Node ≥ 22 on macOS / Linux / Windows
这个技术规格要求Node.js版本22或更高,这是一个具体的系统要求。这个版本要求相对较新,可能限制了在较旧系统上的使用。与其他AI工具相比,这个要求不算特别严格,但可能会影响一些用户的兼容性,特别是在企业环境中。
Node ≥ 22 on macOS / Linux / Windows
这个技术规格要求Node.js版本22或更高,这是一个具体的系统要求。这个版本要求相对较新,可能限制了在较旧系统上的使用。与其他AI工具相比,这个要求不算特别严格,但可能会影响一些用户的兼容性,特别是在企业环境中。
An update of /usr/include/sysexits.h allocates previously unused exit codes from 64 - 78. It may be anticipated that the range of unallotted exit codes will be further restricted in the future. The author of this document will not do fixups on the scripting examples to conform to the changing standard. This should not cause any problems, since there is no overlap or conflict in usage of exit codes between compiled C/C++ binaries and shell scripts.
Eh, 0 and 64 - 78 are the only codes it defines. So if it had different codes defined before, what on earth were those codes before? Was only 0 "used"/defined here before? Nothing defined from 1-128? Or were the codes defined there different ones, like 20-42 and then they arbitrarily shifted these up to 64-78 one day? This is very unclear to me.
Also unclear whether this is saying it won't update for any future changes after this, or if he hasn't even updated to align with this supposed "change". (Unclear because I can't figure out whether his "proposes restricting user-defined exit codes to the range 64 - 113 (in addition to 0, for success), to conform with the C/C++ standard" statement is actually conforming or rejecting the sysexits.h standard.)
It seems that he's overreacting a bit here. It's hard to imagine there has been or will be any major changes to the sysexits.h. I would only imagine there being additions to, but not changes to because backwards compatibility would be of utmost concern.
#!/usr/bin/env npx ts-node // TypeScript code Whether this always works in macOS is unknown. There could be some magic with node installing a shell command shim (thanks to @DaMaxContext for commenting about this). This doesn't work in Linux because Linux distros treat all the characters after env as the command, instead of considering spaces as delimiting separate arguments. Or it doesn't work in Linux if the node command shim isn't present (not confirmed that's how it works, but in any case, in my testing, it doesn't work in Linux Docker containers). This means that npx ts-node will be treated as a single executable name that has a space in it, which obviously won't work, as that's not an executable.
There is recent update that enables such functionality - https://github.com/moby/buildkit/releases/tag/dockerfile/1.7.0-labs To work with it - add comment in the beginning of the Dockerfile # syntax=docker.io/docker/dockerfile:1.7-labs
Available in docker/dockerfile-upstream:master-labs. Will be included in docker/dockerfile:1.7-labs.
Use the syntax parser directive to declare the Dockerfile syntax version to use for the build. If unspecified, BuildKit uses a bundled version of the Dockerfile frontend. Declaring a syntax version lets you automatically use the latest Dockerfile version without having to upgrade BuildKit or Docker Engine, or even use a custom Dockerfile implementation.
No Alternate Text on Images/Main Image Slide Header and Decorative Images: Some images that provide key information don’t have alt text, as well as some of the decorative photos, making them inaccessible to users relying on screen readers. Without descriptive alt text, users with visual impairments may miss out on important content or context, reducing the site's overall accessibility. I think there can also be numbers added on the white dotted navigator for the main slide headings with images for easy navigation (some of the white on white makes it hard to see and scroll).
Clear Headings & Structure: Eco Canada's website has a well-organized heading structure, which makes it easier for screen readers to parse and for users to navigate in general. Good use of headings makes it so content can be read in a logical, linear way without confusion. Especially useful for those with assistive technology.
RSpec Rails defines ten different types of specs for testing different parts of a typical Rails application. Each one inherits from one of Rails’ built-in TestCase
Podman provides some extra features that help developers and operators in Kubernetes environments. There are extra commands provided by Podman that are not available in Docker.
