Aucun élève ou étudiant ne doit subir de faits de harcèlement
10,000 Matching Annotations
- Mar 2024
-
www.legifrance.gouv.fr www.legifrance.gouv.fr
-
-
Les parents d'élèves participent, par leurs représentants aux conseils d'école, aux conseils d'administration des établissements scolaires et aux conseils de classe.
-
Leur participation à la vie scolaire et le dialogue avec les enseignants et les autres personnels sont assurés
-
Les parents d'élèves sont membres de la communauté éducative.
-
Dans chaque école, collège ou lycée, la communauté éducative rassemble les élèves et tous ceux qui, dans l'établissement scolaire ou en relation avec lui, participent à l'accomplissement de ses missions.Elle réunit les personnels des écoles et établissements, les parents d'élèves, les collectivités territoriales, les associations éducatives complémentaires de l'enseignement public ainsi que les acteurs institutionnels, économiques et sociaux, associés au service public de l'éducation.
-
Dans le cadre d'une école inclusive, elle fonde sa cohésion sur la complémentarité des expertises.
-
L'Etat garantit le respect de la personnalité de l'enfant et de l'action éducative des familles
-
La formation scolaire favorise l'épanouissement de l'enfant
-
Tout enfant a droit à une formation scolaire qui, complétant l'action de sa famille, concourt à son éducation
-
L'acquisition d'une culture générale et d'une qualification reconnue est assurée à tous les jeunes, quelle que soit leur origine sociale, culturelle ou géographique.
-
permettre de façon générale aux élèves en difficulté, quelle qu'en soit l'origine, en particulier de santé, de bénéficier d'actions de soutien individualisé.
-
Il veille également à la mixité sociale des publics scolarisés au sein des établissements d'enseignement
-
Il veille à la scolarisation inclusive de tous les enfants, sans aucune distinction
-
Article L111-1Modifié par LOI n°2021-1109 du 24 août 2021 - art. 58L'éducation est la première priorité nationale. Le service public de l'éducation est conçu et organisé en fonction des élèves et des étudiants. Il contribue à l'égalité des chances et à lutter contre les inégalités sociales et territoriales en matière de réussite scolaire et éducative. Il reconnaît que tous les enfants partagent la capacité d'apprendre et de progresser. Il veille à la scolarisation inclusive de tous les enfants, sans aucune distinction. Il veille également à la mixité sociale des publics scolarisés au sein des établissements d'enseignement. Pour garantir la réussite de tous, l'école se construit avec la participation des parents, quelle que soit leur origine sociale. Elle s'enrichit et se conforte par le dialogue et la coopération entre tous les acteurs de la communauté éducative.
-
Elle s'enrichit et se conforte par le dialogue et la coopération entre tous les acteurs de la communauté éducative.
-
-
arkloud.xyz arkloud.xyz
-
Expansion of the ICC to include communication infrastructure development
.. I just got back from dreaming that there could be "all roads lead to rome" in an andromeda plan with a thing called the bridge to the venii "suns" venuses? like the version that has ... coastal-west and the one with the rivers instead of roads in the venice that has a version with the verilonamice verily lo, the mice part of the "of the mice and the mammalian's language code for the one we are speak en ...
-
-
browser.engineering browser.engineering
-
inversion of control,
a framework calls custom code, rather than the usual other way around
like how an event driven framework calls custom GUI code
-
-
urbanreadingforum.com urbanreadingforum.com
-
eligibleslum dwellers
Code yellow - new category!
-
ncremental housing
New category - code yellow!
-
enable slum
A new category, not explicitly mentioned in the list of definitions earlier has emerged. Code Yellow! what does this mean? A Ctrl-F may be in order.
-
-
pressbooks.online.ucf.edu pressbooks.online.ucf.edu
-
Pythagorical symbols
Pythagoras was one of the most influential people in history. His influence on mathematics -- and the world which depends on it -- has been evident hundreds of years after his era. However, it is clear that Pythagoras' mathematics was not what the author is referencing, but rather his, "exhortation to abide by a moral code". This theme is also evident when the author mentions Ovid and Metamorphoses as Ovid was references Pythagoras in many of his writings.
Swanson, Roy Arthur. “Ovid’s Pythagorean Essay.” The Classical Journal, vol. 54, no. 1, 1958, pp. 21–24. JSTOR, http://www.jstor.org/stable/3295324. Accessed 28 Mar. 2024.
-
-
www.legifrance.gouv.fr www.legifrance.gouv.fr
-
Article L111-1Version en vigueur depuis le 26 août 2021Modifié par LOI n°2021-1109 du 24 août 2021 - art. 58L'éducation est la première priorité nationale. Le service public de l'éducation est conçu et organisé en fonction des élèves et des étudiants. Il contribue à l'égalité des chances et à lutter contre les inégalités sociales et territoriales en matière de réussite scolaire et éducative.
-
-
www.defenseurdesdroits.fr www.defenseurdesdroits.fr
-
À la suite de nos analyses, nous avonsconstaté un nombre croissant d'élèves sansaffectation, en augmentation de l’ordre de 30à 40 % par rapport à l’année précédente. Or,conformément au code de l’éducation, quil’érige au rang de première priorité nationale,le service public de l’éducation doit être conçuet organisé en fonction de ses usagers, quisont les élèves
-
-
pressbooks.online.ucf.edu pressbooks.online.ucf.edu
-
Save A Life Pet Rescue
add link to:https://savealifepetrescue.org/home
include a copy of the new logo you designed?
A great place to add personal photos of you volunteering if you have them
you could even include this embed link code to show the video about the organization <iframe width="560" height="315" src="https://www.youtube.com/embed/HjK6Op1yF9g?si=jP1uQej6EsVBz8_-" title="YouTube video player" frameborder="0" allow="accelerometer; autoplay; clipboard-write; encrypted-media; gyroscope; picture-in-picture; web-share" referrerpolicy="strict-origin-when-cross-origin" allowfullscreen></iframe>
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
eLife assessment
This important study provides a new, apparently high-performance algorithm for B cell clonal family inference. The new algorithm is highly innovative and based on a rigorous probabilistic analysis of the relevant biological processes and their imprint on the resulting sequences, however, the strength of evidence regarding the algorithm's performance is incomplete, due to (1) a lack of clarity regarding how different data sets were used for different steps during algorithm development and validation, resulting in concerns of circularity, (2) a lack of detail regarding the settings for competitor programs during benchmarking, and (3) method development, data simulation for method validation, and empirical analyses all based on the B cell repertoire of a single subject. With clarity around these issues and application to a more diverse set of real samples, this paper could be fundamental to immunologists and important to any researcher or clinician utilizing B cell receptor repertoires in their field (e.g., cancer immunology).
We apologize for the long delay in implementing the suggested changes. Some of the co-authors had some personal issues that made it hard to efficiently work on the revision.
We have addressed all the essential points below, as well as all the detailed comments of each reviewer in the following pages.
Due to the journal’s guidelines we have to upload an “all black” version of the manuscript as the main version. We have uploaded a revised manuscript with the changes marked in red as a “Related Manuscript file”, which appears at the very end of the Merged Manuscript File, after all the Figures, and at the end of the list of files on the webpage. We apologize for this inconvenience.
In addition, we have added an extension of HILARy to deal with paired-chain repertoires, and have benchmarked the new method on a recently published synthetic dataset. This new analysis is now presented in new Fig. 5.
Reviewer #1 (Public Review):
Identifying individual BCR/Ab chain sequences that are members of the same clone is a longstanding problem in the analysis of BCR/Ab repertoire sequencing data. The authors propose a new method designed to be scalable for application to huge repertoire data sets without sacrificing accuracy. Their approach utilizes Hamming Distance between CDR3 sequences followed by clustering for a fast, high-precision approach to classifying pairs of sequences as related or not, and then refines the classification using mutation information from germline-encoded regions. They compare their method with other state-of-the-art methods using synthetic data.
The authors address an important problem in an interesting, innovative, and rigorous way, using probabilistic representations of CDR3 differences, frequencies of shared and not-shared mutations, and the relationships between the two under hypotheses of related pairs and unrelated pairs, and from these develop an approach for determining thresholds for classification and lineage assignment. Benchmarking shows that the proposed method, the complete method including both steps, outperforms other methods.
Strengths of the method include its theoretical underpinnings which are consistent with an immunologist's intuition about how related and unrelated sequences would compare with each other in terms of the metrics to use and how those metrics are related to each other.
I have two high-level concerns:
(1) It isn't clear how the real and synthetic data are being used to estimate parameters for the classifier and evaluate the classifier to avoid circularity. It seems like the approach is used to assign lineages in the data from [1], and then properties of this set of lineages are used to estimate parameters that are then used to refine the approach and generate synthetic data that is used to evaluate the approach. This may not be a problem with the approach but rather with its presentation, but it isn't entirely clear what data is being used and where for what purpose. An understanding of this is necessary in order to truly evaluate the method and results.
The reviewer is correct in their understanding of the pipeline. It should be stressed that the lineages used to guide the generation of the synthetic data was done on VJl classes for which the clustering was easy and reliable, and should therefore be largely model independent.
We have added an explanation in the main text of why the re-use of real data lineages inferred by HILARy doesn’t bias the procedure, since it’s done on a subset of lineages within VJl classes that are easy to infer (section “Test on synthetic dataset”).
(2) Regarding the data used for benchmarking - given the intertwined fashion by which the classification approach and synthetic data generation approach appear to have been developed, it is not surprising that the proposed approach outperforms the other methods when evaluated on the synthetic data presented here. It would be better to include in the benchmark the data used by the other methods to benchmark themselves or also generate synthetic data using their data generation procedures.
We agree with the reviewer that a test of the method on an independent synthetic dataset is important for its applicability and to compare to other methods.
We have added a new synthetic dataset from the group that designed the partis method to our benchmark. Our method still performs competitively, on par with partis—which was developed and tested on that dataset—and better than other methods. The results are presented in revised Fig. 4 (panels E-G), and Figure 4–figure supplement 1 as a function of the mutation rate.
In addition, we have used that dataset to benchmark a new version of HILARy that also uses the light chain. We present the results in new Figures 5 and Figure 4–figure supplement 1.
An improved method for BCR/Ab sequence lineage assignment would be a methodologic advancement that would enable more rigorous analyses of BCR/Ab repertoires across many fields, including infectious disease, cancer, autoimmune disease, etc., and in turn, enable advancement in our understanding of humoral immune responses. The methods would have utility to a broad community of researchers.
Reviewer #2 (Public Review):
This manuscript describes a new algorithm for clonal family inference based on V and J gene identity, sequence divergence in the CDR3 region, and shared mutations outside the CDR3. Specifically, the algorithm starts by grouping sequences that have the same V and J genes and the same CDR3 length. It then performs single-linkage clustering on these groups based on CDR3 Hamming distance, then further refines these groups based on shared mutations.
Although there are a number of algorithms that use a similar overall strategy, a couple of aspects make this work unique. First, a persistent challenge for algorithms such as this one is how to set a cutoff for single-linkage clustering: if it is too low, then one separates clusters that should be together, and if too high one joins together clusters that should be separate. Here the authors leverage a rich collection of probabilistic tools to make an optimal choice. Specifically, they model the probability distributions of within- and between-cluster CDR3 Hamming distances, with parameters depending on CDR3 length and the "prevalence" of clonal sequence pairs (i.e. family size distribution). This allows the algorithm to make optimal choices for separating clusters, given the particular chosen distance metric, and assuming the sample in question has been accurately modeled. Second, the algorithm uses a highly efficient means of doing single-linkage clustering on nucleotide sequences.
This leads to a fast and highly performant algorithm on data meant to replicate the original sample used in algorithm design. The ideas are new and beautifully developed. The application to real data is interesting, especially the point about dN/dS.
However, the paper leaves open the question of how this inference algorithm works on samples other than the one used for simulation and as a template for validation. If I understand the simulation procedure correctly - that one takes a collection of inferred trees from the real data, then re-draws the root sequence and the identity of the mutations on the branches - then the simulated data should be very close to the data used to develop the methods in the paper. This consideration seems especially important given that key methods in this paper use mutation counts and overall mutation counts are preserved.
Repertoires come in all shapes and sizes: infants to adults, healthy to cancerous, and naive to memory to plasma-cell-just-after-vaccination. If this is being proposed as a general-purpose clonal inference algorithm rather than one just for this sample, then a more diverse set of validations are needed.
We agree that testing the method on a differently generated dataset is a useful check. We should point out, however, that our synthetic dataset is not as biased as it may seem. In particular, it is based on trees from VJl classes that we predicted are very easy to cluster, which means that they are truly faithful to the data, and not dependent on the particular algorithm used to infer them. The big advantage over this synthetic dataset over others is that it recapitulates the power law statistics of clone size distribution, as well as the diversity of mutation rates. To us, it still represents a more useful benchmark than synthetic datasets generated by population genetics models, which miss most of this very broad variability.
However, to check how the method generalizes to other datasets, we repeated our validation procedure on the dataset used to evaluate Partis in Ralph et al 2022. The new results are discussed in the main text and in new panels of Fig. 4 in the same form as the previous comparisons. We also added a comparison of performance as a function of mutation rate in the new Figure 4–figure supplement 1.
It is unclear how to run the code. The software repo has a nice readme explaining the file layout, dependencies, and input file format, but the repo seems to be lacking an
inference.ipynbmentioned there which runs an analysis. Perhaps this is a typo and refers toinference.py, which in addition to the documented cdr3 clustering, seems to have functions to run both clustering methods. However, it does not seem to have any documentation or help messages about how to run these functions.We have completely overhauled the github to provide a detailed step by step explanation of how to run the code. The code is now easily installable using pip.
The results are not currently reproducible, because the simulated data is not available. The data availability statement says that no data have been generated for this manuscript, however simulated data has been generated, and that is a key aspect of the analysis in the paper.
We have uploaded the simulated data to zenodo, as well as provided scripts in the github to run the benchmarks.
More detail is needed to understand the timing comparisons. The new software is clearly written to use many threads. Were the other software packages run using multiple threads? What type of machine was used for the benchmarks?
All timing comparisons were made based on a single VJl class on a 14 double-threaded CPU computer. HILARy uses all 28 threads, and other methods were run with default settings, with multi-threading allowed.
We have clarified the specifications of the computer.
Reviewer #3 (Public Review):
B cell receptors are produced through a combination of random V(D)J recombination and somatic hypermutation. Identifying clonal lineages - cells that descend from a common V(D)J rearrangement - is an important part of B cell repertoire analysis. Here, the authors developed a new method to identify clonal lineages from BCR data. This method builds off of prior advances in the field and uses both an adaptive clonal distance threshold and shared somatic hypermutation information to group B cells into clonal lineages.
The major strength of this paper is its thorough quantitative treatment of the subject and integration of multiple improvements into the clonal clustering process. By their simulation results, the method is both highly efficient and accurate.
The only notable weakness we identified is that much of the impact of the method will depend on its superiority to existing approaches, and this is not convincingly demonstrated by Fig. 4. In particular, little detail is given on how the other clonal clustering programs were run, and this can significantly impact their performance. More specifically:
We have added a new benchmark to address these concerns, presented in Fig. 4 and in new figure 4 – figure supplement 1 as a function of a controllable mutation rate.
(1) Scoper supports multiple methods for clonal clustering, including both adaptive CDR3 distance thresholds (Nouri and Kleinstein, 2018) and shared V-gene mutations (Nouri and Kleinstein, 2020). It is not clear which method was used for benchmarking. The specific functions and settings used should have been detailed and justified. Spectral clustering with shared V gene mutations would be the most comparable to the authors' method. Similar detail is needed for partis.
In the updated version I use the 2020 version. The 2018 is very similar to simple single linkage so will be removed from the benchmark.
(2) It is not clear how the adaptive thresholds and shared mutation analysis in the authors' method differ from prior approaches such as scoper and partis.
We have changed the paragraph in the discussion section about the benchmark to highlight the innovative aspects and differences with previous approaches.
(3) The scripts for performing benchmarking analyses, as well as the version numbers of programs tested, are not available.
We have added to the github all the scripts used for benchmarking. We have added details about the version numbers in the data and code availability section of the methods.
(4) Similar to above, P. 10 describes single linkage hierarchical clustering with a fixed threshold as a "crude method" that "suffers from inaccuracy as it loses precision in the case of highlymutated sequences and junctions of short length." As far as we could tell, this statement is not backed up by either citations or analyses in the paper. It should not be difficult for the authors to test this though using their simulations, as this method is also implemented in scoper.
We have added this method to our benchmark to support that point. The results are presented in Figure 4 – figure supplement 2.
References
Nouri N, Kleinstein SH. 2020. Somatic hypermutation analysis for improved identification of B cell clonal families from next-generation sequencing data. PLOS Comput Biol 16:e1007977. doi:10.1371/journal.pcbi.1007977
Nouri N, Kleinstein SH. 2018. A spectral clustering-based method for identifying clones from high- throughput B cell repertoire sequencing data. Bioinformatics 34:i341-i349. doi:10.1093/bioinformatics/bty235
We have changed citation [22] to refer to the 2018 paper. The 2020 paper is citation [18].
-
Reviewer #2 (Public Review):
This manuscript describes a new algorithm for clonal family inference based on V and J gene identity, sequence divergence in the CDR3 region, and shared mutations outside the CDR3. Specifically, the algorithm starts by grouping sequences that have the same V and J genes and the same CDR3 length. It then performs single-linkage clustering on these groups based on CDR3 Hamming distance, then further refines these groups based on shared mutations.
Although there are a number of algorithms that use a similar overall strategy, a couple of aspects make this work unique. First, a persistent challenge for algorithms such as this one is how to set a cutoff for single-linkage clustering: if it is too low, then one separates clusters that should be together, and if too high one joins together clusters that should be separate. Here the authors leverage a rich collection of probabilistic tools to make an optimal choice. Specifically, they model the probability distributions of within- and between-cluster CDR3 Hamming distances, with parameters depending on CDR3 length and the "prevalence" of clonal sequence pairs (i.e. family size distribution). This allows the algorithm to make optimal choices for separating clusters, given the particular chosen distance metric, and assuming the sample in question has been accurately modeled. Second, the algorithm uses a highly efficient means of doing single-linkage clustering on nucleotide sequences.
This leads to a fast and highly performant algorithm on data meant to replicate the original sample used in algorithm design. The ideas are new and beautifully developed. The application to real data is interesting, especially the point about dN/dS.
However, the paper leaves open the question of how this inference algorithm works on samples other than the one used for simulation and as a template for validation. If I understand the simulation procedure correctly - that one takes a collection of inferred trees from the real data, then re-draws the root sequence and the identity of the mutations on the branches - then the simulated data should be very close to the data used to develop the methods in the paper. This consideration seems especially important given that key methods in this paper use mutation counts and overall mutation counts are preserved.
