such as negative and positive valence systems, arousal and regulatory systems, social processes, and sensorimotor functions. Moreover, in the context of gerontology research, this effect size is considered relatively large (Brydges et al., 2019).
We conducted a commonality analysis to investigate the unique and shared variance of mental health and neuroimaging phenotypes in explaining cognition. As we discussed in our response to Reviewer 1, comment 2, we were able to account for 48% of the covariation between cognition and mental health using the MRI modalities available in the UK Biobank. The remaining 52% of unexplained variance may arise from several sources.
One possibility is the absence of certain neuroimaging modalities in the UK Biobank dataset, such as task-based fMRI contrasts, positron emission tomography, arterial spin labeling, and magnetoencephalography/electroencephalography. Prior research from our group and others has consistently demonstrated strong predictive performance from specific task-based fMRI contrasts, particularly those derived from tasks like the n-Back working memory task and the face-name episodic memory task, none of which is available in the UK Biobank (Tetereva et al., 2025).
Moreover, there are inherent limitations in using MRI as a proxy for brain structure and function. Measurement error and intra-individual variability, such as differences in a cognitive state between cognitive assessments and MRI acquisition, may also contribute to the unexplained variance. According to RDoC framework, brain circuits represent only one level of neurobiological analysis relevant to cognition. Other levels, including genes, molecules, cells, and physiological processes, may also play a role in the cognition-mental health relationship.
We have now incorporated these considerations into the Discussion section.
Line 481: “Our analysis confirmed the validity of the g-factor [31] as a quantitative measure of cognition [31], demonstrating that it captures nearly half (39%) of the variance across twelve cognitive performance scores, consistent with prior studies [63–68]. Furthermore, we were able to predict cognition from 133 mental health indices, showing a medium-sized relationship that aligns with existing literature [69,70]. Although the observed mental health-cognition association is lower than within-sample estimates in conventional regression models, it aligns with our prior mega-analysis in children [69]. Notably, this effect size is not considered small in gerontology. In fact, it falls around the 70th percentile of reported effects and approaches the threshold for a large effect at r = 0.32 [71]. While we focused specifically on cognition as an RDoC core domain, the strength of its relationship with mental health may be bounded by the influence of other functional domains, particularly in normative, non-clinical samples – a promising direction for future research.”
Line 658: “Although recent debates [18] have challenged the predictive utility of MRI for cognition, our multimodal marker integrating 72 neuroimaging phenotypes captures nearly half of the mental health-explained variance in cognition. We demonstrate that neural markers with greater predictive accuracy for cognition also better explain cognition-mental health covariation, showing that multimodal MRI can capture both a substantial cognitive variance and nearly half of its shared variance with mental health. Finally, we show that our neuromarkers explain a substantial portion of the age- and sex-related variance in the cognition-mental health relationship, highlighting their relevance in modeling cognition across demographic strata.
The remaining unexplained variance in the relationship between cognition and mental health likely stems from multiple sources. One possibility is the absence of certain neuroimaging modalities in the UK Biobank dataset, such as task-based fMRI contrasts, positron emission tomography, arterial spin labeling, and magnetoencephalography/electroencephalography. Prior research has consistently demonstrated strong predictive performance from specific task-based fMRI contrasts, particularly those derived from tasks like the n-Back working memory task and the face-name episodic memory task, none of which is available in the UK Biobank [15,17,61,69,114,142,151].
Moreover, there are inherent limitations in using MRI as a proxy for brain structure and function. Measurement error and intra-individual variability, such as differences in a cognitive state between cognitive assessments and MRI acquisition, may also contribute to the unexplained variance. According to the RDoC framework, brain circuits represent only one level of neurobiological analysis relevant to cognition [14]. Other levels, including genes, molecules, cells, and physiological processes, may also play a role in the cognition-mental health relationship.
Nonetheless, neuroimaging provides a valuable window into the biological mechanisms underlying this overlap – insights that cannot be gleaned from behavioural data alone. Ultimately, our findings validate brain-based neural markers as a fundamental neurobiological unit of analysis, advancing our understanding of mental health through the lens of cognition.”
Wang Y, Anney R, Pat N. The relationship between cognitive abilities and mental health as represented by cognitive abilities at the neural and genetic levels of analysis. eLife. 2025.14:RP105537.
Vieira S, Gong QY, Pinaya WHL, et al. Using Machine Learning and Structural Neuroimaging to Detect First Episode Psychosis: Reconsidering the Evidence. Schizophr Bull. 2020;46(1):17-26.