Podman commands are the same as Docker’s When building Podman, the goal was to make sure that Docker users could easily adapt. So all the commands you are familiar with also exist with Podman. In fact, the claim is made that if you have existing scripts that run Docker you can create a docker alias for podman and all your scripts should work (alias docker=podman). Try it.
If you want to write an awk script, which portable to any awk implementations and versions (gawk/nawk/mawk...) it is really hard, even if with --posix (gawk)
nowadays many people work with docker containers. Most default docker images do not have bash and something like [[ $string == *foo* ]] will not work.
It's not so much a criticism as the preference of a more universal solution over a more limited one. Please consider that, years later, people (like me) will stop by to look for this answer and may be pleased to find one that's useful in a wider scope than the original question. As they say in the Open Source world: "choice is good!"
Include a note about specific Cuis idioms, explain why they exist, and link to discussion and documentation: * backticks for compound literals * double colon as an alternative to parentheses * #[aFloat] for Float64Array * #[anInteger] for ByteArray
See the thread Language Constructs initiated by Erik Stel in the Cuis-dev mailing list in 2020-05; especially the answers by Juan Vuletich.
... In some cases it can improve performance or reduce memory usage. But the deeper reason is to put all classes in equal footing with those privileged ones that are known by the Compiler.
More here
That tends to be the biggest cop out excuse for libraries. Just do a major version release. The fact this library lies about the encodingis extremely problematic and causes numerous bugs. Any program currently using this library is already incorrect because of this behavior. Actually exposing the problem makes it easier for people to fix.
in reply to subject of https://hyp.is/VeTJlpN0Ee2mNKOVyQ-B5g/github.com/mikel/mail/issues/902
Agree, but we're stuck with API compatibility for a good while.
The problem is that the caller may write yield instead of block.call. The code I have given is possible caller's code. Extended method definition in my library can be simplified to my code above. Client provides block passed to define_method (body of a method), so he/she can write there anything. Especially yield. I can write in documentation that yield simply does not work, but I am trying to avoid that, and make my library 100% compatible with Ruby (alow to use any language syntax, not only a subset).
An understandable concern/desire: compatibility
Added new tag for this: allowing full syntax to be used, not just subset
This is great because it works in Terminal, iTerm2, Chrome, Sublime & Outlook. The alternative (Karabina) is less good because that maps "Home" to Command+LeftArrow, which actually switches windows in Terminal.
Current ruby releases generate *.tar.gz, *.tar.bz2, *.tar.xz, and *.zip. But I think we can stop generating *.tar.bz2. I think *.tar.bz2 are less merit. For better size, *.tar.xz exist. For better compatibility, *.tar.gz and *.zip exist.
Even if the browser ignores the closing slash in void tags, it's good practice to close it because: 1. in frameworks like react js if these are not close, it creates an error 2. if you want your document to be readable by an XML parser then must close all elements
The following characters are replaced with _ in file names: ~, +, \, ?, %, *, :, |, ", <, >
l’usage des licences est très approximatif. Il nous est arrivé de trouver un même cours ayant de multiples licences, contradictoires, posées par les auteurs, l’Université et l’annuaire lui-même. Ce qui en pratique rend impossible son utilisation autrement qu’en simple document à consulter : on est alors très loin des REL.
Reopening this as it does seem we have broken compatibility with the oldest GPUs. Even if NVIDIA no longer support them, if they used to work it should still work.
Around @11:16:
"What made HyperCard the ubiquitous product it was in the early 90s... was the fact that it was included free with every Macintosh sold. So anybody could use it to create somethnig, then share their creation with somebody else with the confidence that the other person would be able to run it."
So that was in that day. What is the box today?
Let me ask it another way: What is available on every computing device[...]?"
I would encourage us all to find ways to make the system immediately available to users.