Repertoires come in all shapes and sizes: infants to adults, healthy to cancerous, and naive to memory to plasma-cell-just-after-vaccination. If this is being proposed as a general-purpose clonal inference algorithm rather than one just for this sample, then a more diverse set of validations are needed.
It is unclear how to run the code. The software repo has a nice readme explaining the file layout, dependencies, and input file format, but the repo seems to be lacking an `inference.ipynb` mentioned there which runs an analysis. Perhaps this is a typo and refers to `inference.py`, which in addition to the documented cdr3 clustering, seems to have functions to run both clustering methods. However, it does not seem to have any documentation or help messages about how to run these functions.
The results are not currently reproducible, because the simulated data is not available. The data availability statement says that no data have been generated for this manuscript, however simulated data has been generated, and that is a key aspect of the analysis in the paper.
More detail is needed to understand the timing comparisons. The new software is clearly written to use many threads. Were the other software packages run using multiple threads? What type of machine was used for the benchmarks?
-
-
github.com github.com
-
**helps remove weird code stuff when editing
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
This study reports important evidence that infants' internal factors guide children's attention and that caregivers respond to infants' attentional shifts during caregiver-infant interactions. The authors analyzed EEG data and multiple types of behaviors using solid methodologies that can guide future studies of neural responses during social interaction in infants. However, the analysis is incomplete, as several methodological choices need more adequate justification.
Reviewer #1
Public Review:
The authors bring together multiple study methods (brain recordings with EEG and behavioral coding of infant and caregiver looking, and caregiver vocal changes) to understand social processes involved in infant attention. They test different hypotheses on whether caregivers scaffold attention by structuring a child's behavior, versus whether the child's attention is guided by internal factors and caregivers then respond to infants' attentional shifts. They conclude that internal processes (as measured by brain activation preceding looking) control infants' attention, and that caregivers rapidly modify their behaviors in response to changes in infant attention.
The study is meticulously documented, with cutting-edge analytic approaches to testing alternative models; this type of work provides a careful and well-documented guide for how to conduct studies and process and analyze data for researchers in the relatively new area of neural response in infants in social contexts.
We are very pleased that R1 considers our work an important contribution to this developing field, and we hope that we have now addressed their concerns below.
Some concerns arise around the use of terms (for example, an infant may "look" at an object, but that does not mean the infant is actually "attending); collapsing of different types of looks (to people and objects), and the averaging of data across infants that may mask some of the individual patterns.
We thank the reviewer for this feedback and their related comments below, and we feel that our manuscript is much stronger as a result of the changes we have made. Please see blow for a detailed description of our rationale for defining and analysing the attention data, as well as the textual changes made in response to the author’s comments.
Recommendations For The Authors
This paper is rigorous in method, theoretically grounded, and makes an important contribution to understanding processes of infant attention, brain activity, and the reciprocal temporal features of caregiver-infant interactions. The alternative hypothesis approach sets up the questions well (although authors should temper any wording that suggests attention processes are one or the other. That is, certain bouts of infant attention can be guided by exogenous factors such as social input, and others be endogenous; so averaging across all bouts can actually mask the variation in these patterns). I appreciated the focus on multiple types of behavior (e.g., gaze, vocal fluctuations in maternal speech); the emphasis on contingent responding; and the very clear summaries of takeaways after each section. Furthermore, methods and analyses are well described, details on data processing and so on are very thorough, and visualizations aptly facilitate data interpretation. However, I am not an expert on infant neural responses in EEG and assume that a reviewer with such expertise will weigh in on the treatment and quality of the data; therefore, my comments should be interpreted in light of this lack of knowledge.
We thank R1 for these very positive and insightful comments on our analyses which are the result of a number of years of methodological and technical developmental work.
We do agree with R1 that we should more carefully word parts of our argument in the Introduction to make clear the fact that shifts in infant attention could be driven by a combination of interactive and endogenous influences. As a result of this comment, we have made direct changes to parts of the Introduction; removing any wording that suggests that these processes are ‘alternative’ or ‘separate’, and our overall aim states: ‘Here, recording EEG from infants during naturalistic interactions with their caregiver, we examined the (inter)-dependent influences of infants’ endogenous oscillatory neural activity, and inter-dyadic behavioural contingencies in organising infant attention’.
Examining variability between infant attention episodes in the factors that influence the length and timing of the attention episode is an important area for future investigation. We now include a discussion on this on page 38 of the Discussion section, with suggestions for how this could be examined. Investigating different subtypes of infant attention is methodologically challenging, given the number of infant behaviours that would need to inform such an analysis- all of which are time consuming to code. Developing automated methods for performing these kinds of analyses is an important avenue for future work.
Here, I review various issues that require revision or elaboration based on my reading of what I consider to otherwise be a solid and important research paper.
Problem in the use of the term attention scaffolding. Although there may be literature precedent in the use of this term, it is problematic to narrowly define scaffolding as mother-initiated guidance of attention. A mother who responds to infant behaviors, but expands on the topic or supports continued attention, and so on, is scaffolding learning to a higher level. I would think about a different term because it currently implies a caregiver as either scaffolding OR responding contingently. It is not an either-or situation in conceptual meaning. In fact, research on social contingency (or contingent responsiveness), often views the follow-in responding as a way to scaffold learning in an infant.
Yes, we agree with R1 that the term ‘attention scaffolding’ could be confusing given the use of this term in previous work conducted with children and their caregivers in problem-solving tasks, that emphasise modulations in caregiver behaviour as a function of infant behaviour. As a result of this suggestion, we have made direct edits to the text throughout, replacing the term attentional scaffold with terms such as ‘organise’ and ‘structure’ in relation to the caregiver-leading or ‘didactic’ perspective, and terms such as ‘contingent responding’ and ‘dynamic modulation’ in relation to the caregiver-following perspective. We feel that this has much improved the clarity of the argument in the Introduction and Discussion sections.
Do individual data support the group average trends? My concern with unobservable (by definition) is that EEG data averages may mask what's going on in individual brain response. Effects appear to be small as well, which occurs in such conditions of averaging across perhaps very variable response patterns. In the interest of full transparency and open science, how many infants show the type of pattern revealed by the average graph (e.g., do neural markers of infant engagement forward predict attention for all babies? Majority?). Non-parametric tests on how many babies show a claimed pattern would offer the litmus test of significance on whether the phenomenon is robust across infants or pulled by a few infants with certain patterns of data. Ditto for all data. This would bolster my confidence in the summaries of what is going on in the infant brain. (The same applies as I suggest to attention bouts. To what extent does the forward-predict or backward-predict pattern work for all bouts, only some bouts, etc.?). I recognize that to obtain power, summaries are needed across infants and bouts, but I want to know if what's being observed is systematic.
We thank R1 for this comment and understand their concern that the overall pattern of findings reported in relation to the infants’ EEG data might obscure inter-individual variability in the associations between attention and theta power. Averaging across individual participant EEG responses is, however, the gold standard way to perform both event-locked (Jones et al., 2020) and continuous methods (Attaheri et al., 2020) of EEG analysis that are reported in the current manuscript. EEG data, and, in particular, naturalistic EEG data is inherently noisy, and averaging across participants increases the signal to noise ratio (i.e. inconsistent, and, therefore, non-task-related activity is averaged out of the response (Cohen, 2014; Noreika et al., 2020)). Examining individual EEG responses is unlikely to tell us anything meaningful, given that, if a response is not found for a particular participant, then it could be that the response is not present for that participant, or that it is present, but the EEG recording for that participant is too noisy to show the effect. Computing group-level effects, as is most common in all neuroimaging analyses, is, therefore, most optimal to examining our main research questions.
The findings reported in this analysis also replicate previous work conducted by our lab which showed that infant attention to objects significantly forward-predicted increases in infant theta activity during joint table-top play with their caregiver, involving one toy object (compared to our paradigm which involved 3;Wass et al., 2018). More recent work conducted by our lab has also shown continuous and time-locked associations between infant look durations and infant theta activity when infants play with objects on their own (Perapoch Amadó et al., 2023). To reassure readers of the replicability of the current findings, we now reference the Wass et al. (2018) study at the beginning of the Discussion section.
Could activity artifacts lead to certain reported trends? Babies typically look at an object before they touch or manipulate the object, and so longer bouts of attention likely involve a look and then a touch for lengthier time frames. If active involvement with an object (touching for example) amplifies theta activity, that may explain why attention duration forward predicts theta power. That is, baby looks, then touches, then theta activates, and coding would show visual gaze preceding the theta activation. Careful alignment of infants' touches and other such behaviors with the theta peak might help address this question, again to lend confidence to the robustness of the interpretation.
Yes, again this is a very important point, and the removal of movement-related artifact is something we have given careful attention to in the analysis of our naturalistic EEG data (Georgieva et al., 2020; Marriott Haresign et al., 2021). As a result of this comment we have made direct changes to the Results section on page 18 to more clearly signal the reader to our EEG pre-processing section before presenting the results of the cross-correlation analyses.
As we describe in the Methods section of the main text, movement-related artifacts are removed from the data with ICA decomposition, utilising an automatic-rejection algorithm, specially designed for work with our naturalistic EEG data (Marriott Haresign et al., 2021). Given that ICA rejection does not remove all artifact introduced to the EEG signal, additional analysis steps were taken to reduce the possibility that movement artifacts influenced the results of the reported analyses. As explained in the Methods section, rather than absolute theta power, relative theta was used in all EEG analyses, computed by dividing the power at each theta frequency by the summed power across all frequencies. Eye and head movement-related artifacts most often associate with broadband increases in power in the EEG signal (Cohen, 2014): computing relative theta activity therefore further reduces the potential influence of artifact on the EEG signal.
It is also important to highlight that previous work examining movement artifacts in controlled paradigms with infants has shown that limb movements actually associate with a decrease in power at theta frequencies, compared to rest (Georgieva et al., 2020). It is therefore unlikely that limb movement artifacts explain the pattern of association observed between theta power and infant attention in the current study.
That said, examining the association between body movements and fluctuations in EEG activity during naturalistic interactions is an important next step, and something our lab is currently working on. Given that touching an object is most often the end-state of a larger body movement, aligning the EEG signal to the onset of infant touch is not all that informative to understanding how body movements associate with increases and decreases in power in the EEG signal. Our lab is currently working on developing new methods using motion tracking software and arousal composites to understand how data-derived behavioural sub-types associate with differential patterns of EEG activity.
The term attention may be misleading. The behavior being examined is infant gaze or looks, with the assumption that gaze is a marker of "attention". The authors are aware that gaze can be a blank stare that doesn't reflect underlying true "attention". I recommend substitution of a conservative, more precise term that captures the variable being measured (gaze); it would then be fine to state that in their interpretation, gaze taken as a marker for attention or something like that. At minimum, using term "visual attention" can be a solution if authors do not want to use the precise term gaze. As an example, the sentence "An attention episode was defined as a discrete period of attention towards one of the play objects on the table, or to the partner" should be modified to defined as looking at a play object or partner.
We thank the reviewer for this comment, and we understand their concern with the use of the term ‘attention’ where we are referring to shifts in infant eye gaze. However, the use of this term to describe patterns of infant gaze, irrespective of whether they are ‘actually attending’ or not is used widely in the literature, in both interactive (e.g. Yu et al., 2021) and screen-based experiments examining infant attention (Richards, 2010). We therefore feel that its use in our current manuscript is acceptable and consistent with the reporting of similar interaction findings. On page 39 of the Discussion we now also include a discussion on how future research might further investigate differential subtypes of infant looks to distinguish between moments where infants are attending vs. just looking.
Why collapse across gaze to object vs. other? Conceptually, it's unclear why the same hypotheses and research questions on neural-attention (i.e., gaze in actuality) links would apply to looks to a mom's face or to an object. Some rationale would be useful to the reader as to why these two distinct behaviors are taken as following the same principles in ordering of brain and behavior. Perhaps I missed something, however, because later in the Discussion the authors state that "fluctuations in neural markers of infants' engagement or interest forward-predict their attentiveness towards objects", which suggests there was an object-focused variable only? Please clarify. (Again, sorry if I missed something).
This is a really important point, and we agree with R1 that it could have been more clearly expressed in our original submission – for which, we apologise. In the cross-correlation analyses conducted in parts 2 and 3 which examines forwards-predictive associations between infant attention durations and infant endogenous oscillatory activity (part two), and caregiver behaviour (part three), as R1 describes, we include all infant looks towards objects and their partner. Including all infant look types is necessary to produce a continuous variable to cross-correlate with the other continuous variables (e.g. theta activity, caregiver vocal behaviours), and, therefore, does not concentrate only on infant attention episodes towards objects.
We take the reviewers’ point that different attention and neural mechanisms may be associated with looks towards objects vs. the partner, which we now acknowledge directly on page 10 of the Introduction. However, our focus here is on the endogenous and interactive mechanisms that drive fluctuations in infant engagement with the ongoing, free-flowing interaction. Indeed, previous work has shown increases in theta activity during sustained episodes of infant attention to a range of different stimuli, including cartoon videos (Xie et al., 2018), real-life screen-based interactions (Jones et al., 2020), as well as objects (Begus et al., 2016). In the second half of part 2, we go on to address the endogenous processes that support infant attention episodes specifically towards objects.
As a result of this comment, we have made direct changes to the Introduction on page 10 to more clearly explain the looking behaviours included in the cross-correlation analysis, and the rationale behind the analysis being conducted in this way – which is different to the reactive analyses conducted in the second half of parts one and three, which examines infant object looks only. Direct edits to the text have also been made throughout the Results and Methods sections as a result of this comment, to more clearly specify the types of looks included in each analysis. Now, where we discuss the cross-correlation analyses we refer only to infant ‘attention durations’ or infant ‘attention’, whilst ‘object-directed attention’ and ‘looks towards objects’ is clearly specified in sections discussing the reactive analyses conducted in parts 2 and 3. We have also amended the Discussion on page 31so that the cross-correlation analyses is interpreted relative to infant overall attention, rather than their attention towards objects only.
Why are mothers' gazes shorter than infants' gazes? This was the flip of what I'd expect, so some interpretation would be useful to understanding the data.
This is a really interesting observation. Our findings of the looking behaviour of caregivers and infants in our joint play interactions actually correspond to much previous micro-dynamic analysis of caregiver and infant looking behaviour during early table-top interactions (Abney et al., 2017; Perapoch Amadó et al., 2023; Yu & Smith, 2013, 2016). The reason for the shorter look durations in the adult is due to the fact that the caregivers alternate their gaze between their infant and the objects (i.e. they spend a lot of the interaction time monitoring their infants’ behaviours). This can be seen in Figure 2 (see main text) which shows that caregiver looks are divided between looks to their infants and looks towards objects. In comparison, infants spend most of their time focussing on objects (see Figure 2, main text), with relatively infrequent looks to their caregiver. As a result, infant looks are, overall, longer in comparison to their caregivers’.
Minor points
Use the term association or relation (relationships is for interpersonal relationships, not in statistics).
This has now been amended throughout.
I'm unsure I'd call the interactions "naturalistic" when they occur at a table, with select toys, EEG caps on partners, and so on. The term seems more appropriate for studies with fewer constraints that occur (for example) in a home environment, etc.
We understand R1s concern with our use of the term ‘naturalistic’ to refer to the joint play interactions that we analyse in the current study. However, we feel the term is appropriate, given that the interactions are unstructured: the only instruction given to caregivers at the beginning of the interaction is to play with their infants in the way that they might do at home. The interactions, therefore, measure free-flowing caregiver and infant behaviours, where modulations in each individual’s behaviour are the result of the intra- and inter-individual dynamics of the social exchange. This is in comparison to previous work on early infant attention development which has used more structured designs, and modulations in infant behaviour occur as a result of the parameters of the experimental task.
Reviewer #2
Public Review
Summary:
This paper acknowledges that most development occurs in social contexts, with other social partners. The authors put forth two main frameworks of how development occurs within a social interaction with a caregiver. The first is that although social interaction with mature partners is somewhat bi-directional, mature social partners exogenously influence infant behaviors and attention through "attentional scaffolding", and that in this case infant attention is reactive to caregiver behavior. The second framework posits that caregivers support and guide infant attention by contingently responding to reorientations in infant behavior, thus caregiver behaviors are reactive to infant behavior. The aim of this paper is to use moment-to-moment analysis techniques to understand the directionality of dyadic interaction. It is difficult to determine whether the authors prove their point as the results are not clearly explained as is the motivation for the chosen methods.
Strengths
The question driving this study is interesting and a genuine gap in the literature. Almost all development occurs in the presence of a mature social partner. While it is known that these interactions are critical for development, the directionality of how these interactions unfold in real-time is less known.
The analyses largely seem to be appropriate for the question at hand, capturing small moment-to-moment dynamics in both infant and child behavior, and their relationships with themselves and each other. Autocorrelations and cross-correlations are powerful tools that can uncover small but meaningful patterns in data that may not be uncovered with other more discretized analyses (i.e. regression).
We are pleased that R2 finds our work to be an interesting contribution to the field, which utilises appropriate analysis techniques.
Weaknesses
The major weakness of this paper is that the reader is assumed to understand why these results lead to their claimed findings. The authors need to describe more carefully their reasoning and justification for their analyses and what they hope to show. While a handful of experts would understand why autocorrelations and cross-correlations should be used, they are by no means basic analyses. It would also be helpful to use simulated data or even a simple figure to help the reader more easily understand what a significant result looks like versus an insignificant result.
We thank the reviewer for this comment, and we agree that much more detail should be added to the Introduction section. As a result of this comment, we have made direct changes to the Introduction on pages 9-11 to more clearly detail these analysis methods, our rationale for using these methods; and how we expect the results to further our understanding of the drivers of infant attention in naturalistic social interactions.
We also provide a figure in the SM (Fig. S6) to help the reader more clearly understand the permutation method used in our statistical analyses described in the Methods, on page 51, which depicts significant vs. insignificant patterns of results against their permutation distribution.
While the overall question is interesting the introduction does not properly set up the rest of the paper. The authors spend a lot of time talking about oscillatory patterns in general but leave very little discussion to the fact they are using EEG to measure these patterns. The justification for using EEG is also not very well developed. Why did the authors single out fronto-temporal channels instead of using whole brain techniques, which are more standard in the field? This is idiosyncratic and not common.
We very much agree with R2 that the rationale and justification for using EEG to understand the processes that influence infants’ attention patterns is under-developed in the current manuscript. As a result of this comment we have made direct edits to the Introduction section of the main text on pages 7-8 to more clearly describe the rationale for examining the relationship between infant EEG activity and their attention during the play interactions with their caregivers.