Brydges CR. Effect Size Guidelines, Sample Size Calculations, and Statistical Power in Gerontology. Innovation in Aging. 2019;3(4):igz036.
Tetereva A, Knodt AR, Melzer TR, et al. Improving Predictability, Reliability and Generalisability of Brain-Wide Associations for Cognitive Abilities via Multimodal Stacking. Preprint. bioRxiv. 2025;2024.05.03.589404.
Reviewer 3:
Buianova et al. present a comprehensive analysis examining the predictive value of multimodal neuroimaging data for general cognitive ability, operationalized as a derived g-factor. The study demonstrates that functional MRI holds the strongest predictive power among the modalities, while integrating multiple MRI modalities through stacking further enhances prediction performance. The inclusion of a commonality analysis provides valuable insight into the extent to which shared and unique variance across mental health features and neuroimaging modalities contributes to the observed associations with cognition. The results are clearly presented and supported by highquality visualizations. Limitations of the sample are stated clearly.
Thank you once more for your constructive and encouraging feedback. We appreciate your careful reading and valuable methodological insights. Your expertise has helped us clarify key methodological concepts and improve the overall rigour of our study.
Suggestions for improvement:
(1) The manuscript would benefit from the inclusion of permutation testing to evaluate the statistical significance of the predictive models. This is particularly important given that some of the reported performance metrics are relatively modest, and permutation testing could help ensure that results are not driven by chance.
Thank you, this is an excellent point. We agree that evaluating the statistical significance of our predictive models is essential.
In our original analysis, we assessed model performance by generating a bootstrap distribution of Pearson’s r, resampling the data with replacement 5,000 times (see Figure 3b). In response to your feedback, we have made the following updates:
(1) Improved Figure 3b to explicitly display the 95% confidence intervals.
(2) Supplemented the results by reporting the exact confidence interval values.
(3) Clarified our significance testing procedure in the Methods section.
We considered model performance statistically significant when the 95% confidence interval did not include zero, indicating that the observed associations are unlikely to have occurred by chance.
We chose bootstrapping over permutation testing because, while both can assess statistical significance, bootstrapping additionally provides uncertainty estimates in the form of confidence intervals. Given the large sample size in our study, significance testing can be less informative, as even small effects may reach statistical significance. Bootstrapping offers a more nuanced understanding of model uncertainty.
Line 233: “To evaluate model performance and assess statistical significance, we aggregated the predicted and observed g-factor values from each outer-fold test set. We then computed a bootstrap distribution of Pearson’s correlation coefficient (r) by resampling with replacement 5 000 times, generating 95% confidence intervals (CIs) (Fig. 1). Model performance was considered statistically significant if the 95% CI did not include zero, indicating that the observed associations were unlikely to have occurred by chance.”
(2) Applying and testing the trained models on an external validation set would increase confidence in generalisability of the model.
We appreciate this excellent suggestion. While we considered this approach, implementing it would require identifying an appropriate external dataset with comparable neuroimaging and behavioural measures, along with careful matching of acquisition protocols and variable definitions across sites. These challenges extend beyond the scope of the current study, though we fully agree that this represents an important direction for future research.
Our findings, obtained from one of the largest neuroimaging datasets to date with training and test samples exceeding most previous studies, align closely with existing literature: the predictive accuracy of each neuroimaging phenotype and modality for cognition matches the effect size reported in meta-analyses (r ≈ 0.4; e.g., Vieira et al., 2020). The ability of dwMRI, rsMRI and sMRI to capture the cognition-mental health relationship is, in turn, consistent with our previous work in pediatric populations (Wang et al., 2025; Pat et al., 2022).
Vieira S, Gong QY, Pinaya WHL, et al. Using Machine Learning and Structural Neuroimaging to Detect First Episode Psychosis: Reconsidering the Evidence. Schizophr Bull. 2020;46(1):17-26.
Wang Y, Anney R, Pat N. The relationship between cognitive abilities and mental health as represented by cognitive abilities at the neural and genetic levels of analysis. eLife. 2025.14:RP105537.
Pat N, Wang Y, Anney R, Riglin L, Thapar A, Stringaris A. Longitudinally stable, brain-based predictive models mediate the relationships between childhood cognition and socio-demographic, psychological and genetic factors. Hum Brain Mapp. 2022;43:5520–5542.