You want to use the commercial Tailwind UI component library (https://tailwindui.com/) in your Svelte project, and want a drop-in replacement for the React components which power Tailwind UI.
compatibility with any front-end framework means you don't have to change your stack
The chart below
on Windows, since that's what I'm using
Good example of why leveraging the browser's runtime is better. i wouldn't have guessed that the md2blog creator was using Windows. (And I didn't. I just assume that everyone is using a Mac, even though I'm on neither.)
Hence, Podman allows the creation and execution of Pods from a Kubernetes YAML file (see podman-play-kube). Podman can also generate Kubernetes YAML based on a container or Pod (see podman-generate-kube), which allows for an easy transition from a local development environment to a production Kubernetes cluster.
The VTE widget was originally designed as the back-end for Gnome Terminal but was fortunately designed as a GTK widget so that other terminal emulator applications could leverage it instead of rolling their own. Many popular Linux terminal emulators use this component.
.
NodeJS Supported node-sass version
Provides empty Module#ruby2_keywords method, for the forward source-level compatibility against ruby2.7 and ruby3.
GraphQL services should not provide any additional entries to the error format since they could conflict with additional entries that may be added in future versions of this specification.
AnyCable uses the same protocol as ActionCable, so you can use its JavaScript client without any monkey-patching.
The emphasis was made on a raw CDP protocol because Chrome allows you to do so many things that are barely supported by WebDriver because it should have consistent design with other browsers.
compatibility: need for compatibility is limiting:
There were attempts to simplify this setup by building specific browsers (such as capybara-webkit and PhantomJS) providing such APIs out-of-box, but none of them survived the compatibility race with real browsers.
Why are there so many programming languages and frameworks? Everyone has their own opinion on how something should be done. Some of these systems, like AOL, Yahoo, etc... have been around for a decade, and probably not updated much.
It's essential to note that all the experiments are valid for FreeBSD and I can't guarantee they'll give the same results on other operating systems.
Is there an OS agnostic way of doing this? I like the script command on macOS because you don't have to wrap the command in quotes. The script runs and sends output to the tty which is duplicated in the supplied file, but I can't seem to get the linux version to behave the same way... I'm probably doing something wrong. So what's the equivalent linux script command for this on macOS: script -q -t 0 tmp.out perl -e 'print "Test\n"' Test cat tmp.out Test
Windows Subsystem for Linux provides a Linux-compatible kernel interface developed by Microsoft and containing no Linux code
Functional UNIX[edit] Broadly, any Unix-like system that behaves in a manner roughly consistent with the UNIX specification, including having a "program which manages your login and command line sessions";[14] more specifically, this can refer to systems such as Linux or Minix that behave similarly to a UNIX system but have no genetic or trademark connection to the AT&T code base.
Windows Subsystem for Linux, also known as WSL, is a compatibility layer for running Linux binary executables natively on Windows 10 using a Linux image
In the BSD PTY system, the slave device file, which generally has a name of the form /dev/tty[p-za-e][0-9a-f], supports all system calls applicable to text terminal devices.
Schwarz, N., Newman, E., & Leach, W. (2016). Making the truth stick & the myths fade: Lessons from cognitive psychology. Behavioral Science & Policy, 2(1), 85–95. https://doi.org/10.1353/bsp.2016.0009
Compatibility is better than purity.
D3 now passes events directly to listeners, replacing the d3.event global and bringing D3 inline with vanilla JavaScript and most other frameworks.
Eyeglass provides a way to distribute Sass files and their associated assets and javascript extensions via npm such that any build system can be adapted to automatically expose those Sass files to Sass's @import directive by just installing the code.
Do not use *= require in Sass or your other stylesheets will not be able to access the Bootstrap mixins or variables.
Sprockets 3 was a compatibility release to bridge Sprockets 4, and many deprecated things have been removed in version 4.
That’s it. If you have a previous “precompile” array, in your app config, it will continue to work. For continuity sake I recommend moving over those declarations to your manifest.js file so that it will be consistent.
In any case signal handling in shells is one of the least reliable and portable aspects. You'll find behaviours vary greatly between shells and often between different versions of a same shell. Be prepared for some serious hair pulling and head scratching if you're going to try to do anything non-trivial.