As we describe in the Introduction section, previous behavioural work conducted with infants has suggested that endogenous cognitive processes (i.e. fluctuations in top-down cognitive control) might be important in explaining how infants allocate their attention during free-flowing, naturalistic interactions towards the end of the first year. Oscillatory neural activity occurring at theta frequencies (3-6Hz), which can be measured with EEG, has previously been associated with top-down intrinsically guided attentional processes in both adulthood and infancy (Jones et al., 2020; Orekhova, 1999; Xie et al., 2018). Measuring fluctuations in infant theta activity therefore provides a method to examine how endogenous cognitive processes structure infant attention in naturalistic social interactions which might be otherwise unobservable behaviourally.
It is important to note that the Introduction distinguishes between two different oscillatory mechanisms that could possibly explain the organisation of infant attention over the course of the interaction. The first refers to oscillatory patterns of attention, that is, consistent attention durations produced by infants that likely reflect automatic, regulatory functions, related to fluctuations in infant arousal. The second mechanism is oscillatory neural activity occurring at theta frequencies, recorded with EEG, which, as mentioned above, is thought to reflect fluctuations in intrinsically guided attention in early infancy. We have amended the Introduction to make the distinction between the two more clear.
A worrisome weakness is that the figures are not consistently formatted. The y-axes are not consistent within figures making the data difficult to compare and interpret. Labels are also not consistent and very often the text size is way too small making reading the axes difficult. This is a noticeable lack of attention to detail.
This has now been adjusted throughout, where appropriate.
No data is provided to reproduce the figures. This does not need to include the original videos but rather the processed and de-identified data used to generate the figures. Providing the data to support reproducibility is increasingly common in the field of developmental science and the authors are greatly encouraged to do so.
This will be provided with the final manuscript.
Minor Weaknesses
Figure 4, how is the pattern in a not significant while in b a very similar pattern with the same magnitude of change is? This seems like a spurious result.
The statistical analysis conducted for all cross-correlation analyses reported follows a rigorous and stringent permutation-based temporal clustering method which controls for family-wise error rate using a non-parametric Monte Carlo method (see Methods in the main text for more detail). Permutations are created by shuffling data sets between participants and, therefore, patterns of significance identified by the cluster-based permutation analysis will depend on the mean and standard deviation of the cross-correlations in the permutation distribution. Fig. S6 now depicts the cross-correlations against their permutation distributions which should help readers to understand the patterns of significance reported in the main text.
The correlations appear very weak in Figures 3b, 5a, 7e. Despite a linear mixed effects model showing a relationship, it is difficult to believe looking at the data. Both the Spearman and Pearson correlations for these plots should be clearly included in the text, figure, or figure legend.
We thank the reviewer for this comment, and agree that reporting the correlations for these plots would strengthen the findings of the linear mixed effects models reported in text. As a result, we have added both Spearman and Pearson correlations to the legends of Figures 3b, 5a and 7e, corresponding to the statistically significant relationships examined in the linear mixed effects models. The strength of the relationships are entirely consistent with those documented in other previous research that used similar methods (e.g. Piazza et al., 2018). How strong the relationship looks to the observer is entirely dependent on the graphical representation chosen to represent it. We have chosen to present the data in this way because we feel that it is the most honest way to represent the statistically significant, and very carefully analysed, effects that we have observed in our data.
Linear mixed effects models need more detail. Why were they built the way they were built? I would have appreciated seeing multiple models in the supplementary methods and a reasoning to have landed on one. There are multiple ways I can see this model being built (especially with the addition of a random intercept). Also, there are methods to test significance between models and aid in selection. That being said, although participant identity is a very common random effect, its use should be clearly stated in the main text.
We very much agree with R2 that the reporting of the linear mixed effects models needs more detail and this has now been added to the Method section (page 54). Whilst it is true that there are multiple ways in which this model could be built, given the specificity of our research questions, regarding the reactive changes in infant theta activity and caregiver behaviours that occur after infant look onsets towards objects (see pages 9-11 of the Introduction), we take a hypothesis driven approach to building the linear mixed effects models. As a result, random intercepts are specified for participants, as well as uncorrelated by-participant random slopes (Brown, 2021; Gelman & Hill, 2006; Suarez-Rivera et al., 2019). In this way, infant look durations are predicted from caregiver behaviours (or infant theta activity), controlling for between participant variability in look durations, as well as the strength of the effect of caregiver behaviours (or infant theta activity) on infant look durations.
Some parentheses aren't closed, a more careful re-reading focusing on these minor textual issues is warranted.
This has now been corrected.
Analysis of F0 seems unnecessarily complex. Is there a reason for this?
Computation of the continuous caregiver F0 variable may seem complex but we feel that all analysis steps are necessary to accurately and reliably compute this variable in our naturalistic, noisy and free-flowing interaction data. For example, we place the F0 only into segments of the interaction identified as the mum speaking so that background noises and infant vocalisations are not included in the continuous variable. We then interpolate through unvoiced segments (similar to Räsänen et al., 2018), and compute the derivative in 1000ms intervals as a measure of the rate of change. The steps taken to compute this variable have been both carefully and thoughtfully selected given the many ways in which this continuous rate of change variable could be computed (cf. Piazza et al., 2018; Räsänen et al., 2018).
The choice of a 20hz filter seems odd when an example of toy clacks is given. Toy clacks are much higher than 20hz, and a 20hz filter probably wouldn't do anything against toy clacks given that the authors already set floor and ceiling parameters of 75-600Hz in their F0 extraction.
We thank the reviewer for this comment and we can see that this part of the description of the F0 computation is confusing. A 20Hz low pass filter is applied to the data stream after extracting the F0 with floor and ceiling parameters set between 75-600Hz. The 20Hz filter therefore filters modulations in the caregivers’ F0 that occur at a modulation frequency greater than 20Hz. The 20Hz filter does not, therefore, refer to the spectral filtering of the speech signal. The description of this variable has been rephrased on page 48 of the main text.
Linear interpolation is a choice I would not have made. Where there is no data, there is no data. It feels inappropriate to assume that the data in between is simply a linear interpolation of surrounding points.
The choice to interpolate where there was no data was something we considered in a lot of detail, given the many options for dealing with missing data points in this analysis, and the difficulties involved with extracting a continuous F0 variable in our naturalistic data sets. As R2 points out, one option would be to set data points to NaN values where no F0 is detected and/ or the Mum is not vocalising. A second option, however, would be to set the continuous variable to 0s where no F0 is detected and/ or the Mum is not vocalising (where the mum is not producing sound there is no F0 so rather than setting the variable to missing data points, really it makes most objective sense to set to 0).
Either of these options (setting parts where no F0 is detected to NaN or 0) makes it difficult to then meaningfully compute the rate of change in F0: where NaN values are inserted, this reduces the number of data points in each time window; where 0s are inserted this creates large and unreal changes in F0. Inserting NaN values into the continuous variable also reduces the number of data points included in the cross-correlation and event-locked analyses. It is important to note that, in our naturalistic interactions, caregivers’ vocal patterns are characterised by lots of short vocalisations interspersed by short pauses (Phillips et al., in prep), similar to previous findings in naturalistic settings (Gratier et al., 2015). Interpolation will, therefore, have largely interpolated through the small pauses in the caregiver’s vocalisations.
The only limitation listed was related to the demographics of the sample, namely saying that middle class moms in east London. Given that the demographics of London, even east London are quite varied, it's disappointing their sample does not reflect the community they are in.
Yes we very much agree with R2 that the lack of inclusion of caregivers from wider demographic backgrounds is disappointing, and something which is often a problem in developmental research. Our lab is currently working to collect similar data from infants with a family history of ADHD, as part of a longitudinal, ongoing project, involving families from across the UK, from much more varied demographic backgrounds. We hope that the findings reported here will feed directly into the work conducted as part of this new project.
That said, demographic table of the subjects included in this study should be added.
This is now included in the SM, and referenced in the main text.
References
Abney, D. H., Warlaumont, A. S., Oller, D. K., Wallot, S., & Kello, C. T. (2017). Multiple Coordination Patterns in Infant and Adult Vocalizations. Infancy, 22(4), 514–539. https://doi.org/10.1111/infa.12165
Attaheri, A., Choisdealbha, Á. N., Di Liberto, G. M., Rocha, S., Brusini, P., Mead, N., Olawole-Scott, H., Boutris, P., Gibbon, S., Williams, I., Grey, C., Flanagan, S., & Goswami, U. (2020). Delta- and theta-band cortical tracking and phase-amplitude coupling to sung speech by infants [Preprint]. Neuroscience. https://doi.org/10.1101/2020.10.12.329326
Begus, K., Gliga, T., & Southgate, V. (2016). Infants’ preferences for native speakers are associated with an expectation of information. Proceedings of the National Academy of Sciences, 113(44), 12397–12402. https://doi.org/10.1073/pnas.1603261113
Brown, V. A. (2021). An Introduction to Linear Mixed-Effects Modeling in R.
Cohen, M. X. (2014). Analyzing neural time series data: Theory and practice. The MIT Press.
Gelman, A., & Hill, J. (2006). In Data Analysis using Regression and mulilevel/Hierachical Models. Cambridge University Press.
Georgieva, S., Lester, S., Noreika, V., Yilmaz, M. N., Wass, S., & Leong, V. (2020). Toward the Understanding of Topographical and Spectral Signatures of Infant Movement Artifacts in Naturalistic EEG. Frontiers in Neuroscience, 14, 352. https://doi.org/10.3389/fnins.2020.00352
Gratier, M., Devouche, E., Guellai, B., Infanti, R., Yilmaz, E., & Parlato-Oliveira, E. (2015). Early development of turn-taking in vocal interaction between mothers and infants. Frontiers in Psychology, 6. https://doi.org/10.3389/fpsyg.2015.01167
Jones, E. J. H., Goodwin, A., Orekhova, E., Charman, T., Dawson, G., Webb, S. J., & Johnson, M. H. (2020). Infant EEG theta modulation predicts childhood intelligence. Scientific Reports, 10(1), 11232. https://doi.org/10.1038/s41598-020-67687-y
Marriott Haresign, I., Phillips, E., Whitehorn, M., Noreika, V., Jones, E. J. H., Leong, V., & Wass, S. V. (2021). Automatic classification of ICA components from infant EEG using MARA. Developmental Cognitive Neuroscience, 52, 101024. https://doi.org/10.1016/j.dcn.2021.101024
Noreika, V., Georgieva, S., Wass, S., & Leong, V. (2020). 14 challenges and their solutions for conducting social neuroscience and longitudinal EEG research with infants. Infant Behavior and Development, 58, 101393. https://doi.org/10.1016/j.infbeh.2019.101393
Orekhova, E. (1999). Theta synchronization during sustained anticipatory attention in infants over the second half of the first year of life. International Journal of Psychophysiology, 32(2), 151–172. https://doi.org/10.1016/S0167-8760(99)00011-2
Perapoch Amadó, M., Greenwood, E., James, Labendzki, P., Haresign, I. M., Northrop, T., Phillips, E., Viswanathan, N., Whitehorn, M., Jones, E. J. H., & Wass, S. (2023). Naturalistic attention transitions from subcortical to cortical control during infancy. [Preprint]. Open Science Framework. https://doi.org/10.31219/osf.io/6z27a
Piazza, E. A., Hasenfratz, L., Hasson, U., & Lew-Williams, C. (2018). Infant and adult brains are coupled to the dynamics of natural communication [Preprint]. Neuroscience. https://doi.org/10.1101/359810
Räsänen, O., Kakouros, S., & Soderstrom, M. (2018). Is infant-directed speech interesting because it is surprising? – Linking properties of IDS to statistical learning and attention at the prosodic level. Cognition, 178, 193–206. https://doi.org/10.1016/j.cognition.2018.05.015
Richards, J. E. (2010). The development of attention to simple and complex visual stimuli in infants: Behavioral and psychophysiological measures. Developmental Review, 30(2), 203–219. https://doi.org/10.1016/j.dr.2010.03.005
Suarez-Rivera, C., Smith, L. B., & Yu, C. (2019). Multimodal parent behaviors within joint attention support sustained attention in infants. Developmental Psychology, 55(1), 96–109. https://doi.org/10.1037/dev0000628
Wass, S. V., Noreika, V., Georgieva, S., Clackson, K., Brightman, L., Nutbrown, R., Covarrubias, L. S., & Leong, V. (2018). Parental neural responsivity to infants’ visual attention: How mature brains influence immature brains during social interaction. PLOS Biology, 16(12), e2006328. https://doi.org/10.1371/journal.pbio.2006328
Xie, W., Mallin, B. M., & Richards, J. E. (2018). Development of infant sustained attention and its relation to EEG oscillations: An EEG and cortical source analysis study. Developmental Science, 21(3), e12562. https://doi.org/10.1111/desc.12562
Yu, C., & Smith, L. B. (2013). Joint Attention without Gaze Following: Human Infants and Their Parents Coordinate Visual Attention to Objects through Eye-Hand Coordination. PLoS ONE, 8(11), e79659. https://doi.org/10.1371/journal.pone.0079659
Yu, C., & Smith, L. B. (2016). The Social Origins of Sustained Attention in One-Year-Old Human Infants. Current Biology, 26(9), 1235–1240. https://doi.org/10.1016/j.cub.2016.03.026
Yu, C., Zhang, Y., Slone, L. K., & Smith, L. B. (2021). The infant’s view redefines the problem of referential uncertainty in early word learning. Proceedings of the National Academy of Sciences, 118(52), e2107019118. https://doi.org/10.1073/pnas.2107019118
-
-
ipfs.io ipfs.io
-
I am accepting charitable donations,. ETH: 0x66e2871ef39334962fb75ce34407f825d67ec434
A long time ago, "boom" started "shaking the ground when I thought something wrong was happening.