(3) The rationale for selecting a 5-by-10-fold cross-validation scheme is not clearly explained. Clarifying why this structure was preferred over more commonly used alternatives, such as 10-by-10 or 5-by-5 cross-validation, would strengthen the methodological transparency.
Thank you for this important methodological question. Our choice of a 5-by-10-fold crossvalidation scheme was motivated by the need to balance robust hyperparameter tuning with computational efficiency, particularly memory and processing time. Retaining five outer folds allowed us to rigorously assess model performance across multiple data partitions, leading to an outer-fold test set at least n = 4 000 and providing a substantial amount of neuroimaging data involved in model training. In contrast, employing ten inner folds ensured robust and stable hyperparameter tuning that maximizes the reliability of model selection. Thus, the 5-outer-fold with our large sample provided sufficient out-of-sample test set size for reliable model evaluation and efficient computation, while 10 inner folds enabled robust hyperparameter tuning. We now provide additional rationale for this design decision on Page 10.
Line 188: “We employed nested cross-validation to predict cognition from mental health indices and 72 neuroimaging phenotypes (Fig. 1). Nested cross-validation is a robust method for evaluating machine-learning models while tuning their hyperparameters, ensuring that performance estimates are both accurate and unbiased. Here, we used a nested cross-validation scheme with five outer folds and ten inner folds.
We started by dividing the entire dataset into five outer folds. Each fold took a turn being held out as the outerfold test set (20% of the data), while the remaining four folds (80% of the data) were used as an outer-fold training set. Within each outer-fold training set, we performed a second layer of cross-validation – this time splitting the data into ten inner folds. These inner folds were used exclusively for hyperparameter tuning: models were trained on nine of the inner folds and validated on the remaining one, cycling through all ten combinations.
We then selected the hyperparameter configuration that performed best across the inner-fold validation sets, as determined by the minimal mean squared error (MSE). The model was then retrained on the full outer-fold training set using this hyperparameter configuration and evaluated on the outer-fold test set, using four performance metrics: Pearson r, the coefficient of determination ( R<sup>2</sup>), the mean absolute error (MAE), and the MSE. This entire process was repeated for each of the five outer folds, ensuring that every data point is used for both training and testing, but never at the same time. We opted for five outer folds instead of ten to reduce computational demands, particularly memory and processing time, given the substantial volume of neuroimaging data involved in model training. Five outer folds led to an outer-fold test set at least n = 4 000, which should be sufficient for model evaluation. In contrast, we retained ten inner folds to ensure robust and stable hyperparameter tuning, maximising the reliability of model selection.”
(4) A more detailed discussion of which specific brain regions or features within each neuroimaging modality contributed most strongly to the prediction of cognition would enhance neurobiological relevance of the findings.
Thank you for this thoughtful suggestion. To address this point, we have included feature importance plots for the top-performing neuroimaging phenotypes within each modality (Figure 5 and Figures S2–S4), demonstrating the relative contributions of individual features to the predictive models. While we maintain our primary focus on cross-modality performance comparisons in the main text, as this aligns with our central aim of evaluating multimodal MRI markers at the integrated level, we outline the contribution of neuroimaging features with the highest predictive performance for cognition in the revised Results and Discussion.
Methods
Line 255: “To determine which neuroimaging features contribute most to the predictive performance of topperforming phenotypes within each modality, while accounting for the potential latent components derived from neuroimaging, we assessed feature importance using the Haufe transformation [62]. Specifically, we calculated Pearson correlations between the predicted g-factor and scaled and centred neuroimaging features across five outer-fold test sets. We also examined whether the performance of neuroimaging phenotypes in predicting cognition per se is related to their ability to explain the link between cognition and mental health. Here, we computed the correlation between the predictive performance of each neuroimaging phenotype and the proportion of the cognition-mental health relationship it captures. To understand how demographic factors, including age and sex, contribute to this relationship, we also conducted a separate set of commonality analyses treating age, sex, age<sup>2</sup>, age×sex, and age<sup>2</sup>×sex as an additional set of explanatory variables (Fig. 1).”