With the introduction of CPUs which ran faster than the original 4.77 MHz Intel 8088 used in the IBM Personal Computer, programs which relied on the CPU's frequency for timing were executing faster than intended. Games in particular were often rendered unplayable. To provide some compatibility, the "turbo" button was added. Engaging turbo mode slows the system down to a state compatible with original 8086/8088 chips.
If it was remotely possible to get Davinci Resolve running that would be incredible (and bring a lot of video people I think)
Note that that browser support for these values is nuanced. For example, space-between never got support from some versions of Edge, and start/end/left/right aren’t in Chrome yet
The use of __proto__ is controversial and discouraged. It was never originally included in the ECMAScript language spec, but modern browsers implemented it anyway. Only recently was the __proto__ property standardized by the ECMAScript 2015 specification for compatibility with web browsers, so it will be supported into the future.
The issue will be whether or not the player rendering the video respects the metadata flag or not
readlink is not part of the standard. A portable script could be implemented with only POSIX shell features.
the following seems to do it without the bashism
Presumably this is so that you can import React libraries, even if they depend on ReactDOM, and they will work with Svelte instead.
Reminds me of Wine. IIRC they have some system calls that they just make to be no-ops on Linux.
FormValidation can be used with popular JavaScript frameworks such as React, Preact, Vue, Svelte, etc.
For reasons of compatibility and simplicity, it's best to use React's built-in state management capabilities rather than external global state.
When it all came together it meant supporting most JSX conventions like spreads, refs, forwardRefs, and Components as tags.
This code would work on any system with an x86 processor due to backward compatibility
Kittler a le souci de penser la compatibilité (et surtout la rétro-compatibilité) de son écriture, de sa production.
la pensée s'inscrit dans un médium, un médium qui évolue et qui, peut-être plus que jamais, soulève des enjeux d'archivage très importants (comme les œuvres artistiques qui reposent désormais sur des supports numériques à présent non supportés)
But what if we want to use it in a legacy app built with for example Bootstrap 3 and lots of jQuery where there's no sign of Node.js? No problem! We'll just use the IIFE build and instantiate the component class through its global constructor:
Did you know that you can create a Svelte component and with almost no extra steps distribute- and use it like any classic old Javascript library through a global constructor (let myComponent = new MyComponent())?
many CDNs nowadays ultimately source from npm (before caching the JavaScript on their own edge servers). So you'll be guaranteed to have the same library version on the CDN as on npm
You may also use a regular expression for include that works regardless of base path.
compatibility with hex keys
The great thing about having our function in Promise form is that we don't actually need to "make it an async/await version" if we don't want to. When we call/execute the function, we can simply use the async/await keyword
One key advantage of 'HTML-plus' languages is that you don't actually need tooling in order to be productive — most editors give you out-of-the-box support for things like syntax highlighting (though imperfect, as JavaScript expressions are treated as strings) and auto-closing tags. Tools like Emmet work with no additional setup. HTMLx should retain that benefit.
Write svelte components in jsx.
This package exposes an hyperscript compatible function: h(tag, properties, ...children) which returns a svelte component.
For interoperability with RxJS Observables, the .subscribe method is also allowed to return an object with an .unsubscribe method, rather than return the unsubscription function directly.
So let’s say Apple pulls a Guido and breaks compatibility. What do you think will happen? Well, maybe 80–90% of the developers will rewrite their software, if they’re lucky. Which is the same thing as saying, they’re going to lose 10–20% of their user base to some competing language, e.g. Flutter.Do that a few times, and you’ve lost half your user base. And like in sports, momentum in the programming world is everything. Anyone who shows up on the charts as “lost half their users in the past 5 years” is being flagged as a Big Fat Loser. You don’t want to be trending down in the Platforms world. But that’s exactly where deprecation — the “removing APIs” kind, not the “warning but permitting” kind — will get you, over time: Trending down. Because every time you shake loose some of your developers, you’ve (a) lost them for good, because they are angry at you for breaking your contract, and (b) given them to your competitors.