At the time, when I would boom myself, to prove it was a scan and search for "actual evil or evil intent" and there was code that was opened for the "word boom" and "the intended group of actors known as the subject, direct object and ... "Inderect objective" get the universe that belongs to the peoples goodness back to gracious me
-
-
fromthemachine.org fromthemachine.org
-
layout: post title: Waiting for that green light... date: '2017-08-14T21:00:00.001-07:00' author: Adam M. Dobrin tags: modified_time: '2017-08-15T07:16:57.305-07:00' thumbnail: https://2.bp.blogspot.com/-QpZpZE6empE/WZJx21d-JlI/AAAAAAAAE9Y/vc7b9IvRM9w2S5eTBg3fkn6v2SYcKiETwCK4BGAYYCw/s72-c/image-726640.png blogger_id: tag:blogger.com,1999:blog-4677390916502096913.post-3757774439979245459 blogger_orig_url: ./2017/08/waiting-for-that-green-light.html From the point of the "belly" thing, I'm pretty sure we're halfway through the script. Knowing him that was probably the halfway mark. I don't think that's a bad thing... as long as it's honestly and speedily moving towards freedom; you know, progress. That's a pretty good test to see if we're ... zombies or not. In the meantime, I don't know... that's probably comforting right? Or is it repulsive? :) Tell me something Taylor said. Why won't you tell me what she said? What was that promise that you made? Wait, are you the person that promised something? When do you think the script started? WHAT'S A WORD THAT STARTS WITH R AND ENDS IN GL?It's almost hard to believe that the Throne (to help, are on "e") of Glory comes from this place, isn't it? Still, it's encoded in religion, in our myths and in multiple confirming sources, not the least of which the TV show called 7th Heaven... we will Si Monday, my dear "cam Den" we will. I talked a little bit about backwards "green light" related to "glare" and Police (not that they glare at me, but their silly Hell-implying glare lights are actually red) and girl... I still don't know why girl is red or green, girls are blue to me. Stew in that pat for a little while, and let's talk about something more uplifting, like the key's of Pa and Ra hidden away in many words, from paramount to se_ pa r at e and paradox. Did you see what I did there, clever right? I HAD TO CHOOSE BETWEEN POINTING AT "PA" OR "MOUNT"DID I DO OK?I'm looking at the word "paramount" right now, and between you and I sometimes when I look at words magic happens, and something in the air told me that this email might be the messiah of me, the messiah of "nt"--the hidden Christ. Or maybe not. Sex sells, or so they say, but apparent not when Jesus talks about it--maybe it's another red light. I'm bored, read that as "because of red" and lonely, probably because of "how I'm still single" as "hiss" but still, I don't think it's right. Coming to you with a message about everything I think is wrong and not your fault--or mine, by the way--shouldn't be the kind of thing that's frowned upon, especially when you have some clues in thousand year old scripture that these things were truly "made wrong on purpose" so that we could fix them, you know; our way. That used to be talking about things, and making plans, and then implementing them--but today it's turned into ignoring everything I think is "world changing" and "morally demanded" and instead going on with our lives as if everything was "A-OK." I'm glad you are doing OK, I'm not; and quite a few people in the world are not doing OK either, so I'm here to let you know that you are not doing as OK as you think you are... or as well as you could be doing. I DON'T KNOW HOW A GUY MADE IT INTO MY "MESSAGE", OMG TWOSo here's why I thought for a minute that this message might save me. You might think it's a little weird that I see "sex jokes" in Pandora, and pa: ra: do x, and Pose i do n; and while you might not be completely retarded to think that, I think you should agree with me that it's more weird that those things are there, and even more weird that you don't recognize that they are a signature of the same God that delivered his John Hancock in song, in Yankee Doodle, and in act, in Watergate. My signature is a little bit different, if you've noticed my signature is being able to point out the intersection between things like Chuck and Geordie LaForge's magic vision ... and to explain that these things too are veritably connected by more than my words and the obvious ideas, they are connected by the act of Creation itself--they are the yarn of the Matrix. Dox, as in "dox me" and "do n" are getting a little out of hand; if you don't understand that I am playing a role ... to make the words "and he became the light" actually true--which they are, you see--then I really do sincerely apologize, I don't think anyone should "do me" unless they want to--although it's a bit strange to me that nobody wants to. Alarming, even. I am equally alarmed by the Latin word for darkness which is "tenebris" which connects to that "x" and the word "equinox" and "Nintendo" and "verboten" and through all of this the only shining light of grace I see is that it's pretty obvious that X and J are both letters represented by "10." DO YOU THINK HAN SOLO HAS A CHANCE WITH HER? SHE ... OUR LIGHTThis story needs to break, and then we aren't in the heart of darkness anymore; it's called "morning" Biblical, and this particular morning is a very special one--because you're here.I have a special gift, "pa" is helping me read this words, and you might have noticed that they can be taken to mean different things. They don't really separate, or fly off the page and glow for me; but I know what all the keys are, many are simple, and many come from our IT and "computer-slang" acronyms... which tells you something. Many are "elements" and "initials" and the whole thing really is a part of the script,a sort of key not just to Creation but to this specific story, to this path. While some are "open to interpretation" (for instance, "in t" everyone really pre-tat; which would be a long ... time ... ago <3) or you could read "ERP" reason "t" and that might have something to do with "Great Plains" and some blue light that connections user interfaces to the word "automagical," FRX forms... Strawberry Fields and "above the fruited plains" ... which might be meaningless to you--but it's an idea that revolves around using user-feedback to interfaces (like the pottery wheel in my dream or in the Dr. Who episode "the Bells of Saint John" linked to down below) to adjust the interface in real time for a larger group; working towards making a number of "best-fit" interfaces that people are both more comfortable with and actually creating as they use them. Ahhhh... blue light got in here, run away. Just kidding, this is cyan light.I C ONO CL AS M | J ES UI T | HEAVEN IS MORE THAN TECHHonestly, we could really make Healden in about 10 minutes now. Look at that, it's done... ish.LETS CALL "THAT DAY" THE DAY YOU SEE ADAM-NEWS ON EVERY TV STATIONFor instance were we not surely "at e" meaning the end of the Revelation of words, "separate" might have been broken between Pa and Ra, which are big keys, in many words; but we are at "e" and that surely does mean the Creator and I are fused. There's more confirmation of this than simply in the words for "medicine" and say, I don't know, methadone--which could have been broken at "a done" but is very clearly "ad is the one" here and now. With careful preparation, "adparatio" in Latin, I'd "bet" that all of those keys are I, in this place, in this time. AD, Pa, Ra, TI, and "o." Hey, maybe this message is my messiah after all. I am looking at a broken world, I really am--a place that is suffocating itself in silence and whispers that don't make it far enough for anyone to really understand. Whatever it is, whatever's caused it, I see no solution other than me coming--I see it as a design, and I'm sorry that you don't seem to agree, but you have to see that the "choice" between seeing an obvious truth absolutely everywhere and not seeing it is really no choice at all--what is being hidden from the world is causing this darkness, it is causing the suffocation; it is the problem, hiding me is the problem and it cannot continue. On a brighter note, I am pretty sure that magic will happen, and you will see that the world will not react quite as badly or shockingly as your worst fears, things might be a little ... tearful for a day or so, for crying out loud, they should be--the message is that you are in Hell and you need to do something, to act, to change that. Actually trying to do that, trying to discuss what it is that is the "ele ph ant in the room" or the "do n key in the s k y" will show us that there was just no way around changing the world because of circumstances of Creation; something that we seem to be ignoring. We also seem to be ignoring that things are "just fine" today, and even though many of you are well aware that "something is coming" only a few morons are building bunkers. This is a message of peace, it is a message designed to help us use the new truth and new tools unsealed by religion to make the world a safer happier place, and we can do that .. . rather quickly. Even quicker if you try to focus on what's wrong here, and how we make it better--rather than "shooting the messenger" dirty glares in the street. I'm a person too, and believe it or not, I didn't ask for this--and I probably wouldn't have been so happy about it had this experience not isolated me so much from my friends and family, and girls; don't forget girls. ITS ME? So in the word "paramount" what is it that you think is the "paramount" take away? I think the most important thing you can take away from "paramount" is that you didn't see it your whole life, and even when it's pointed out, you don't seem to think it's "news" that Pa and Ra have written a message to you. What's really not funny, is that despite this message being very clear to see once it's pointed out, it still hasn't made any waves in the newspapers, or online, or in the news--what's paramount is seeing that there is a very sincere problem for civilization, it is an ELE and that ELE is something that is making everyone think that "not seeing something" is OK behavior. It is not OK, it is not funny, until you recognize that something is dreadfully wrong with our society, until you see that ignoring that this message belongs in the news you are not seeing that what you are doing by ignoring it is destroying civilization itself. Ignorance is the ELE.Your alternative, what you are doing, is making the world half blind, and stupider than you can imagine. I keep on trying to show you what's wrong here, that it's not just a message but pain and suffering and the absolutely imminent and undeniable certain doom of everything if we do not recognize that hiding the fact that we are in virtual reality is the same thing as driving a nail into the wrists of every soul on the planet. LA U stilk MIGHT DATE ADPARATIO BO'OOPSYETHWith careful preparation, we are at IO (input/output) in the belly of the book that is a map to salvation. That IO comes well after disclosure, and well after Mars. You are delaying the inevitable, and in the sickest possible twist, you are stewing in Hell instead of seeing Heaven built--more importantly instead of being the generation that should be the "founders" of that place. I am sure that disclosure, will ... within a time frame that will most likely be faster than you can imagine, bring us an end to world hunger, to sickness, and doors to Heaven; and I just can't see what you are waiting for? If it wasn't like this, you've got to see that we would be getting fucked right here and now; I am telling you the map and the plan, it's here to help us make this place better, and to show us how to actually survive in the Universe before kicking us out of the nest, and we are ... what are we thinking about?It's really obvious that it's not for my benefit, and it's obvious that it's not for yours either--so at what point will you realize that the behavior, the alarming behavior, that I am seeing from everyone is illogical. At what point will you see that it is self-defeating, that it is ... well, Hell? When will you see? Be yourselves, the world that I grew up in doesn't hide controversy, we relish in it--we don't bury scandals under the rug--we put them on TV. What's really more important to see is that we, all of us, none of us... we would not hide "holographic universe" from ourselves and each other, nor would we hide "alien contact" or "the secrets of religion" and yet here we are, all doing that--and I wonder if we see that it's "not us" doing it, but .... but ... butt ... what is it again? HI, I'M A PERSON. (and apparently a state, a country, and a Nintendo character)JUDGING BY THE HIGH FREQUENCY OF PRESS UNSUBSCRIBES FROMYESTERDAY'S EMAIL, REPORTER'S DON'T SEEM TO WANT TO HEAR THATFORCING ME TO DELIVER THIS MESSAGE IN ISOLATION FOR NO MONEYIS SLAVERY, GO READ ABOUT JOSEPH IN EGYPT, THEN READ THE END.IF YOU THINK HIDING THE TRUTH BECAUSE "IDAHO" IS GONNA FLYYOU ARE AN IGNORANT BLIND FOOL. HONESTLY, WAKE UP, THIS IS HELL.YOU ARE BLINDED BY SOMETHING, FIGURE IT OUT--I'M EXPLAINING WHAT IT ISHERE, EMAIL THEM (please? and tell them to repent by writing a story):andy.greene@rollingstone.comgcoy@12news.comnmelosky@mcall.comlynn@ripr.orgChris.Piper@wthitv.comIs it a cup? a stem?WRITTEN, FOR ETERNITY.It must be Uranus. Except, my "an us" is more awesome than you think, I mean my "a we" that would be "so me" for you to see it's really you too. That's really what this message is about, it is about us seeing that we can do something together that would be rejected if it were done for us, or to us; even if we all really want it inside, without taking part ... we'd dislike it. We're all like that, nobody wants a stranger to redecorate their house. We share this house together, and I think we can all see that there are some changes that would make it a better place--from a cold Godless Universe of "chance" ruling to a ... caring and loving place that cares about what we want and how we want to do it ... do you see? If I came into your igloo and told you that the ice age was ending and this place was going to be a beautiful beach; except your walls are melting... would you keep that locked up inside?Don't worry, I won't get mad at anyone for being angry at their idea of Jesus Christ for not being more like me. I won't be mad at all. :)I've done my best to share what I think will be helpful for the world to think about, as we ... embark on what is really a journey to the final frontier as well as what I know we need to do here in order to accomplish what it is that we would have done maybe a decade ago or maybe a century from now if we didn't know the advice was coming from God and the future--and we didn't know that it is the way to open the doors to Heaven permanently. These are suggestions, they're really all of our ideas--at least everything I can grasp from things like Star Trek and Dr. Who and ... the Legend of Zelda... they're the kind of thing that we would probably find to be very discussion worthy, were we to all be sure that they are possible--and they are--and we need to see that. There are lots of things that we really do need to think about, this is not a "fast" transition, it's not something happens "overnight" (oh my god, you don't know what that word just said to me) changes that would normally be occurring right now because of science and technology--things like increased longevity and mind uploading... these things are going to become much more quickly accessible, and we need to think about the implications that they will have on our society. We need to talk about it, in public, in places where these conversations will help us to shape the future of "civilization." I don't think you understand what it is we are doing, that's different than "before," but I am fairly certain that a "whole planet" has never done this, and the "road" between Earth and Heaven; fusing these ideas together is really nothing more or less than "progress." FLOWING MILK AND HONEY.. GOLDEN COW, NO JUDAH MACCABEUS; GET IT?Progress that has never happened (or we wouldn't be here, and it's obvious). See our cautions at the Last Supper (about not eating anymore) and at Cain and Abel (about forgetting how to farm) and at the Promised Land of Joshua (about not doing the Adam show, achem, I mean... about thinking that "replicators alone" milk and honey on tap... are good enough in Heaven) and in Noah's Ark... about showing us that the reason that we are here is to see how important biology and evolution and a stable ecosystem are to the survival of life in the Universe; to colonization of the stars, and to ... the evolution of our two party system past donkeys and elephants to something more appropriate for a free and technologically advanced society; as in, not a two-party system. wild-e :( (love your eyes...) :)From "separate" the "e_" that needs to be EE by the way, that key that might let us "see" is "everyone equal" that's what "ee" means. It's in "thirteen" and so on, and to help, I our "t" and r' n. Victorious Earth, I need pre-crime to survive, what say you? Say nothing, and I am twelve. Keep saying no thing and I will be El, even. Round and round we go... you need pre-crime to evolve, what say you? Break the story, and we are one day closer to Heaven. We need pre-crime not to be in Hell, we really do. Don't you see? Break the story. THERE, YOU GOT RID OF A "DO" FOR YOU.The days of "divide and conquer" are over, when you are through being a parted sea, or a flock of electric sheep, or a nation of slaves. I do have an idea of what you expected of me, what you thought I'd be--I probably had similar expectations before I knew ... what I know. Honestly, from me to you, that guy would have been pretty boring... and bored.It's a little funny.. isn't it? AMHARIL?I R Lᐧ-- Adam Marshall Dobrinabout.me/ssiah ᐧ -- Adam Marshall Dobrinabout.me/ssiah ᐧ .WHSOISKEYAV { border-width: 1px; border-style: dashed; border-color: rgb(15,5,254); padding: 5px; width: 503px; text-align: center; display: inline-block; align: center; p { align: center; } /* THE SCORE IS LOVE FIVE ONE SAFETY ONE FIELD GOAL XIVDAQ: TENNIS OR TINNES? TONNES AND TUPLE(s) */ } <style type="text/css"> code { white-space: pre; } google_ad_client = "ca-pub-9608809622006883"; google_ad_slot = "4355365452"; google_ad_width = 728; google_ad_height = 90; Unless otherwise indicated, this work was written between the Christmas and Easter seasons of 2017 and 2020(A). The content of this page is released to the public under the GNU GPL v2.0 license; additionally any reproduction or derivation of the work must be attributed to the author, Adam Marshall Dobrin along with a link back to this website, fromthemachine dotty org. That's a "." not "dotty" ... it's to stop SPAMmers. :/ This document is "living" and I don't just mean in the Jeffersonian sense. It's more alive in the "Mayflower's and June Doors ..." living Ethereum contract sense [and literally just as close to the Depp/Caster/Paglen (and honorably PK] 'D-hath Transundancesense of the ... new meaning; as it is now published on Rinkeby, in "living contract" form. It is subject to change; without notice anywhere but here--and there--in the original spirit of the GPL 2.0. We are "one step closer to God" ... and