Results
dwMRI
Line 331: “Overall, models based on structural connectivity metrics performed better than TBSS and probabilistic tractography (Fig. 3). TBSS, in turn, performed better than probabilistic tractography (Fig. 3 and Table S13). The number of streamlines connecting brain areas parcellated with aparc MSA-I had the best predictive performance among all dwMRI neuroimaging phenotypes (R<sup>2</sup><sub>mean</sub> = 0.052, r<sub>mean</sub> = 0.227, 95% CI [0.212, 0.235]). To identify features driving predictions, we correlated streamline counts in aparc MSA-I parcellation with the predicted g_factor values from the PLSR model. Positive associations with the predicted _g-factor were strongest for left superior parietal-left caudal anterior cingulate, left caudate-right amygdala, and left putamen-left hippocampus connections. The most marked negative correlations involved left putamen-right posterior thalamus and right pars opercularis-right caudal anterior cingulate pathways (Fig. 5 and Supplementary Fig. S2).”
rsMRI
Line 353: “Among RSFC metrics for 55 and 21 ICs, tangent parameterization matrices yielded the highest performance in the training set compared to full and partial correlation, as indicated by the cross-validation score. Functional connections between the limbic (IC10) and dorsal attention (IC18) networks, as well as between the ventral attention (IC15) and default mode (IC11) networks, displayed the highest positive association with cognition. In contrast, functional connectivity between the limbic (IC43, the highest activation within network) and default mode (IC11) and limbic (IC45) and frontoparietal (IC40) networks, between the dorsal attention (IC18) and frontoparietal (IC25) networks, and between the ventral attention (IC15) and frontoparietal (IC40) networks, showed the highest negative association with cognition (Fig. 5 and Supplementary Fig. S3 and S4)”
sMRI
Line 373: “FreeSurfer subcortical volumetric subsegmentation and ASEG had the highest performance among all sMRI neuroimaging phenotypes (R<sup>2</sup><sub>mean</sub> = 0.068, r<sub>mean</sub> = 0.244, 95% CI [0.237, 0.259] and R<sup>2</sup><sub>mean</sub> = 0.059, r<sub>mean</sub> = 0.235, 95% CI [0.221, 0.243], respectively). In FreeSurfer subcortical volumetric subsegmentation, volumes of all subcortical structures, except for left and right hippocampal fissures, showed positive associations with cognition. The strongest relations were observed for the volumes of bilateral whole hippocampal head and whole hippocampus (Fig. 5 and Supplementary Fig. S5 for feature importance maps). Grey matter morphological characteristics from ex vivo Brodmann Area Maps showed the lowest predictive performance (R<sup>2</sup><sub>mean</sub> = 0.008, r<sub>mean</sub> = 0.089, 95% CI [0.075, 0.098]; Fig. 3 and Table S15).”
Discussion
dwMRI
Line 562: “Among dwMRI-derived neuroimaging phenotypes, models based on structural connectivity between brain areas parcellated with aparc MSA-I (streamline count), particularly connections with bilateral caudal anterior cingulate (left superior parietal-left caudal anterior cingulate, right pars opercularis-right caudal anterior cingulate), left putamen (left putamen-left hippocampus, left putamen-right posterior thalamus), and amygdala (left caudate-right amygdala), result in a neural indicator that best reflects microstructural resources associated with cognition, as indicated by predictive modeling, and more importantly, shares the highest proportion of the variance with mental health-g, as indicated by commonality analysis.”
rsMRI
Line 583: “We extend findings on the superior performance of rsMRI in predicting cognition, which aligns with the literature [15, 28], by showing that it also explains almost a third of the variance in cognition that mental health captures. At the rsMRI neuroimaging phenotype level, this performance is mostly driven by RSFC patterns among 55 ICA-derived networks quantified using tangent space parameterization. At a feature level, these associations are best captured by the strength of functional connections among limbic, dorsal attention and ventral attention, frontoparietal and default mode networks. These functional networks have been consistently linked to cognitive processes in prior research [127–130].”
sMRI
Line 608: “Integrating information about brain anatomy by stacking sMRI neuroimaging phenotypes allowed us to explain a third of the link between cognition and mental health. Among all sMRI neuroimaging phenotypes, those that quantified the morphology of subcortical structures, particularly volumes of bilateral hippocampus and hippocampal head, explain the highest portion of the variance in cognition captured by mental health. Our findings show that, at least in older adults, volumetric properties of subcortical structures are not only more predictive of individual variations in cognition but also explain a greater portion of cognitive variance shared with mental health than structural characteristics of more distributed cortical grey and white matter. This aligns with the Scaffolding Theory that proposes stronger compensatory engagement of subcortical structures in cognitive processing in older adults [138–140].”
(5) The formatting of some figure legends could be improved for clarity - for example, some subheadings were not formatted in bold (e.g., Figure 2 c)
Thank you for noticing this. We have updated the figures to enhance clarity, keeping subheadings plain while bolding figure numbers and MRI modality names.