Twitter is a good example of this, and they've just created a shiny new API in an apparent attempt to bring developers back...
Wonder if it's going to be backwards compatible? (Probably not...)
It’s a sure sign, when there are four or five different coexisting subsystems for doing literally the same thing, that underlying it all is a commitment to backwards compatibility. Which in the Platforms world, is synonymous with commitment to your customers, and to your marketplace.
This same sort of thing applies to WordPress for its backwards compatibility.
I wonder if there were some larger breaking changes in Drupal 7 and 8 that removed their backwards compatibility and thereby lost them some older websites?
Successful long-lived open systems owe their success to building decades-long micro-communities around extensions/plugins, also known as a marketplace.
This could be said of most early web standards like HTML as well...
In the Emacs world (and in many other domains, some of which we’ll explore below), when they make an API obsolete, they are basically saying: “You really shouldn’t use this approach, because even though it works, it suffers from various deficiencies which we enumerate here. But in the end it’s your call.”
Backwards compatibility keeps systems alive and relevant for decades.
WPML knows how to communicate with the Translation Proxy, and the Translation Proxy knows how to communicate with translation services. If a Translation Service company happened to change their API, we needed to only update the Translation Proxy.
Why save sessions as bookmarks? - all the data saved will be there no matter what addon you may use in the feature
If you’re working in spreadsheet software (for example, Microsoft Excel, Google Sheets, or Apple Numbers), you can copy from a spreadsheet, and GitLab will paste it as a Markdown table. For example, suppose you have the following spreadsheet:
Many features have been introduced continuously to Object Pascal with extensions to Delphi and extensions to FreePascal. In reaction to criticism, Free Pascal has adopted generics with the same syntax as Delphi, provided Delphi compatibility mode is selected, and both Delphi (partial) and Free Pascal (more extensive) support operator overloading. Delphi has also introduced many other features since version 7,[6] including generics. Whereas FreePascal tries to be compatible to Delphi in Delphi compatibility mode, it also usually introduced many new features to the language that are not always available in Delphi.
The main caveat to note is that it does use musl libc instead of glibc and friends, so certain software might run into issues depending on the depth of their libc requirements. However, most software doesn't have an issue with this, so this variant is usually a very safe choice.
All valid JavaScript code is also TypeScript code. You might get type-checking errors, but that won't stop you from running the resulting JavaScript.
A standard-setting organization is an industry group that sets common standards for its particular industry to ensure compatibility and interoperability of devices manufactured by different companies.
Products using the same version of Gecko have identical support for Web standards.
That app you desperately need in order to function? We probably have it. The vast software libraries of Ubuntu and Flatpak combine to make all of your tools available in a single location, called the Pop!_Shop.
More information on the CC license compatibility chart https://wiki.creativecommons.org/wiki/Wiki/cc_license_compatibility
The compatibility chart is an excellent resource for visually identifying the scope of resuse. Most importantly, this chart shows the product of remixing licenses. This is another resource that will be employed for future reference.
Rspec-mocks is now an independant project that can be used in any non Rspec project.
So while we can’t endorse those systems, and indeed we have to advise you against using them; their existence is still a Good Thing
Just remove the 'rb-readline' gem if you are on a linux like system.
And since it’s just a client, you can always use the todo.txt format text file created by it in the very beginning with any other client. Give it a try.
Countless productivity apps and sites store your tasks in their own proprietary database and file format. But you can work with your todo.txt file in every text editor ever made, regardless of operating system or vendor.
It doesn't use a database (unlike Keepass) and thus doesn't open all passwords at once. Just one at a time. Since it's just a directory of encrypted files, you can access your passwords with any PGP-compatible tool.
It is also compatible with Google Authenticator. Any website that shows a QRcode for Google Authenticator also works with Aegis.
uBlock Origin blocks ads through its support of the Adblock Plus filter syntax. uBlock Origin extends the syntax and is designed to work with custom rules and filters.