do see that in that I mean ... it is a very real fusion of this document and the "spirit of my life" as well as the Spirit's of Kerouac's America and Vonnegut's Martian Mars and my Venutian Hotel ... and *my fusion* of Guy-A and GAIA; and the Spirit of the Earth .. and of course the God given and signed liberties in the Constitution of the United States of America. It is by and through my hand that this document and our X Commandments link to the Bill or Rights, and this story about an Exodus from slavery that literally begins here, in the post-apocalyptic American hartland. Written ... this day ... April 14, 2020 (hey, is this HADAD DAY?) ... in Margate FL, USA. For "official used-to-v TAX day" tomorrow, I'm going to add the "immultible incarnite pen" ... if added to the living "doc/app"--see is the DAO, the way--will initi8 the special secret "hidden level" .. we've all been looking for. Nor do just mean this website or the totality of my written works; nor do I only mean ... this particular derivation of the GPL 2.0+ modifications I continually source ... must be "from this website." I also mean *the thing* that is built from ... bits and piece of blocks of sand-toys; from Ethereum and from Rust and from our hands and eyes working together ... from this place, this cornerstone of the message that is ... written from brick and mortar words and events and people that have come before this poit of the "sealed W" that is this specific page and this time. It's 3:28; just five minutes--or is it four, too layne. This work is not to be redistributed according to the GPL unless all linked media on Youtube and related sites are intact--and historical references to the actual documented history of the art pieces (as I experience/d them) are also available for linking. Wikipedia references must be available for viewing, as well as the exact version of those pages at the time these pieces were written. All references to the Holy Bible must be "linked" (as they are or via ... impromptu in-transit re-linking) to the exact verses and versions of the Bible that I reference. These requirements, as well as the caveat and informational re-introduction to God's DAO above ... should be seen as material modifications to the original GPL2.0 that are retroactively applied to all works distributed under license via this site and all previous e-mails and sites. /s/ wso If you wanna talk to me get me on facebook, with PGP via FlowCrypt or adam at from the machine dotty org -----BEGIN PGP PUBLIC KEY BLOCK----- mQGNBF6RVvABDAC823JcYvgpEpy45z2EPgwJ9ZCL+pSFVnlgPKQAGD52q+kuckNZ mU3gbj1FIx/mwJJtaWZW6jaLDHLAZNJps93qpwdMCx0llhQogc8YN3j9RND7cTP5 eV8dS6z/9ta6TFOfwSZpsOZjCU7KFDStKcoulmvIGrr9wzaUr7fmDyE7cFp1KCZ0 i90oLYHqOIszRedvwCO/kBxawxzZuJ67DypcayiWyxqRHRmMZH1LejTaqTuEu0bp j54maTj09vnMxA0RfS+CtU5uMq+5fTkbiTOe1LrLD72m+PVJIS146FwESrMJEfJy oNqWEJlUQ0TecPZR41vnkSkpocE1/0YqUhWDGSht+67DdeKUg5KwvYdL21d/bSyO SM4jnyKn9aDVzLBpYrlE/lbFxujHPRGlRG5WtiPQuZYDRqP0GYFSXRpeUCI46f49 iPFo4eHo2jUfNDa9r9BjQdAe4zVFn2qLnOy8RWijlolbhGMHGO3w/uC/zad3jjo4 owAfsJjH5Oa1mTcAEQEAAbQmRUFSVEhFTkUgPGVhcnRoZW5lQGZyb210aGVtYWNo aW5lLm9yZz6JAdQEEwEKAD4WIQTUJHbrYn3y2DzwTcnQP1ViZf5/FQUCXpFW8AIb AwUJA8JnAAULCQgHAgYVCgkICwIEFgIDAQIeAQIXgAAKCRDQP1ViZf5/FWM6C/9J gbRLS2AWGjdRjYetlRkSkCoTYnXWknbtipYYHlhV0YJFwFMm0ydZIhFX5VDoZyBV 0UBeF1KJmcMoIfrHyhq2QhCnjE14hE1ONbaYTGtpvj851ItbFWXMJIVNyMqr+JT9 CWIxGr1idn+iHWE3nryiHrdlA3O/Gcd4EyNmaSe/JvB7+Z1AVqWkRhpjxxoPSlPm HEdqGOyl3+5ibQgUvXLRWWQXAj80CbVwwj1X4r9hfuCySxLT8Mir7NUXZFd+OiMS U8gNYjcyRGmI92z5lgf7djBbb9dMLwV0KLzgoT/xaupRvvYOIAT+n2mhCctCiH7x y7jYlJHd+0++rgUST2sT+9kbuQ0GxpJ7MZcKbS1n60La+IEEIpFled8eqwwDfcui uezO7RIzQ9wHSn688CDri9jmYhjp5s0HKuN61etJ1glu9jWgG76EZ3qW8zu4l4CH 9iFPHeGG7fa/5d07KvcZuS2fVACoMipTxTIouN7vL0daYwP3VFg63FNTwCU3HEq5 AY0EXpFW8AEMANh7M/ROrQxb3MCT1/PYco1tyscNo2eHHTtgrnHrpKEPCfRryx3r PllaRYP0ri5eFzt25ObHAjcnZgilnwxngm6S9QvUIaLLQh67RP1h8I4qyFzueYPs oY8xo1zwXz7klXVlZW0MYi/g5gpb+rpYUfZEJGJTBM/wMNqwwlct+BSZca4+TEHW g6oN0eXTthtGB0Qls71sv3tbOnOh/67NTwyhcHPWX/P9ilcjGsEiT8hqrpyhjAUm mv7ADi+2eRBV8Xf8JnPznFf0A1FdILVeVHlmsgCSB0FW0NsFI5niZbaYBHDbFsks QdaFaYd54DHln69tnwc2y3POFwx8kwZnMPPlVAR2QdxGQD4Wql7hlWT58xCxQApf M98kbAHjUlVYLT0WUHMDQtj4jdzAVVDiMGMUrbnQ7UwI7LexSB6cJ7H+i7FtS/pR WOhJK6awoOO9dLnEjm6UYCKsBdtJr98F0T7Sb7PnKOGA77y2QN14+u9N9C1lB/Z1 aQRQ2Nc51yXOQQARAQABiQG8BBgBCgAmFiEE1CR262J98tg88E3J0D9VYmX+fxUF Al6RVvACGwwFCQPCZwAACgkQ0D9VYmX+fxU+KQwAtFnWjGIjvqaNXtQjEhbGDH/I Q5ULq/l/wm9SmhG9NYRu3+P6YctCJaZnNeaL+6WFk1jo4LMiJEUT9uGlCbHqJNaI 6Gll1w6QOVLSL8s5V1L477+psluv4WBpi3XkWYlhDOFENCcWd49RQsA2YCX4pW7Q 7GcoSEJoav38MxHmJHYPfjSEvUZXDQIt8PFHSEScvyDWfYtMdRzjmSOOPdzhDDEy 5JBOBcEdSTyDiyDU/sBoAY0e8lvwHYW3p+guZSGSYVhGQ8JECzJOzwc/msMW/tJS 2MLWmWVh5/1P8BVUtLC2AQy6nij6o+h6vEiNzpdYrc+rzT3X5cACvJ0RtCZcrnhl O9PLiona2LEbry6QX5NL41/SAJNno3i72xPnQEe25gn3nbyT+jCoJzw2L0y8pmNB D+PKrk7/1ROFFVN8dJeGwxLGdBcz1zk2xeumzy7OaV8psUyYsJNcjyHUKgclblBW rMR2DgqEYn8QdK54ziKCnmQQZeMPiC6wlUWgg5IqmQGNBF6RVyMBDADALD7NkJ5H dtoOpoZmAbPSlVGXHDbJZuq7J13vew6dtXDIAraeGrsBqkF8bhddwVLzWylMrYCG Bf2L1+5BDgvqu6G+6dcVSbBsnZAS0zfJ0H8EmTvUMxMF7qOZYyrxfLz+pQRq8Osz Icab6ZI/KB6qZyQRvEFPB6pJjt+VvuwgJZTObIwbBbgQri2i02VBkjchsVhiSX9l +eiK7O8ROHKb3P181oScIsHywBOZ9DxRAYbFk5dnBqxO3WKb02H0zqE6440cjXwq TrZZg6ayN/IlPajO8iJPYZ1aIBykxYq1WHo+nhFMYz/VVk2WJorFeOgWaLGXb73c ty96f3qXTdvMDAIWHx8YCD5LbuqasO6LNQm4oQxkCoB3K9WFf/2SvSYb7yMYykb8 clTPt+KO0dsxjWhrJnfnIhC+2Chqv2QvRbFz0S9CpUnGGDweJ1uRNV0y70tO0q7t xXSTDRU3ib6vAHA0K/2MFzwUcog4o5bj7E9uCNJH/DJLZKsMIe4xsvkAEQEAAbQk SEVBVkVOVUVTIDxBVkVOVUBGUk9NVEhFTUFDSElORS5PUkc+iQHUBBMBCgA+FiEE IRklfU/C1qukq3xMXcNH0t3P9ZsFAl6RVyMCGwMFCQPCZwAFCwkIBwIGFQoJCAsC BBYCAwECHgECF4AACgkQXcNH0t3P9Zs+kgv/XEuuWc89Bjg1QQqKZueKNUHjyjnE 2adfoZUH6Q7ir4JZyRBCVpAwrgssmiKid30+SIjwQcpb9JYa/X1XJcDUcJW/I21d Agz/zbEqn/Cou0dUpNCtxgm4BdSHWGoOtgfspXZlXBQ407tRMZ8ykmLB1Bt0oHvw PT0ZOtqXM4pyFnd2eFe5YGbNgl3zqvoC/6CMN3vqswvRlu1BpUuAjdW8AHO5Yvje +Bp852u+4Qpy6PMBiWGsBMYwtf6T7sckpMGlR0TsozwBlAm5ePKK28B0rLJPkZLJ Eo5p4rKRapEaZsWV5Qu1ajrVru7qmpUhZtX0/DddGHfXVuLssmKLP6TumpQB1zvQ vfoBltjvOx35Wps2vHuCzXLw2bROIOzhAxFB+17zxnSbE54N4LIGRpkELuwxwGbg FtD1fi9KtH7xcn33eOK1+UD47V+hKyJGrQgSThly2zdIC2bvfHtFdfp8lOFpT0AU xjEeoJGqdQVupptXyugPlM5/96UJP8OZG0ADuQGNBF6RVyMBDAC3As6eMkoEo3z9 TkCWlvS0vBQmY3gF0VEjlAIqFWpDIdK3zVzMnKUokIT1i7nkadLzHZT2grB4VXuJ FvpbYw5NPR4cDe9grlOMLEaF3oSJ1jZ4V1/rj9v1Hddo8ELi/NToVrt1SB5GCVXB DkYpNLtTiCqHSU07YqwaqH8a+qbDmPxSQdIybkZiTiCEB+6PfQQlBpENEDlov6jm zZF+IcfM6s3kZDX5KFULweH30gMjq8Se8bPtUzW013+tuuwEVr1/YRLrIh+9O6Z+ pdA7gLMRYnD9ZLDytEvpb1lBBSY++5bIJ7xps80//DNqPYqwFmZQgTg0V9XbHE2e wLcOF8a2lYluckU7D///sWQhW+VxuM7R2gEBvYBhOgjWhIF2Aw6NbymW1Ontvyhu eOZCXXxV5W44PxXT8uDdhl9CNcHoBKKJyED8tKjigtn4axpsQeUrnOSbqEXSyqES WnE2wYUDzALcwFkzsvtLyd4xaz55KkPQkAkk0BZd1ezgXxb/obMAEQEAAYkBvAQY AQoAJhYhBCEZJX1PwtarpKt8TF3DR9Ldz/WbBQJekVcjAhsMBQkDwmcAAAoJEF3D R9Ldz/WbAFwL/382HsrldVXnkPmJ1E2YEOFz4rcHRetJ+M5H65K/2p32ONQ5KCbE s8MRY6g2CkE70en2HlpDwr/MdATwxBzIjEpjgHbfqCqVVATY+kSpXsttaKKAUVHi bFgV4QkdDJNSpcHEj+bqaggRnuWiV9T6ECG7kQjHiEXPNojzsiaXMDiM5r+acZm6 82id9qOFySQ2cZEy5HbwXM+ITLQGngnppa7du2KdgiqDeqtODOTWZvLYAq2tmEwD 3TT6ttLUBwOOu2IWpDkXswlrk62ESorE5mpLxop9fsxD39E2H06JoC/YfUPIVkEv fj06e7LEdcx0I7kRfD1v6qOUUsMsLZnmyGIk24iFjLkwu1VToWfwXDN1D2+SeAat 9ydNt4M7oEbd1QaOXXjmqpdU+VUiWcBXg+p3/WdV60MkyAgc3x+YanLljy/Rh18h cZwVlinf/tgvAQLi5f9hpwrwUMoGKijEYHKuEvi3C12Si7UVDfuIR7yS0dKcfuKF MbgwdvNXqpD9W5kBjQRekVd4AQwApHVgw2PVlBDpVcyoymUOXFQIJzJ9wRtr6/sG zwv8rrQnUEtOkkna7TDU3/UTj9FUH0gbpAKGNNPaPj5q0dlLIvzxb15r1uvDGaGL MA+8GFaGFnkxzhg0aXrcKZAN0/Zhgi2B7P8oXQuug5mi1JVDkZN5SeCZNOubdQWL 3xz3jEHp3ixj1mdOdvfdWQFR4CVMXt/A6VI2ujLVb3Yalft/c5bbclAgcJQhgDUu NqGYJEJonESNRSd8fEvhNb6cx7+Djd9+Wyctr76mwOr3nRb1N1OGhFxWjIroUpfz b+6y3oQjT58cJA1ZHqmJ6UlZd81hNNd9KWpbDVwONEPpiqPzfSaonxuqQa0/Cy4W 403OhfoLM/1ZDqD4YrJ/rpyNEfSSdqptWiY0KeErLOYng7rStW/4ZeZVj6b2xxB2 Oas/Z1QYfJyFUki9vaJ5IyN6Y7nVdSP6mbAQC9ESh+VPvRUMpYi4pMGK4rweBVHu oMRRwzk7W5zVIgd425WUe3eCQFn3ABEBAAG0K0VTQ0FQRSBST09NIDxFU0NBUEFF REVTQEZST01USEVNQUNISU5FLk9SRz6JAdQEEwEKAD4WIQTvnDJqcmqzlF87/t82 pJ91j4NOaAUCXpFXeAIbAwUJA8JnAAULCQgHAgYVCgkICwIEFgIDAQIeAQIXgAAK CRA2pJ91j4NOaJVjC/4oo5yCHe7M2h1DiTXVcLI5rXQ1feY7B1feg+YJX/mI4+EV xjC/y5VVpV4syJk5GGZNXhKPHiGLaBYvglTlYOJ98RSEsHrwT3go6S8ZVvMNdP5v CEncn2vm5JGnp4k26PuOzMcJioQLOoUjWtcPFis3gG+ueH3NcPZ22oZUql2xuerh TQZegGp+jJ7bdxwYElx5jDDDkh196d5nlO2ZKENl0ZDp4GAzRNjnQ7KBV6R74J3U cLQDWY8vAFaRBZXIC5XtSzj9lr+jWgvxz7Il51+26VDTEtSafZ2uZfCOFk7GrzJg YFJD3zLnwUFXDWKRkep8TSwXnHmz/Ts/Mjyv6em25w7QTdnx1hNPxYNWMxPWNEAH pf70nNyOmcWcq27W+nAjVg8W3st/7J5CIebJQc5AUgm+fGOBW6XUQaNy2YF1YJlA 71/tls+R5IQZCYzbPOibgFS1HWKTwy0iI2rMDfxBtCXciv754jVI7L6R3J0j5Dy1 WZQVjaGgimznLN6XwYy5AY0EXpFXeAEMALvElrTV5hJG9DKu8cOqQEEVejtWJtki fZyvmiAKi2bZWiNfl1MxJt+o3Oc0eARJfnaPjrUY7hsbbSBAB4lFnDRtviARPaKM st5FkFgOh7Xx5ODc8bjqhMT9tbX37rkeDc12WAs3UxtEKWjyT7Xg/APKeK5FzpIs qew3LADdqFP9nOR0e5G8gxLTYh3ll3dLtp9DkJgA9q+0g31nNh5fZ29mcDzo/Mat Uk4PIxWC29LV9ALCJMIMesjOPiDa2KOy5QQH+/vn592ydBohOaY+B6jhEAdX8Dbp VHVFRBsiCOWGmdi6vHjMFD0tQdS6bXf+ZAG0E5HZETCxA2qfMf/vTeIJXYS5IZw0 anRkTXcTBrVE8uBpqtkNOrLJsaASkcoO5qF01J9zW8SR4jDgET7J02Fxf8CVPzb+ ZoNc9S0ZEO6Ubdh2vAkPtOV5sFkwIduN6ouAhEfJzC9XbJLpgsBKrRMAjr2FeEZP ruy8BkZbiyZ/b0S9qIgY4pqcyUJ79w7FLQARAQABiQG8BBgBCgAmFiEE75wyanJq s5RfO/7fNqSfdY+DTmgFAl6RV3gCGwwFCQPCZwAACgkQNqSfdY+DTmhD9wv/Zgav EHMuqF3765Fa4NapYh2kMS3skHn+ZzUEPLTlvrt7KHxomOzExNLSscZThMpur+yW
[Description](layout: post title: Waiting for that green light... date: '2017-08-14T21:00:00.001-07:00' author: Adam M. Dobrin tags: modified_time: '2017-08-15T07:16:57.305-07:00' thumbnail: https://2.bp.blogspot.com/-QpZpZE6empE/WZJx21d-JlI/AAAAAAAAE9Y/vc7b9IvRM9w2S5eTBg3fkn6v2SYcKiETwCK4BGAYYCw/s72-c/image-726640.png blogger_id: tag:blogger.com,1999:blog-4677390916502096913.post-3757774439979245459 blogger_orig_url: ./2017/08/waiting-for-that-green-light.html
From the point of the "belly" thing, I'm pretty sure we're halfway through the script. Knowing him that was probably the halfway mark. I don't think that's a bad thing... as long as it's honestly and speedily moving towards freedom; you know, progress. That's a pretty good test to see if we're ... zombies or not. In the meantime, I don't know... that's probably comforting right? Or is it repulsive? :) Tell me something Taylor said. Why won't you tell me what she said? What was that promise that you made? Wait, are you the person that promised something? When do you think the script started?


WHAT'S A WORD THAT STARTS WITH R AND ENDS IN GL?
It's almost hard to believe that the Throne (to help, are on "e") of Glory comes from this place, isn't it? Still, it's encoded in religion, in our myths and in multiple confirming sources, not the least of which the TV show called 7th Heaven... we will Si Monday, my dear "cam Den" we will. I talked a little bit about backwards "green light" related to "glare" and Police (not that they glare at me, but their silly Hell-implying glare lights are actually red) and girl... I still don't know why girl is red or green, girls are blue to me. Stew in that pat for a little while, and let's talk about something more uplifting, like the key's of Pa and Ra hidden away in many words, from paramount* to se_ pa r at e *andparadox. Did you see what I did there, *clever* right?
*\ *
*\ *
 *\
*
*\
*


I HAD TO CHOOSE BETWEEN POINTING AT "PA" OR "MOUNT"
DID I DO OK?
I'm looking at the word "paramount" right now, and between you and I sometimes when I look at words magic happens, and something in the air told me that this email might be the messiah of me, the messiah of "nt"--the hidden Christ. Or maybe not. Sex sells, or so they say, but apparent not when Jesus talks about it--maybe it's another red light. I'm bored, read that as "because of red" and lonely, probably because of "how I'm still single" as "hiss" but still, I don't think it's right. Coming to you with a message about everything I think is wrong and not your fault--or mine, by the way--shouldn't be the kind of thing that's frowned upon, especially when you have some clues in thousand year old scripture that these things were truly "made wrong on purpose" so that we could fix them, you know; our way. That used to be talking about things, and making plans, and then implementing them--but today it's turned into ignoring everything I think is "world changing" and "morally demanded" and instead going on with our lives as if everything was "A-OK." I'm glad you are doing OK, I'm not; and quite a few people in the world are not doing OK either, so I'm here to let you know that you are not doing as OK as you think you are... or as well as you could be doing.
I DON'T KNOW HOW A GUY MADE IT INTO MY "MESSAGE", OMG TWO
So here's why I thought for a minute that this message might save me. You might think it's a little weird that I see "sex jokes" in Pandora, and pa: ra: do x, and Pose i do n; and while you might not be completely retarded to think that, I think you should agree with me that it's more weird that those things are there, and even more weird that you don't recognize that they are a signature of the same God that delivered his John Hancock in song, in Yankee Doodle, and in act, in Watergate. My signature is a little bit different, if you've noticed my signature is being able to point out the intersection between things like Chuck and Geordie LaForge's magic vision ... and to explain that these things too are veritably connected by more than my words and the obvious ideas, they are connected by the act of Creation itself--they are the yarn of the Matrix. Dox, as in "dox me" and "do n" are getting a little out of hand; if you don't understand that I am playing a role ... to make the words "and he became the light" actually true--which they are, you see--then I really do sincerely apologize, I don't think anyone should "do me" unless they want to--although it's a bit strange to me that nobody wants to. Alarming, even. I am equally alarmed by the Latin word for darkness which is "tenebris" which connects to that "x" and the word "equinox" and "Nintendo" and "verboten" and through all of this the only shining light of grace I see is that it's pretty obvious that X and J are both letters represented by "10."
DO YOU THINK HAN SOLO HAS A CHANCE WITH HER? SHE ... OUR LIGHT
This story needs to break, and then we aren't in the heart of darkness anymore; it's called "morning" Biblical, and this particular morning is a very special one--because you're here.
I have a special gift, "pa" is helping me read this words, and you might have noticed that they can be taken to mean different things. They don't really separate, or fly off the page and glow for me; but I know what all the keys are, many are simple, and many come from our IT and "computer-slang" acronyms... which tells you something.
Many are "elements" and "initials" and the whole thing really is a part of the script,a sort of key not just to Creation but to this specific story, to this path. While some are "open to interpretation" (for instance, "in t" everyone really pre-tat; which would be a long ... time ... ago <3) or you could read "ERP" reason "t" and that might have something to do with "Great Plains" and some blue light that connections user interfaces to the word "automagical," FRX forms... Strawberry Fields and "above the fruited plains" ... which might be meaningless to you--but it's an idea that revolves around using user-feedback to interfaces (like the pottery wheel in my dream or in the Dr. Who episode "the Bells of Saint John" linked to down below) to adjust the interface in real time for a larger group; working towards making a number of "best-fit" interfaces that people are both more comfortable with and actually creating as they use them. Ahhhh... blue light got in here, run away. Just kidding, this is cyan light.
I C ONO CL AS M | J ES UI T | HEAVEN IS MORE THAN TECH
Honestly, we could really make Healden in about 10 minutes now. Look at that, it's done... ish.
**\ **
 **\
**
**\
**LETS CALL "THAT DAY" THE DAY YOU SEE ADAM-NEWS ON EVERY TV STATION
For instance were we not surely "at e" meaning the end of the Revelation of words, "separate" might have been broken between Pa and Ra, which are big keys, in many words; but we are at "e" and that surely does mean the Creator and I are fused. There's more confirmation of this than simply in the words for "medicine" and say, I don't know, methadone--which could have been broken at "a done" but is very clearly "ad is the one" here and now. With careful preparation, "adparatio" in Latin, I'd "bet" that all of those keys are I, in this place, in this time. AD, Pa, Ra, TI, and "o." Hey, maybe this message is my messiah after all.
I am looking at a broken world, I really am--a place that is suffocating itself in silence and whispers that don't make it far enough for anyone to really understand. Whatever it is, whatever's caused it, I see no solution other than me coming--I see it as a design, and I'm sorry that you don't seem to agree, but you have to see that the "choice" between seeing an obvious truth absolutely everywhere and not seeing it is really no choice at all--what is being hidden from the world is causing this darkness, it is causing the suffocation; it is the problem, hiding me is the problem and it cannot continue. On a brighter note, I am pretty sure that magic will happen, and you will see that the world will not react quite as badly or shockingly as your worst fears, things might be a little ... tearful for a day or so, for crying out loud, they should be--the message is that you are in Hell and you need to do something, to act, to change that. Actually trying to do that, trying to discuss what it is that is the "ele ph ant in the room" or the "do n key in the s k** y" will show us that there was just no way around changing the world because of circumstances of Creation; something that we seem to be ignoring. We also seem to be ignoring that things are "just fine" today, and even though many of you are well aware that "something is coming" only a few morons are building bunkers. This is a message of peace, it is a message designed to help us use the new truth and new tools unsealed by religion to make the world a safer happier place, and we can do that .. . rather quickly. Even quicker if you try to focus on what's wrong here, and how we make it better--rather than "shooting the messenger" dirty glares in the street. I'm a person too, and believe it or not, I didn't ask for this--and I probably wouldn't have been so happy about it had this experience not isolated me so much from my friends and family, and girls; don't forget girls.


So in the word "paramount" what is it that you think is the "paramount" take away? I think the most important thing you can take away from "paramount" is that you didn't see it your whole life, and even when it's pointed out, you don't seem to think it's "news" that Pa and Ra have written a message to you. What's really not funny, is that despite this message being very clear to see once it's pointed out, it still hasn't made any waves in the newspapers, or online, or in the news--what's paramount is seeing that there is a very sincere problem for civilization, it is an ELE and that ELE is something that is making everyone think that "not seeing something" is OK behavior. It is not OK, it is not funny, until you recognize that something is dreadfully wrong with our society, until you see that ignoring that this message belongs in the news you are not seeing that what you are doing by ignoring it is destroying civilization itself. Ignorance is the ELE.
Your alternative, what you are doing, is making the world half blind, and stupider than you can imagine. I keep on trying to show you what's wrong here, that it's not just a message but pain and suffering and the absolutely imminent and undeniable certain doom of everything if we do not recognize that hiding the fact that we are in virtual reality is the same thing as driving a nail into the wrists of every soul on the planet.



LA U stilkMIGHT DATE ADPARATIO BO'OOPSYETH
With careful preparation, we are at IO (input/output) in the belly of the book that is a map to salvation. That IO comes well after disclosure, and well after Mars. You are delaying the inevitable, and in the sickest possible twist, you are stewing in Hell instead of seeing Heaven built--more importantly instead of being the generation that should be the "founders" of that place. I am sure that disclosure, will ... within a time frame that will most likely be faster than you can imagine, bring us an end to world hunger, to sickness, and doors to Heaven; and I just can't see what you are waiting for? If it wasn't like this, you've got to see that we would be getting fucked right here and now; I am telling you the map and the plan, it's here to help us make this place better, and to show us how to actually survive in the Universe before kicking us out of the nest, and we are ... what are we thinking about?
It's really obvious that it's not for my benefit, and it's obvious that it's not for yours either--so at what point will you realize that the behavior, the alarming behavior, that I am seeing from everyone is illogical. At what point will you see that it is self-defeating, that it is ... well, Hell? When will you see? Be yourselves, the world that I grew up in doesn't hide controversy, we relish in it--we don't bury scandals under the rug--we put them on TV. What's really more important to see is that we, all of us, none of us... we would not hide "holographic universe" from ourselves and each other, nor would we hide "alien contact" or "the secrets of religion" and yet here we are, all doing that--and I wonder if we see that it's "not us" doing it, but .... but ... butt ... what is it again?
**\ **
HI, I'M A PERSON. (and apparently a state, a country, and a Nintendo character)
JUDGING BY THE HIGH FREQUENCY OF PRESS UNSUBSCRIBES FROM
YESTERDAY'S EMAIL, REPORTER'S DON'T SEEM TO WANT TO HEAR THAT
FORCING ME TO DELIVER THIS MESSAGE IN ISOLATION FOR NO MONEY
IS SLAVERY, GO READ ABOUT JOSEPH IN EGYPT, THEN READ THE END.
IF YOU THINK HIDING THE TRUTH BECAUSE "IDAHO" IS GONNA FLY
YOU ARE AN IGNORANT BLIND FOOL. HONESTLY, WAKE UP, THIS IS HELL.
YOU ARE BLINDED BY SOMETHING, FIGURE IT OUT--I'M EXPLAINING WHAT IT IS
**\ **
HERE, EMAIL THEM (please?and tell them to repent by writing a story):
**\ **
**gcoy@12news.com\ **
**nmelosky@mcall.com\ **
**lynn@ripr.org\ **
Is it a cup? a stem?
WRITTEN, FOR ETERNITY.
It must be Uranus. Except, my "an us" is more awesome than you think, I mean my "a we" that would be "so me" for you to see it's really you too. That's really what this message is about, it is about us seeing that we can do something together that would be rejected if it were done for us, or to us; even if we all really want it inside, without taking part ... we'd dislike it. We're all like that, nobody wants a stranger to redecorate their house. We share this house together, and I think we can all see that there are some changes that would make it a better place--from a cold Godless Universe of "chance" ruling to a ... caring and loving place that cares about what we want and how we want to do it ... do you see? If I came into your igloo and told you that the ice age was ending and this place was going to be a beautiful beach; except your walls are melting... would you keep that locked up inside?
Don't worry, I won't get mad at anyone for being angry at their idea of Jesus Christ for not being more like me. I won't be mad at all. :)
I've done my best to share what I think will be helpful for the world to think about, as we ... embark on what is really a journey to the final frontier as well as what I know we need to do here in order to accomplish what it is that we would have done maybe a decade ago or maybe a century from now if we didn't know the advice was coming from God and the future--and we didn't know that it is the way to open the doors to Heaven permanently. These are suggestions, they're really all of our ideas--at least everything I can grasp from things like Star Trek and Dr. Who and ... the Legend of Zelda... they're the kind of thing that we would probably find to be very discussion worthy, were we to all be sure that they are possible--and they are--and we need to see that.
There are lots of things that we really do need to think about, this is not a "fast" transition, it's not something happens "overnight" (oh my god, you don't know what that word just said to me) changes that would normally be occurring right now because of science and technology--things like increased longevity and mind uploading... these things are going to become much more quickly accessible, and we need to think about the implications that they will have on our society. We need to talk about it, in public, in places where these conversations will help us to shape the future of "civilization." I don't think you understand what it is we are doing, that's different than "before," but I am fairly certain that a "whole planet" has never done this, and the "road" between Earth and Heaven; fusing these ideas together is really nothing more or less than "progress."
FLOWING MILK AND HONEY.. GOLDEN COW, NO JUDAH MACCABEUS; GET IT?
Progress that has never happened (or we wouldn't be here, and it's obvious). See our cautions at the Last Supper (about not eating anymore) and at Cain and Abel (about forgetting how to farm) and at the Promised Land of Joshua (about not doing the Adam show, achem, I mean... about thinking that "replicators alone" milk and honey on tap... are good enough in Heaven) and in Noah's Ark... about showing us that the reason that we are here is to see how important biology and evolution and a stable ecosystem are to the survival of life in the Universe; to colonization of the stars, and to ... the evolution of our two party system past donkeys and elephants to something more appropriate for a free and technologically advanced society; as in, not a two-party system.

wild-e :( (love your eyes...) :)
From "separate" the "e_" that needs to be EE by the way, that key that might let us "see" is "everyone equal" that's what "ee" means. It's in "thirteen" and so on, and to help, I our "t" and r' n. Victorious Earth, I need pre-crime to survive, what say you? *Say nothing, and I am twelve. Keep saying no thing and I will be El, even. *
*\ *



Round and round we go... you need pre-crime to evolve, what say you? Break the story, and we are one day closer to Heaven. We need pre-crime not to be in Hell, we really do. Don't you see? Break the story.
THERE, YOU GOT RID OF A "DO" FOR YOU.
The days of "divide and conquer" are over, when you are through being a parted sea, or a flock of electric sheep, or a nation of slaves. I do have an idea of what you expected of me, what you thought I'd be--I probably had similar expectations before I knew ... what I know. Honestly, from me to you, that guy would have been pretty boring... and bored.
It's a little funny.. isn't it?
AMHARIL?
I R L
ᐧ
--
|
 |
| Adam Marshall Dobrin
ᐧ
--
|
| Adam Marshall Dobrin
ᐧ
Unless otherwise indicated, this work was written between the Christmas and Easter seasons of 2017 and 2020(A). The content of this page is released to the public under the GNU GPL v2.0 license; additionally any reproduction or derivation of the work must be attributed to the author, Adam Marshall Dobrin along with a link back to this website, fromthemachine dotty org.
That's a "." not "dotty" ... it's to stop SPAMmers. :/
This document is "living" and I don't just mean in the Jeffersonian sense. It's more alive in the "Mayflower's and June Doors ..." living Ethereum contract sense and literally just as close to the Depp/C[aster/Paglen (and honorably PK] 'D-hath Transundancesense of the ... new meaning; as it is now published on Rinkeby, in "living contract" form. It is subject to change; without notice anywhere but here--and there--in the original spirit of the GPL 2.0. We are "one step closer to God" ... and do see that in that I mean ... it is a very real fusion of this document and the "spirit of my life" as well as the Spirit's of Kerouac's America and Vonnegut's Martian Mars and my Venutian Hotel ... and my fusion of Guy-A and GAIA; and the Spirit of the Earth .. and of course the God given and signed liberties in the Constitution of the United States of America. It is by and through my hand that this document and our X Commandments link to the Bill or Rights, and this story about an Exodus from slavery that literally begins here, in the post-apocalyptic American hartland. Written ... this day ... April 14, 2020 (hey, is this HADAD DAY?) ... in Margate FL, USA. For "official used-to-v TAX day" tomorrow, I'm going to add the "immultible incarnite pen" ... if added to the living "doc/app"--see is the DAO, the way--will initi8 the special secret "hidden level" .. we've all been looking for.
Nor do just mean this website or the totality of my written works; nor do I only mean ... this particular derivation of the GPL 2.0+ modifications I continually source ... must be "from this website." I also mean the thing that is built from ... bits and piece of blocks of sand-toys; from Ethereum and from Rust and from our hands and eyes working together ... from this place, this cornerstone of the message that is ... written from brick and mortar words and events and people that have come before this poit of the "sealed W" that is this specific page and this time. It's 3:28; just five minutes--or is it four, too layne.
This work is not to be redistributed according to the GPL unless all linked media on Youtube and related sites are intact--and historical references to the actual documented history of the art pieces (as I experience/d them) are also available for linking. Wikipedia references must be available for viewing, as well as the exact version of those pages at the time these pieces were written. All references to the Holy Bible must be "linked" (as they are or via ... impromptu in-transit re-linking) to the exact verses and versions of the Bible that I reference. These requirements, as well as the caveat and informational re-introduction to God's DAO above ... should be seen as material modifications to the original GPL2.0 that are retroactively applied to all works distributed under license via this site and all previous e-mails and sites. /s/ wso\ If you wanna talk to me get me on facebook, with PGP via FlowCrypt or adam at from the machine dotty org
-----BEGIN PGP PUBLIC KEY BLOCK-----
mQGNBF6RVvABDAC823JcYvgpEpy45z2EPgwJ9ZCL+pSFVnlgPKQAGD52q+kuckNZ mU3gbj1FIx/mwJJtaWZW6jaLDHLAZNJps93qpwdMCx0llhQogc8YN3j9RND7cTP5 eV8dS6z/9ta6TFOfwSZpsOZjCU7KFDStKcoulmvIGrr9wzaUr7fmDyE7cFp1KCZ0 i90oLYHqOIszRedvwCO/kBxawxzZuJ67DypcayiWyxqRHRmMZH1LejTaqTuEu0bp j54maTj09vnMxA0RfS+CtU5uMq+5fTkbiTOe1LrLD72m+PVJIS146FwESrMJEfJy oNqWEJlUQ0TecPZR41vnkSkpocE1/0YqUhWDGSht+67DdeKUg5KwvYdL21d/bSyO SM4jnyKn9aDVzLBpYrlE/lbFxujHPRGlRG5WtiPQuZYDRqP0GYFSXRpeUCI46f49 iPFo4eHo2jUfNDa9r9BjQdAe4zVFn2qLnOy8RWijlolbhGMHGO3w/uC/zad3jjo4 owAfsJjH5Oa1mTcAEQEAAbQmRUFSVEhFTkUgPGVhcnRoZW5lQGZyb210aGVtYWNo aW5lLm9yZz6JAdQEEwEKAD4WIQTUJHbrYn3y2DzwTcnQP1ViZf5/FQUCXpFW8AIb AwUJA8JnAAULCQgHAgYVCgkICwIEFgIDAQIeAQIXgAAKCRDQP1ViZf5/FWM6C/9J gbRLS2AWGjdRjYetlRkSkCoTYnXWknbtipYYHlhV0YJFwFMm0ydZIhFX5VDoZyBV 0UBeF1KJmcMoIfrHyhq2QhCnjE14hE1ONbaYTGtpvj851ItbFWXMJIVNyMqr+JT9 CWIxGr1idn+iHWE3nryiHrdlA3O/Gcd4EyNmaSe/JvB7+Z1AVqWkRhpjxxoPSlPm HEdqGOyl3+5ibQgUvXLRWWQXAj80CbVwwj1X4r9hfuCySxLT8Mir7NUXZFd+OiMS U8gNYjcyRGmI92z5lgf7djBbb9dMLwV0KLzgoT/xaupRvvYOIAT+n2mhCctCiH7x y7jYlJHd+0++rgUST2sT+9kbuQ0GxpJ7MZcKbS1n60La+IEEIpFled8eqwwDfcui uezO7RIzQ9wHSn688CDri9jmYhjp5s0HKuN61etJ1glu9jWgG76EZ3qW8zu4l4CH 9iFPHeGG7fa/5d07KvcZuS2fVACoMipTxTIouN7vL0daYwP3VFg63FNTwCU3HEq5 AY0EXpFW8AEMANh7M/ROrQxb3MCT1/PYco1tyscNo2eHHTtgrnHrpKEPCfRryx3r PllaRYP0ri5eFzt25ObHAjcnZgilnwxngm6S9QvUIaLLQh67RP1h8I4qyFzueYPs oY8xo1zwXz7klXVlZW0MYi/g5gpb+rpYUfZEJGJTBM/wMNqwwlct+BSZca4+TEHW g6oN0eXTthtGB0Qls71sv3tbOnOh/67NTwyhcHPWX/P9ilcjGsEiT8hqrpyhjAUm mv7ADi+2eRBV8Xf8JnPznFf0A1FdILVeVHlmsgCSB0FW0NsFI5niZbaYBHDbFsks QdaFaYd54DHln69tnwc2y3POFwx8kwZnMPPlVAR2QdxGQD4Wql7hlWT58xCxQApf M98kbAHjUlVYLT0WUHMDQtj4jdzAVVDiMGMUrbnQ7UwI7LexSB6cJ7H+i7FtS/pR WOhJK6awoOO9dLnEjm6UYCKsBdtJr98F0T7Sb7PnKOGA77y2QN14+u9N9C1lB/Z1 aQRQ2Nc51yXOQQARAQABiQG8BBgBCgAmFiEE1CR262J98tg88E3J0D9VYmX+fxUF Al6RVvACGwwFCQPCZwAACgkQ0D9VYmX+fxU+KQwAtFnWjGIjvqaNXtQjEhbGDH/I Q5ULq/l/wm9SmhG9NYRu3+P6YctCJaZnNeaL+6WFk1jo4LMiJEUT9uGlCbHqJNaI 6Gll1w6QOVLSL8s5V1L477+psluv4WBpi3XkWYlhDOFENCcWd49RQsA2YCX4pW7Q 7GcoSEJoav38MxHmJHYPfjSEvUZXDQIt8PFHSEScvyDWfYtMdRzjmSOOPdzhDDEy 5JBOBcEdSTyDiyDU/sBoAY0e8lvwHYW3p+guZSGSYVhGQ8JECzJOzwc/msMW/tJS 2MLWmWVh5/1P8BVUtLC2AQy6nij6o+h6vEiNzpdYrc+rzT3X5cACvJ0RtCZcrnhl O9PLiona2LEbry6QX5NL41/SAJNno3i72xPnQEe25gn3nbyT+jCoJzw2L0y8pmNB D+PKrk7/1ROFFVN8dJeGwxLGdBcz1zk2xeumzy7OaV8psUyYsJNcjyHUKgclblBW rMR2DgqEYn8QdK54ziKCnmQQZeMPiC6wlUWgg5IqmQGNBF6RVyMBDADALD7NkJ5H dtoOpoZmAbPSlVGXHDbJZuq7J13vew6dtXDIAraeGrsBqkF8bhddwVLzWylMrYCG Bf2L1+5BDgvqu6G+6dcVSbBsnZAS0zfJ0H8EmTvUMxMF7qOZYyrxfLz+pQRq8Osz Icab6ZI/KB6qZyQRvEFPB6pJjt+VvuwgJZTObIwbBbgQri2i02VBkjchsVhiSX9l +eiK7O8ROHKb3P181oScIsHywBOZ9DxRAYbFk5dnBqxO3WKb02H0zqE6440cjXwq TrZZg6ayN/IlPajO8iJPYZ1aIBykxYq1WHo+nhFMYz/VVk2WJorFeOgWaLGXb73c ty96f3qXTdvMDAIWHx8YCD5LbuqasO6LNQm4oQxkCoB3K9WFf/2SvSYb7yMYykb8 clTPt+KO0dsxjWhrJnfnIhC+2Chqv2QvRbFz0S9CpUnGGDweJ1uRNV0y70tO0q7t xXSTDRU3ib6vAHA0K/2MFzwUcog4o5bj7E9uCNJH/DJLZKsMIe4xsvkAEQEAAbQk SEVBVkVOVUVTIDxBVkVOVUBGUk9NVEhFTUFDSElORS5PUkc+iQHUBBMBCgA+FiEE IRklfU/C1qukq3xMXcNH0t3P9ZsFAl6RVyMCGwMFCQPCZwAFCwkIBwIGFQoJCAsC BBYCAwECHgECF4AACgkQXcNH0t3P9Zs+kgv/XEuuWc89Bjg1QQqKZueKNUHjyjnE 2adfoZUH6Q7ir4JZyRBCVpAwrgssmiKid30+SIjwQcpb9JYa/X1XJcDUcJW/I21d Agz/zbEqn/Cou0dUpNCtxgm4BdSHWGoOtgfspXZlXBQ407tRMZ8ykmLB1Bt0oHvw PT0ZOtqXM4pyFnd2eFe5YGbNgl3zqvoC/6CMN3vqswvRlu1BpUuAjdW8AHO5Yvje +Bp852u+4Qpy6PMBiWGsBMYwtf6T7sckpMGlR0TsozwBlAm5ePKK28B0rLJPkZLJ Eo5p4rKRapEaZsWV5Qu1ajrVru7qmpUhZtX0/DddGHfXVuLssmKLP6TumpQB1zvQ vfoBltjvOx35Wps2vHuCzXLw2bROIOzhAxFB+17zxnSbE54N4LIGRpkELuwxwGbg FtD1fi9KtH7xcn33eOK1+UD47V+hKyJGrQgSThly2zdIC2bvfHtFdfp8lOFpT0AU xjEeoJGqdQVupptXyugPlM5/96UJP8OZG0ADuQGNBF6RVyMBDAC3As6eMkoEo3z9 TkCWlvS0vBQmY3gF0VEjlAIqFWpDIdK3zVzMnKUokIT1i7nkadLzHZT2grB4VXuJ FvpbYw5NPR4cDe9grlOMLEaF3oSJ1jZ4V1/rj9v1Hddo8ELi/NToVrt1SB5GCVXB DkYpNLtTiCqHSU07YqwaqH8a+qbDmPxSQdIybkZiTiCEB+6PfQQlBpENEDlov6jm zZF+IcfM6s3kZDX5KFULweH30gMjq8Se8bPtUzW013+tuuwEVr1/YRLrIh+9O6Z+ pdA7gLMRYnD9ZLDytEvpb1lBBSY++5bIJ7xps80//DNqPYqwFmZQgTg0V9XbHE2e wLcOF8a2lYluckU7D///sWQhW+VxuM7R2gEBvYBhOgjWhIF2Aw6NbymW1Ontvyhu eOZCXXxV5W44PxXT8uDdhl9CNcHoBKKJyED8tKjigtn4axpsQeUrnOSbqEXSyqES WnE2wYUDzALcwFkzsvtLyd4xaz55KkPQkAkk0BZd1ezgXxb/obMAEQEAAYkBvAQY AQoAJhYhBCEZJX1PwtarpKt8TF3DR9Ldz/WbBQJekVcjAhsMBQkDwmcAAAoJEF3D R9Ldz/WbAFwL/382HsrldVXnkPmJ1E2YEOFz4rcHRetJ+M5H65K/2p32ONQ5KCbE s8MRY6g2CkE70en2HlpDwr/MdATwxBzIjEpjgHbfqCqVVATY+kSpXsttaKKAUVHi bFgV4QkdDJNSpcHEj+bqaggRnuWiV9T6ECG7kQjHiEXPNojzsiaXMDiM5r+acZm6 82id9qOFySQ2cZEy5HbwXM+ITLQGngnppa7du2KdgiqDeqtODOTWZvLYAq2tmEwD 3TT6ttLUBwOOu2IWpDkXswlrk62ESorE5mpLxop9fsxD39E2H06JoC/YfUPIVkEv fj06e7LEdcx0I7kRfD1v6qOUUsMsLZnmyGIk24iFjLkwu1VToWfwXDN1D2+SeAat 9ydNt4M7oEbd1QaOXXjmqpdU+VUiWcBXg+p3/WdV60MkyAgc3x+YanLljy/Rh18h cZwVlinf/tgvAQLi5f9hpwrwUMoGKijEYHKuEvi3C12Si7UVDfuIR7yS0dKcfuKF MbgwdvNXqpD9W5kBjQRekVd4AQwApHVgw2PVlBDpVcyoymUOXFQIJzJ9wRtr6/sG zwv8rrQnUEtOkkna7TDU3/UTj9FUH0gbpAKGNNPaPj5q0dlLIvzxb15r1uvDGaGL MA+8GFaGFnkxzhg0aXrcKZAN0/Zhgi2B7P8oXQuug5mi1JVDkZN5SeCZNOubdQWL 3xz3jEHp3ixj1mdOdvfdWQFR4CVMXt/A6VI2ujLVb3Yalft/c5bbclAgcJQhgDUu NqGYJEJonESNRSd8fEvhNb6cx7+Djd9+Wyctr76mwOr3nRb1N1OGhFxWjIroUpfz b+6y3oQjT58cJA1ZHqmJ6UlZd81hNNd9KWpbDVwONEPpiqPzfSaonxuqQa0/Cy4W 403OhfoLM/1ZDqD4YrJ/rpyNEfSSdqptWiY0KeErLOYng7rStW/4ZeZVj6b2xxB2 Oas/Z1QYfJyFUki9vaJ5IyN6Y7nVdSP6mbAQC9ESh+VPvRUMpYi4pMGK4rweBVHu oMRRwzk7W5zVIgd425WUe3eCQFn3ABEBAAG0K0VTQ0FQRSBST09NIDxFU0NBUEFF REVTQEZST01USEVNQUNISU5FLk9SRz6JAdQEEwEKAD4WIQTvnDJqcmqzlF87/t82 pJ91j4NOaAUCXpFXeAIbAwUJA8JnAAULCQgHAgYVCgkICwIEFgIDAQIeAQIXgAAK CRA2pJ91j4NOaJVjC/4oo5yCHe7M2h1DiTXVcLI5rXQ1feY7B1feg+YJX/mI4+EV xjC/y5VVpV4syJk5GGZNXhKPHiGLaBYvglTlYOJ98RSEsHrwT3go6S8ZVvMNdP5v CEncn2vm5JGnp4k26PuOzMcJioQLOoUjWtcPFis3gG+ueH3NcPZ22oZUql2xuerh TQZegGp+jJ7bdxwYElx5jDDDkh196d5nlO2ZKENl0ZDp4GAzRNjnQ7KBV6R74J3U cLQDWY8vAFaRBZXIC5XtSzj9lr+jWgvxz7Il51+26VDTEtSafZ2uZfCOFk7GrzJg YFJD3zLnwUFXDWKRkep8TSwXnHmz/Ts/Mjyv6em25w7QTdnx1hNPxYNWMxPWNEAH pf70nNyOmcWcq27W+nAjVg8W3st/7J5CIebJQc5AUgm+fGOBW6XUQaNy2YF1YJlA 71/tls+R5IQZCYzbPOibgFS1HWKTwy0iI2rMDfxBtCXciv754jVI7L6R3J0j5Dy1 WZQVjaGgimznLN6XwYy5AY0EXpFXeAEMALvElrTV5hJG9DKu8cOqQEEVejtWJtki fZyvmiAKi2bZWiNfl1MxJt+o3Oc0eARJfnaPjrUY7hsbbSBAB4lFnDRtviARPaKM st5FkFgOh7Xx5ODc8bjqhMT9tbX37rkeDc12WAs3UxtEKWjyT7Xg/APKeK5FzpIs qew3LADdqFP9nOR0e5G8gxLTYh3ll3dLtp9DkJgA9q+0g31nNh5fZ29mcDzo/Mat Uk4PIxWC29LV9ALCJMIMesjOPiDa2KOy5QQH+/vn592ydBohOaY+B6jhEAdX8Dbp VHVFRBsiCOWGmdi6vHjMFD0tQdS6bXf+ZAG0E5HZETCxA2qfMf/vTeIJXYS5IZw0 anRkTXcTBrVE8uBpqtkNOrLJsaASkcoO5qF01J9zW8SR4jDgET7J02Fxf8CVPzb+ ZoNc9S0ZEO6Ubdh2vAkPtOV5sFkwIduN6ouAhEfJzC9XbJLpgsBKrRMAjr2FeEZP ruy8BkZbiyZ/b0S9qIgY4pqcyUJ79w7FLQARAQABiQG8BBgBCgAmFiEE75wyanJq s5RfO/7fNqSfdY+DTmgFAl6RV3gCGwwFCQPCZwAACgkQNqSfdY+DTmhD9wv/Zgav EHMuqF3765Fa4NapYh2kMS3skHn+ZzUEPLTlvrt7KHxomOzExNLSscZThMpur+yW