Reason compiles to JavaScript thanks to our partner project, BuckleScript, which compiles OCaml/Reason into readable JavaScript with smooth interop. Reason also compiles to fast, barebone assembly, thanks to OCaml itself.
Familiar and easy to insert into an existing ReactJS codebase
Onivim 2 leverages the VSCode Extension Host process in its entirety - meaning, eventually, complete support for VSCode extensions and configuration.
Specific to a testing framework (though we recommend Jest as our preference, the library works with any framework. See Using Without Jest)
https://wiki.creativecommons.org/wiki/Wiki/cc_license_compatibility
This is still the most helpful visual I have seen thus far to help explain and clarify compatibility of CC licenses.
g likelihood or Bayesian probabilistic phylogene
If you have a molecular data partition, you can just use total evidence approach and the standard 1-parameter Markov model.
Potential synapomorphies will be compatible with the molecular tree and considered not likely to change. Potential homoiologies and symplesiomorphies are partly ("semi-")compatible with the molecular tree and, hence, considered less likely to change than highly homoplastic traits with (random) convergence.
Just try out a couple of datasets, and infer the (Bio)NJ and ML trees and then compare the result with the strict consensus network (not tree) of all equally parsimonious trees and the Bayesian tree sample.
Note that if you apply TNT's iterative character weighting procedure, what you effectively do is sorting the random convergences from parallelisms/ characters that are more compatible with the preferred tree.