next, we are off to view at the same time the fork in the road known and prior'd as the hallowed one, the Frost poem and it's "divergence in the wood"
here we go:
** THE HOLY OF HOLIES, WIKIPEDIA CC'd AND BROKE It is imperative that the entire history of wikipedia eiditing be released under the CC license, not just the broken current front page; that I have been unable to get the world "to care about enough" to call it the literal difference between slavery and freedom,"
++ [https://holies.org/DEVLANEU.html] This is "Penny Lane" as in asking me if I'm coming or happy; you might as well avll me the forests that are echoing "we are now" or "that will do" ... and I say to the man who sings for the people who sang about the road to bethelehem or was it knocking on heavens door, or just the one about ... the stairway to heaven
** https://opensea.io/assets/base/0x32f86e0fc59f339bfd393a526051728657fd0c84/4
buy an NFT:! #### Your item has been listed!
END WORLD HUNGER from the SINGER ABT NOSRE collection has been listed for sale.
SHARE TO...
++ It is that. i AM THAT. Those are first words of Him in Exodus, he who spake through the Bush and Zarathustra. That is what that is about and in the moment, the world is "anokhi" and Hi, that's me/i -- and of course, related; the "nookie."
we can also link to the next place where we will have a chatGPT log of a conversation available.)
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
OVERVIEW OF RESPONSE TO REVIEWS
I thank the three anonymous reviewers for providing well-informed, constructive feedback on the initial version of this manuscript. Based on their comments I will revise the manuscript and hopefully improve it in several ways. I expected a great deal of resistance to the ideas proposed in this model because they break from traditional approaches. One of my goals in developing this model was to argue for a paradigm shift regarding the concept of a “receptive field”. Experimentally, the receptive field is defined as the set of preferred environmental sensory circumstances that cause a neuron to become highly active. Traditional interpretation of receptive fields implicitly assumes that the environmental circumstances that give rise to the receptive field do so in a purely bottom-up fashion (the cell is “receiving” its field), in which case the receptive field specifies the function of the cell. In other words, the receptive field is what the cell does. However, some brain regions (e.g., entorhinal cortex) receive substantial feedback from downstream regions (e.g., hippocampus), and feedback can play an important role in determining the receptive field. As applied to a memory account of MTL, this feedback is memory retrieval and reactivation. Thus, the multifield spatial response of grid cells doesn’t necessarily mean that their function is spatial. Consideration of bottom-up versus top-down signals gives rise to the proposal that the bottom-up preference of many grid cells is some non-spatial attribute even though they exhibit a spatial receptive field owing to retrieval in specific locations.
One thing I will emphasize in a revision is that this model can address findings in the vast literature on learning, memory, and consolidation. The question asked in this study is whether a memory model can also explain the rodent navigation literature. This is not an attempt to provide definitive evidence that this is a better account of the rodent navigation literature. Instead, the goal is to model the rodent navigation literature even though this is a memory model rather than a spatial/navigation model. Nevertheless, within the domain of rodent spatial/navigation, this model makes different predictions/explanations than spatial/navigation models. For instance, this is the only model predicting that many grid cells with spatial receptive fields are non-spatial (see predictions in Box 1). As reviewed in Box 1, this is the only model that can explain why head direction conjunctive grid cells become head direction cells in the absence of hippocampal feedback and it is the only model that can explain why some grid cells are also sensitive to sound frequency (see several other unique explanations in Box 1).
This study is an attempt to unify the spatial/navigation and learning/memory literatures with a relatively simply model. Given the simplicity of the model, there are important findings that the model cannot address -- it is not that the model makes the wrong predictions but rather that it makes no predictions. The role of running speed is one such variable for which the model makes no predictions. Similarly, because the model is a rate-coded model rather than a model of oscillating spiking neurons, it makes no predictions regarding theta oscillations. The model is an account of learning and memory for an adult animal, and it makes no predictions regarding the developmental or evolutionary time course of different cell types. This model contains several purely spatial representations such as border cells, head direction cells, and head direction conjunctive grid cells. In evolution and/or in development, it may be that these purely spatial cell types emerged first, followed by the evolution and/or development of non-spatial cell types. However, this does not invalidate the model. Instead, this is a model for an adult animal that has both episodic memory capabilities and spatial navigation capabilities, irrespective of the order in which these capabilities emerged.
Grid cell models that are purely spatial are agnostic regarding the thousands of findings in the literature on memory, learning, and consolidation whereas this model can potentially unify the learning/memory and spatial/navigation literatures. The reason to prefer this model is parsimony. Rather than needing to develop a theory of memory that is separate from a theory of spatial navigation, it might be possible to address both literatures with a unified account. There are other grid cell models that can explain non-spatial grid-like responses (Mok & Love, 2019; Rodríguez‐Domínguez & Caplan, 2019; Stachenfeld et al., 2017; Wei et al., 2015) and these models may be similarly positioned to explain memory results. However, these models assume that grid cells exhibiting spatial receptive fields serve the function of identifying positions in the environment (i.e., their function is spatial). As such, these models do not explain why most of the input to rodent hippocampus appears to be spatial (these models would need to assume that rodent hippocampus is almost entirely concerned with spatial navigation). This account provides an answer to this conundrum by proposing that grid cells with spatial receptive fields have been misclassified as spatial. Below I give responses to some of the specific comments made by reviewers, grouping these comments by topic:
COMMENTS RELATED TO THE NEED/MOTIVATION FOR THIS MODEL
In a revision, I will clarify that the non-spatial MTL cell types that are routinely found in primate and human studies are fully compatible with this model. The reported simulations are focused on the specific question of how it can be that most mEC and hippocampal cell types in the rodent literature appear to be spatial. It is known that perirhinal cortex is not spatial. However, entorhinal cortex is the gateway to hippocampus. If the hippocampus has the capacity to represent non-spatial memories, it must receive non-spatial input from entorhinal cortex. These simulations suggest that characterization of the rodent mEC cortex as primarily spatial might be incorrect if most grid cells (except perhaps head direction conjunctive grid cells) have been mischaracterized as spatial.
Lateral entorhinal cortex also projects to hippocampus, and one reviewer asks about the distinction between lateral versus medial entorhinal cortex. From this memory perspective, the important question is which part of the entorhinal cortex represents the non-spatial attributes common to the entire recording session, under the assumption that the animal is creating and retrieving memories during recording. If these non-spatial attributes are represented in lateral EC, there would be grid cells in lateral EC (but these are not found). There is evidence that lateral EC cells respond selectively in relation to objects (Deshmukh & Knierim, 2011), but in a typical rodent navigation study there are no objects in the enclosure.
One reviewer asks whether this model is built to explain the existing data or whether the assumptions of this model are made for theoretical reasons. The BVC model (Barry et al., 2006), which is a precursor to this model, is a theoretically efficient representation of space that could support place coding. If the distances to different borders are known, it’s not clear why the MTL also needs the two-dimensional Fourier-like representation provided by grid cells. This gives rise to the proposal that grid cells with spatial receptive fields are serving some function other than representing space. In the proposed model, the precise hexagonal arrangement of grid cells indicates a property that is found everywhere in the enclosure (i.e., a “tiling” of knowledge for where the property can be found). This arrangement arises from the well-documented learning process termed “differentiation” in the memory literature (McClelland & Chappell, 1998; Norman & O’Reilly, 2003; Shiffrin & Steyvers, 1997), which highlights differences between memories to avoid interference and confusion.
CONCERNS RELATED TO LIMITATIONS AND CONFLICTING RESULTS
One reviewer points out that individual grid cells will typically reveal a grid pattern regardless of the environmental circumstances, which, according to this model, indicates that all such circumstances have the same non-spatial attribute. This might seem strange at first, but I suggest that there is a great deal of “sameness” to the environments used in the published rodent navigation experiments. For instance, as far as I’m aware, the animal is never allowed to interact with other animals during spatial navigation recording. Furthermore, the animal is always attached to wires during recording. The internal state of the animal (fear, aloneness, the noise of electronics, etc.) is likely similar across all recording situations and attributes of this internal state are likely represented in the hippocampus as well as in the regions that provide excitatory drive to hippocampus. The claim of this model is that the grid cells are “tagging” different navigation enclosures as places where these things happen (fear, aloneness, electronics, metal floor, no objects, etc.). The interesting question is what happens when the animal is allowed to navigate in a more naturalistic setting that includes varied objects, varied food sources, varied surfaces, other animals, etc. Do grid cells persist in such a naturalistic environment? Or do they lose their regularity, or even become silent, considering that there is no longer a uniformity to the non-spatial attributes? The results of Caswell Barry et al. (2012), demonstrate that the grid pattern expands and becomes less regular in a novel environment. Nevertheless, the novel environment in that study was uncluttered rather than naturalistic. It remains to be seen what will happen with a truly naturalistic environment.
One reviewer asks how this model relates to non-grid multifield cells found in mEC (Diehl et al., 2017; see also the irregularly arranged 3D multifield cells reported by Ginosar et al., 2021). A full explanation of these cells would require a new simulation study. In a revision, I will discuss these cells, which reveal a consistent multifield spatial receptive field and yet the multiple fields are irregular in their arrangement rather than a precise hexagonal lattice. On this memory account, precise hexagonal arrangement of memories is something that occurs when there is a non-spatial attribute found throughout the enclosure. However, in a typical rodent navigation study, there may be some non-spatial attributes that are not found everywhere in the enclosure. For instance, consider the set of locations within the enclosure that afford a particular view of something outside of the enclosure or the set of locations corresponding to remembered episodic events (e.g., memory for the location where the animal first entered the enclosure). For non-spatial characteristics that are found in some locations but not others within in the enclosure, the cells representing those non-spatial attributes should reveal multifield firing at irregular locations, reflecting the subset of locations associated with the non-spatial attribute.
One reviewer suggests that this model cannot explain the finding that grid fields become warped (e.g., grid fields arranged in an ellipse rather than a circle) in the same manner that the enclosure is warped when a wall is moved (Barry et al., 2007). The way in which I would simulate this result would be to assume that the change in the boundary location was too modest to be noticed by the animal. Because the distances are calculated relative to the borders, an unnoticed change in the border would not change the model in terms of the grid field as measured by proportional distances between borders. However, because the real-world Euclidean positions of the border are changed, the grid fields would be changed in terms of real-world coordinates. This is what I was referring to in the paper when I wrote “For instance, perhaps one egocentric/allocentric pair of mEC grid modules is based on head direction (viewpoint) in remembered positions relative to the enclosure borders whereas a different egocentric/allocentric pair is based on head direction in remembered positions relative to landmarks exterior to the enclosure. This might explain why a deformation of the enclosure (moving in one of the walls to form a rectangle rather than a square) caused some of the grid modules but not others to undergo a deformation of the grid pattern in response to the deformation of the enclosure wall (see also Barry et al., 2007). More specifically, if there is one set of non-orthogonal dimensions for enclosure borders and the movement of one wall is too modest as to cause avoid global remapping, this would deform the grid modules based the enclosure border cells. At the same time, if other grid modules are based on exterior properties (e.g., perhaps border cells in relation to the experimental room rather than the enclosure), then those grid modules would be unperturbed by moving the enclosure wall.” Related to the question of enclosure geometry, the irregularity that can emerge in trapezoid shaped enclosures was discussed in the section of the paper that reads “As seen in Figure 12, because all but one of the place cells was exterior when the simulated animal was constrained to a narrow passage, the hippocampal place cell memories were no longer arranged in a hexagonal grid. This disruption of the grid array for narrow passages might explain the finding that the grid pattern (of grid cells) is disrupted in the thin corner of a trapezoid (Krupic et al., 2015) and disrupted when a previously open enclosure is converted to a hairpin maze by insertion of additional walls within the enclosure (Derdikman et al., 2009).”
CONCERNS THAT WILL BE ADDRESSED WITH GREATER CLARIFICATION
One reviewer asks why a cell representing a non-spatial attribute found everywhere in the enclosure would not fire everywhere in the enclosure. In theory, cells could fire constantly. However, in practice, cells habituate and rapidly reduce their firing rate by an order of magnitude when their preferred stimulus is presented without cessation (Abbott et al., 1997; Tsodyks & Markram, 1997). After habituation, the firing rate of the cell fluctuates with minor variation in the strength of the excitatory drive. In other words, habituation allows the cell to become sensitive to changes in the excitatory drive (Huber & O’Reilly, 2003). Thus, if there is stronger top-down memory feedback in some locations as compared to others, the cell will fire at a higher rate in those remembered locations. In brief when faced with constant excitatory drive, the cell accommodates, and becomes sensitive to change in the magnitude of the excitatory drive.