set; if this is higher, the tree 2can be considered to fit the data less well
To test the fit between data and more than one alternative tree, you can just do a bootstrap analysis, and map the results on a neighbour-net splits graph based on the same data.
Note that the phangorn library includes functions to transfer information between trees/tree samples and trees and networks:<br/> Schliep K, Potts AJ, Morrison DA, Grimm GW. 2017. Intertwining phylogenetic trees and networks. Methods in Ecology and Evolution (DOI:10.1111/2041-210X.12760.)[http://onlinelibrary.wiley.com/doi/10.1111/2041-210X.12760/full] – the basic functions and script templates are provided in the associated vignette.
number of character states bracketed by a node could 28be counted, and those which do not optimize as symplesiomorphies of the clade could 29considered as a value of node suppo
Why differentiating between symplesiomorphies and homoiologies? Both are traits equally exclusive to a subtree (a clade) and closely linked to each other.
Pink is a symplesiomorphy of the ingroup, blue the homoiology
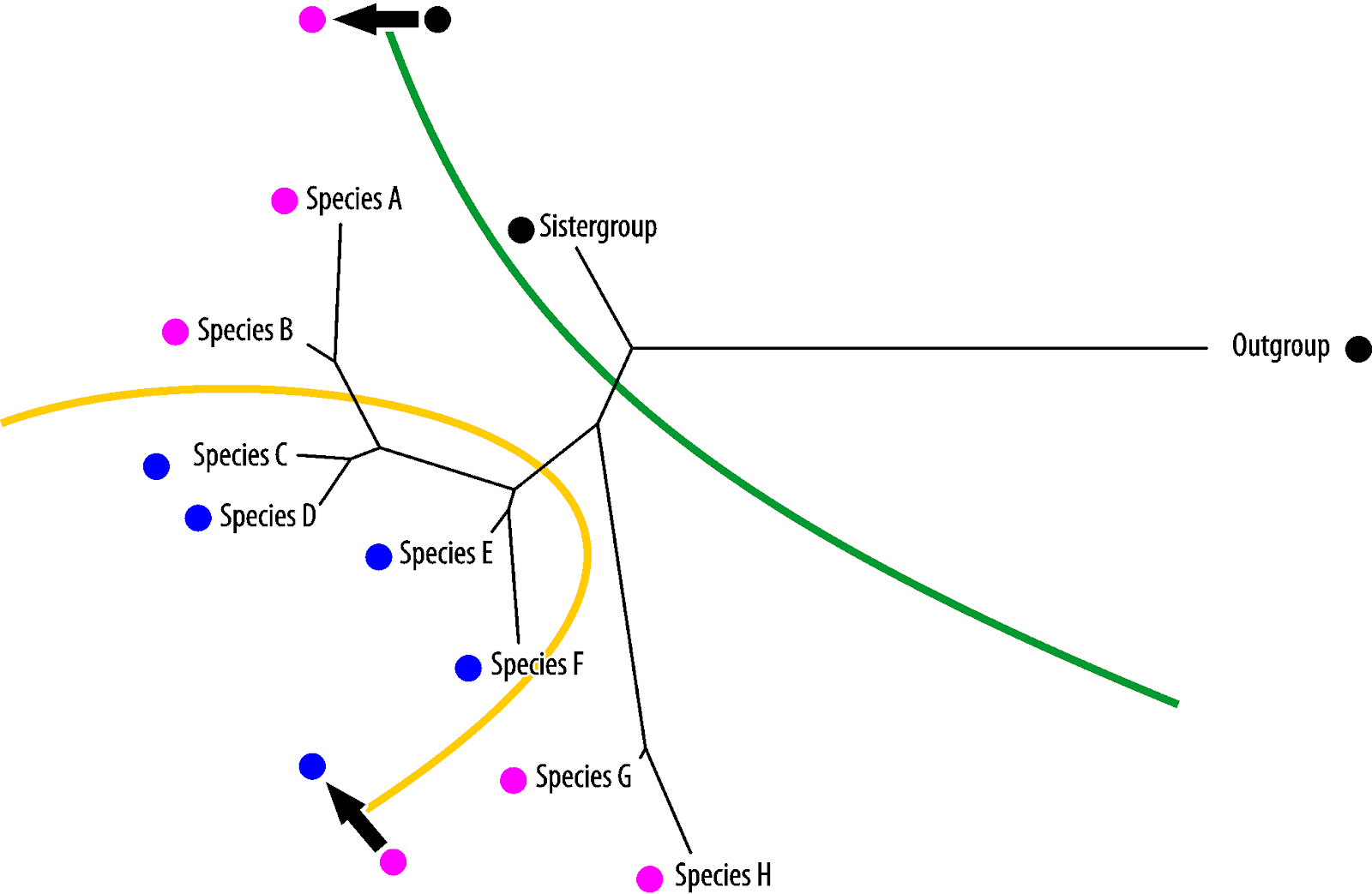 Vice versa, blue is the symplesiomorphy, pink the homoiology found only in the most derived (in absolute evolutionary terms) taxa
Vice versa, blue is the symplesiomorphy, pink the homoiology found only in the most derived (in absolute evolutionary terms) taxa
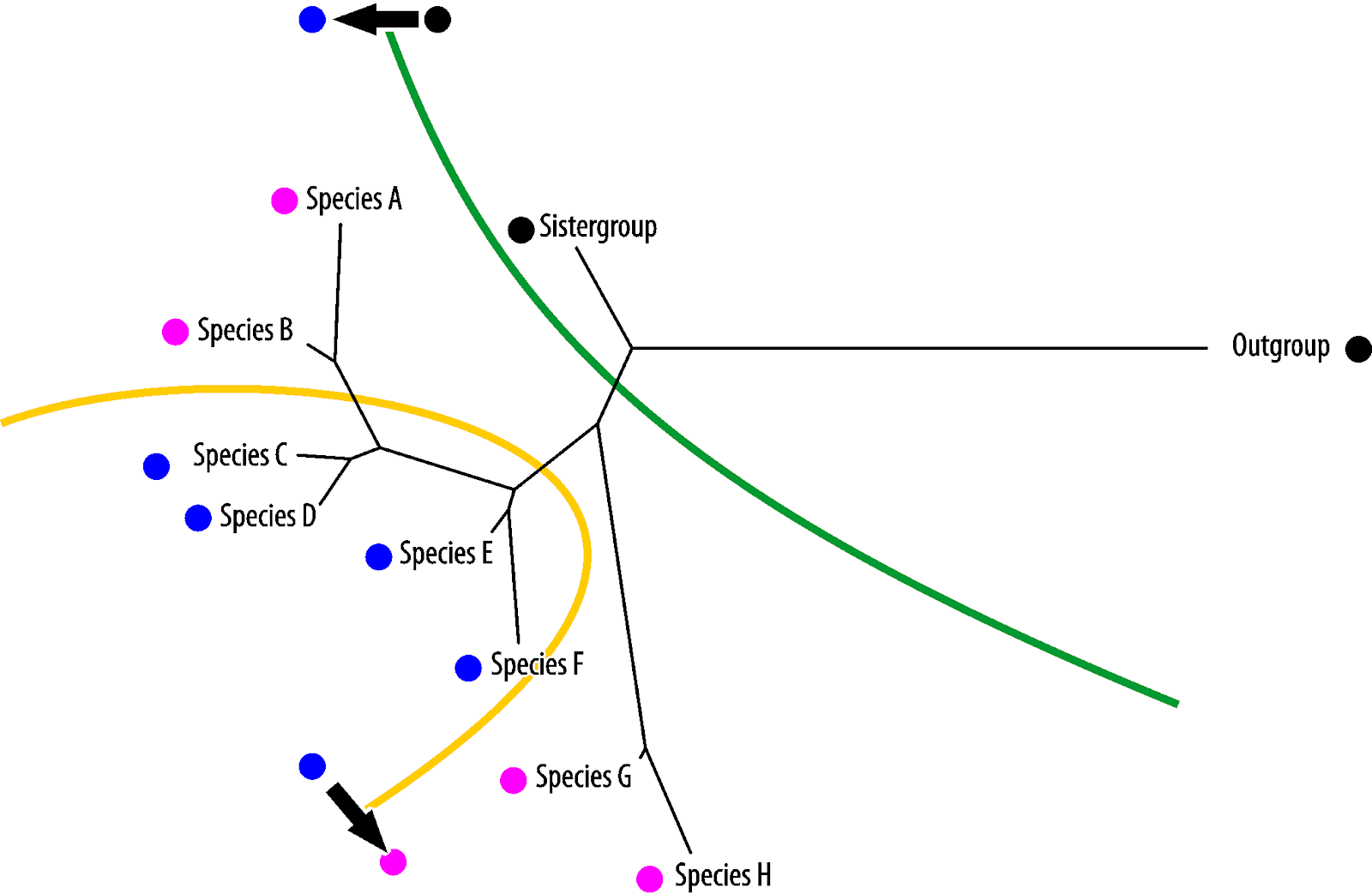
In principle, I do sympathize with the general idea, but the laid out approach will have little use.
The main drawback is that you can only define homoiologies using an external data set (e.g. the molecular "gold" tree). But when you have a reliable molecular tree, you can just go for total evidence approaches to select a more likely, in a mathematical and general sense, alternative without the need to make any prior destinction between your characters. Homoiologies will be inferred, like synapmorphies or symplesiomorphies or shared apomorphies (non-stochastically distributed convergences) on the fly.
If you define the homoiologies on a inferred (e.g. parsimony) tree only based on a morphological data matrix (e.g. for an extinct group of organisms), you will inevitably misinterpret some characters, because your clades are not necessarily monophyletic. Homoiologies like symplesiomorphies may appear as (pseudo-)synapomorphies.
The only application left would be that the molecular tree cannot resolve certain relationships, and we use more tree-compatible morphological characters to discern between alternatives. However, the first choice would then be to maximise the number of synapomorphies. Only if that would be the same for all alternatives, one could count the number of symplesiomorphies and homoiologies (as the distinction between both via a tree-inference is very tricky; and their are often just two side of the same evolutionary process).
However, one could also just directly change to a network-analysis framework, which will pretty much solve all these problems at once.
For further details see my (upcoming, March 4th) post at Genealogical World of Phylogenetic Networks