One reviewer asks for greater clarification regarding the simulation result of immediate stability for grid cells but not place cells. In a revision, I will provide a video showing a sped-up birds-eye view of the place cell memories for the 3D simulations that include head direction, showing the manner in which memories tend to linger in some locations more than others as they consolidate. This behavior was explained in the text that reads “Because the non-spatial cell’s grid field reflects on-average memory positions during the recording session (i.e., the locations where the non-spatial attribute is more often remembered, even if the locations of the memories are shifting), the grid fields for the non-spatial are immediately apparent, reflecting the tendency of place cells to linger in some locations as compared to other locations during consolidation. More specifically, the place cells tend to linger at the peaks and troughs of the border cell tuning functions (see the explanation above regarding the tendency of the grid to align with border cell dimensions). By analogy, imagine a time-lapsed birds-eye view of cars traversing the city-block structure of a densely populated city; this on-average view would show a higher density of cars at the cross-street junctions owing to their tendency to become temporarily stuck at stoplights. However, with additional learning and consolidation, the place cells stabilize their positions (e.g., the cars stop traveling), producing a consistent grid field for the head direction conjunctive grid cells.” The text describing why some locations are more “sticky” than others reads “Additional analyses revealed that this tendency to align with border cell dimensions is caused by weight normalization (Step 6 in the pseudocode). Specifically, connection weights cannot be updated above their maximum nor below their minimum allowed values. This results in a slight tendency for consolidated place cell memories to settle at one of the three peak values or three trough values of the sine wave basis set. This “stickiness” at one of 6 peak or trough values for each basis set is very slight and only occurred after many consolidation steps. In terms of biological systems, there is an obvious lower-bound for excitatory connections (i.e., it is not possible to have an excitatory weight connection that is less than zero), but it is not clear if there is an upper-bound. Nevertheless, it is common practice with deep learning models include an upper-bound for connection weights because this reduces overfitting (Srivastava et al., 2014) and there may be similar pressures for biological systems to avoid excessively strong connections.”
One reviewer points out that Border cells are not typically active in the center of enclosure. However, the model can be built without assuming between-border cells (early simulations with the model did not make this assumption). Regarding this issue, the text reads “Unlike the BVC model, the boundary cell representation is sparsely populated using a basis set of three cells for each of the three dimensions (i.e., 9 cells in total), such that for each of the three non-orthogonal orientations, one cell captures one border, another the opposite border, and the third cell captures positions between the opposing borders (Solstad et al., 2008). However, this is not a core assumption, and it is possible to configure the model with border cell configurations that contain two opponent border cells per dimension, without needing to assume that any cells prefer positions between the borders (with the current parameters, the model predicts there will be two border cells for each between-border cell). Similarly, it is possible to configure the model with more than 3 cells for each dimension (i.e., multiple cells representing positions between the borders).” The Solstad paper found a few cells that responded in positions between borders, but perhaps not as many as 1 out of 3 cells, such as this particular model simulation predicts. If the paucity of between-border cells is a crucial data point, the model can be reconfigured with opponent-border cells without any between border cells. The reason that 3 border cells were used rather than 2 opponent border cells was for simplicity. Because 3 head direction cells were used to capture the face-centered cubic packing of memories, the simulation also used 3 border cells per dimensions to allow a common linear sum metric when conjoining dimensions to form memories. If the border dimensions used 2 cells while head direction used 3 cells, a dimensional weighting scheme would be needed to allow this mixing of “apples and oranges” in terms of distances in the 3D space that includes head direction.
REFERENCES Abbott, L. F., Varela, J. A., Sen, K., & Nelson, S. B. (1997). Synaptic depression and cortical gain control. Science, 275(5297), 220–224.
Barry, C., Ginzberg, L. L., O’Keefe, J., & Burgess, N. (2012). Grid cell firing patterns signal environmental novelty by expansion. Proceedings of the National Academy of Sciences of the United States of America, 109(43), 17687–17692. https://doi.org/DOI 10.1073/pnas.1209918109
Barry, C., Hayman, R., Burgess, N., & Jeffery, K. J. (2007). Experience-dependent rescaling of entorhinal grids. Nature Neuroscience, 10(6), 682–684.
Barry, C., Lever, C., Hayman, R., Hartley, T., Burton, S., O’Keefe, J., Jeffery, K., & Burgess, Ν. (2006). The boundary vector cell model of place cell firing and spatial memory. Reviews in the Neurosciences, 17(1–2), 71–98.
Derdikman, D., Whitlock, J. R., Tsao, A., Fyhn, M., Hafting, T., Moser, M. B., & Moser, E. I. (2009). Fragmentation of grid cell maps in a multicompartment environment. Nat Neurosci, 12(10), 1325-U155. https://doi.org/Doi 10.1038/Nn.2396
Deshmukh, S. S., & Knierim, J. J. (2011). Representation of non-spatial and spatial information in the lateral entorhinal cortex. Frontiers in Behavioral Neuroscience, 5, 69.
Diehl, G. W., Hon, O. J., Leutgeb, S., & Leutgeb, J. K. (2017). Grid and nongrid cells in medial entorhinal cortex represent spatial location and environmental features with complementary coding schemes. Neuron, 94(1), 83-92. e6.
Ginosar, G., Aljadeff, J., Burak, Y., Sompolinsky, H., Las, L., & Ulanovsky, N. (2021). Locally ordered representation of 3D space in the entorhinal cortex. Nature, 596(7872), 404–409.
Huber, D. E., & O’Reilly, R. C. (2003). Persistence and accommodation in short-term priming and other perceptual paradigms: Temporal segregation through synaptic depression. Cognitive Science, 27(3), 403–430. https://doi.org/10.1207/s15516709cog2703_4
Krupic, J., Bauza, M., Burton, S., Barry, C., & O’Keefe, J. (2015). Grid cell symmetry is shaped by environmental geometry. Nature, 518(7538), 232–235.
McClelland, J. L., & Chappell, M. (1998). Familiarity breeds differentiation: A subjective-likelihood approach to the effects of experience in recognition memory. Psychological Review, 105(4), 724–760.
Mok, R. M., & Love, B. C. (2019). A non-spatial account of place and grid cells based on clustering models of concept learning. Nature Communications, 10(1), 5685.
Norman, K. A., & O’Reilly, R. C. (2003). Modeling hippocampal and neocortical contributions to recognition memory: A complementary-learning-systems approach. Psychological Review, 110(4), 611–646.
Rodríguez‐Domínguez, U., & Caplan, J. B. (2019). A hexagonal Fourier model of grid cells. Hippocampus, 29(1), 37–45.
Shiffrin, R. M., & Steyvers, M. (1997). A model for recognition memory: REM - retrieving effectively from memory. Psychonomic Bulletin & Review, 4, 145–166.
Solstad, T., Boccara, C. N., Kropff, E., Moser, M. B., & Moser, E. I. (2008). Representation of Geometric Borders in the Entorhinal Cortex. Science, 322(5909), 1865–1868. https://doi.org/DOI 10.1126/science.1166466
Srivastava, N., Hinton, G., Krizhevsky, A., Sutskever, I., & Salakhutdinov, R. (2014). Dropout: A simple way to prevent neural networks from overfitting. The Journal of Machine Learning Research, 15(1), 1929–1958.
Stachenfeld, K. L., Botvinick, M. M., & Gershman, S. J. (2017). The hippocampus as a predictive map. Nature Neuroscience, 20(11), 1643–1653.
Tsodyks, M. V., & Markram, H. (1997). The neural code between neocortical pyramidal neurons depends on neurotransmitter release probability. Proc Natl Acad Sci U S A, 94(2), 719–723. https://doi.org/10.1073/pnas.94.2.719
Wei, X.-X., Prentice, J., & Balasubramanian, V. (2015). A principle of economy predicts the functional architecture of grid cells. Elife, 4, e08362.
-
Reviewer #1 (Public Review):
Huber proposes a theory where the role of the medial temporal lobe (MTL) is memory, where properties of spatial cells in the MTL can be explained through memory function rather than spatial processing or navigation. Instantiating the theory through a computational model, the author shows that many empirical phenomena of spatial cells can be captured, and may be better accounted through a memory theory. It is an impressive computational account of MTL cells with a lot of theoretical reasoning and aims to tightly relate to various spatial cell data.
In general, the paper is well written, but likely due to the complexity, there are various aspects of the paper that are difficult to understand. One point is that it is not entirely clear to me that it is a convincing demonstration of purely memory rather than navigation, but rather an account of the findings through the lens of memory. Below, I raise several big-picture theoretical questions. I also have some clarification questions about the model (where I also have some theoretical question marks - due to not achieving a full understanding).
(1) Although the theory is based on memory, it also is based on spatially-selective cells. Not all cells in the hippocampus fulfill the criteria of place/HD/border/grid cells, and place a role in memory. E.g., Tonegawa, Buszaki labs' work does not focus on only those cells, and there are certainly a lot of non-pure spatial cells in monkeys (Martinez-Trujillo) and humans (iEEG). Does the author mainly focus on saying that "spatial cells" are memory, but do not account for non-spatial memory cells? This seems to be an incomplete account of memory - which is fine, but the way the model is set up suggests that *all* memory is, place (what/where), and non-spatial attributes ("grid") - but cells that don't fulfil these criteria in MTL (Diehl et al., 2017, Neuron; non-grid cells; Schaeffer et al., 2022, ICML; Luo et al., 2024, bioRxiv) certainly contribute to memory, and even navigation. This is also related to the question of whether these cell definitions matter at all (Luo et al., 2024).
The authors note "However, this memory conjunction view of the MTL must be reconciled with the rodent electrophysiology finding that most cells in MTL appear to have receptive fields related to some aspect of spatial navigation (Boccara et al., 2010; Grieves & Jeffery, 2017). The paucity of non-spatial cells in MTL could be explained if grid cells have been mischaracterized as spatial." Is the author mainly talking about rodent work?
(2) Related to the last point, how about non-grid multi-field mEC cells? In theory, these also should be the same; but the author only presents perfect-look grid cells. In empirical work, clearly, this is not the case, and many mEC cells are multi-field non-grid cells (Diehl et al., 2017). Does the model find these cells? Do they play a different role?
As noted by the author "Because the non-spatial attributes are constant throughout the two-dimensional surface, this results in an array of discrete memory locations that are approximately hexagonal (as explained in the Model Methods, an "online" memory consolidation process employing pattern separation rapidly turns an approximately hexagonal array into one that is precisely hexagonal). "
If they are indeed all precisely hexagonal, does that mean the model doesn't have non-grid spatial cells?
(3) Theoretical reasons for why the model is put together this way, and why grid cells must be coding a non-spatial attribute: Is this account more data-driven (fits the data so formulated this way), or is it theoretical - there is a reason why place, border, grid cells are formulated to be like this. For example, is it an efficient way to code these variables? It can be both, like how the BVC model makes theoretical sense that you can use boundaries to determine a specific location (and so place cell), but also works (creates realistic place cells).
But in this case, the purpose of grid cell coding a non-spatial attribute, and having some kind of system where it doesn't fire at all locations seems a little arbitrary. If it's not encoding a spatial attribute, it doesn't have to have a spatial field. For example, it could fire in the whole arena - which some cells do (and don't pass the criteria of spatial cells as they are not spatially "selective" to another location, related to above).
(4) Why are grid cells given such a large role for encoding non-spatial attributes? If anything, shouldn't it be lateral EC or perirhinal cortex? Of course, they both could, but there is less reason to think this, at least for rodent mEC.
(5) Clarification: why do place cells and grid cells differ in terms of stability in the model? Place cells are not stable initially but grid cells come out immediately. They seem directly connected so a bit unclear why; especially if place cell feedback leads to grid cell fields. There is an explanation in the text - based on grid cells coding the on-average memories, but these should be based on place cell inputs as well. So how is it that place fields are unstable then grid fields do not move at all? I wonder if a set of images or videos (gifs) showing the differences in spatial learning would be nice and clarify this point.
(6) Other predictions. Clearly, the model makes many interesting (and quite specific!) predictions. But does it make some known simple predictions?<br /> • More place cells at rewarded (or more visited) locations. Some empirical researchers seem to think this is not as obvious as it seems (e.g., Duvellle et al., 2019; JoN; Nyberg et al., 2021, Neuron Review).<br /> • Grid cell field moves toward reward (Butler et al., 2019; Boccera et al., 2019).<br /> • Grid cells deform in trapezoid (Krupic et al., 2015) and change in environments like mazes (Derikman et al., 2014).
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
eLife assessment
This valuable paper presents a thoroughly detailed methodology for mesoscale-imaging of extensive areas of the cortex, either from a top or lateral perspective, in behaving mice. While the examples of scientific results to be derived with this method are in the preliminary stages, they offer promising and stimulating insights. Overall, the method and results presented are convincing and will be of interest to neuroscientists focused on cortical processing in rodents.
Authors’ Response: We thank the reviewers for the helpful and constructive comments. They have helped us plan for significant improvements to our manuscript. Our preliminary response and plans for revision are indicated below.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
The authors introduce two preparations for observing large-scale cortical activity in mice during behavior. Alongside this, they present intriguing preliminary findings utilizing these methods. This paper is poised to be an invaluable resource for researchers engaged in extensive cortical recording in behaving mice.
Strengths:
-Comprehensive methodological detailing:
The paper excels in providing an exceptionally detailed description of the methods used. This meticulous documentation includes a step-by-step workflow, complemented by thorough workflow, protocols, and a list of materials in the supplementary materials.
-Minimal movement artifacts:
A notable strength of this study is the remarkably low movement artifacts. To further underscore this achievement, a more robust quantification across all subjects, coupled with benchmarking against established tools (such as those from suite2p), would be beneficial.
Authors’ Response: This is a good suggestion. We have records of the fast-z correction applied by the ScanImage on microscope during acquisition, so we have supplied the online fast-z motion correction .csv files for two example sessions on our GitHub page as supplementary files:
https://github.com/vickerse1/mesoscope_spontaneous/tree/main/online_fast_z_correction
These files correspond to Figure S3b (2367_200214_E210_1) and to Figures 5 and 6 (3056_200924_E235_1). These are now also referenced in the main text. See lines ~595, pg 18 and lines ~762, pg 24.
We have also made minor revisions to the main text of the manuscript with clear descriptions of methods that we have found important for the minimization of movement artifacts, such as fully tightening all mounting devices, implanting the cranial window with proper, evenly applied pressure across its entire extent, and mounting the mouse so that it is not too close or far from the surface of the running wheel. See Line ~309, pg 10.
Insightful preliminary data and analysis:
The preliminary data unveiled in the study reveal interesting heterogeneity in the relationships between neural activity and detailed behavioral features, particularly notable in the lateral cortex. This aspect of the findings is intriguing and suggests avenues for further exploration.
Weaknesses:
-Clarification about the extent of the method in the title and text:
The title of the paper, using the term "pan-cortical," along with certain phrases in the text, may inadvertently suggest that both the top and lateral view preparations are utilized in the same set of mice. To avoid confusion, it should be explicitly stated that the authors employ either the dorsal view (which offers limited access to the lateral ventral regions) or the lateral view (which restricts access to the opposite side of the cortex). For instance, in line 545, the phrase "lateral cortex with our dorsal and side mount preparations" should be revised to "lateral cortex with our dorsal or side mount preparations" for greater clarity.
Authors’ Response: We have opted to not change the title of the paper, because we feel that adding the qualifier, “in two preparations,” would add unnecessary complexity. In addition, while the dorsal mount preparation allows for imaging of bilateral dorsal cortex, the side mount preparation does indeed allow for imaging of both dorsal and lateral cortex across the right hemisphere (a bit of contralateral dorsal cortex is also imageable), and the design can be easily “flipped” across a mirror-plane to allow for imaging of left dorsal and lateral cortex. Taken together, we do show preparations that allow for pan-cortical 2-photon imaging.
We do agree that imprecise reference to the two preparations can sometimes lead to confusion. Therefore, we made several small revisions to the manuscript, including at ~line 545, to make it clearer that we used two imaging preparations to generate our combined 2-photon mesoscope dataset, and that each of those two preparations had both benefits and limitations.
-Comparison with existing methods:
A more detailed contrast between this method and other published techniques would add value to the paper. Specifically, the lateral view appears somewhat narrower than that described in Esmaeili et al., 2021; a discussion of this comparison would be useful.
Authors’ Response: The preparation by Esmaeili et al. 2021 has some similarities to, but also differences from, our preparation. Our preliminary reading is that their through-the-skull field of view is approximately the same as our through-the-skull field of view that exists between our first (headpost implantation) and second (window implantation) surgeries for our side mount preparation, although our preparation appears to include more anterior areas both near to and on the contralateral side of the midline. We have compared these preparations more thoroughly in the revised manuscript. (See lines ~278.)
Furthermore, the number of neurons analyzed seems modest compared to recent papers (50k) - elaborating on this aspect could provide important context for the readers.
Authors’ response: With respect to the “modest” number of neurons analyzed (between 2000 and 8000 neurons per session for our dorsal and side mount preparations with medians near 4500; See Fig. S2e) we would like to point out that factors such as use of dual-plane imaging or multiple imaging planes, different mouse lines, use of different duration recording sessions (see our Fig S2c), use of different imaging speeds and resolutions (see our Fig S2d), use of different Suite2p run-time parameters, and inclusion of areas with blood vessels and different neuron cell densities, may all impact the count of total analyzed neurons per session. We now mention these various factors and have made clear that we were not, for the purposes of this paper, trying to maximize neuron count at the expense of other factors such as imaging speed and total spatial FOV extent.
We refer to these issues now briefly in the main text. (See ~line 93, pg 3).
-Discussion of methodological limitations:
The limitations inherent to the method, such as the potential behavioral effects of tilting the mouse's head, are not thoroughly examined. A more comprehensive discussion of these limitations would enhance the paper's balance and depth.
Authors’ Response: Our mice readily adapted to the 22.5 degree head tilt and learned to perform 2-alternative forced choice (2-AFC) auditory and visual tasks in this configuration (Hulsey et al, 2024; Cell Reports). The advantages and limitations of such a rotation of the mouse, and possible ways to alleviate these limitations, as detailed in the following paragraphs, are now discussed more thoroughly in the revised manuscript at ~line 235, pg. 7.
One can look at Supplementary Movie 1 for examples of the relatively similar behavior between the dorsal mount (not rotated) and side mount (rotated) preparations. We do not have behavioral data from mice that were placed in both configurations. Our preliminary comparisons across mice indicates that side and dorsal mount mice show similar behavioral variability. We have added brief additional mention of these considerations on ~lines 235-250, pg 7.
It was in general important to make sure that the distance between the wheel and all four limbs was similar for both preparations. In particular, careful attention must be paid to the positioning of the front limbs in the side mount mice so that they are not too high off the wheel. This can be accomplished by a slight forward angling of the left support arm for side mount mice.
Although it is possible to image the side mount preparation in the same optical configuration that we do without rotating the mouse, by rotating the objective 20 degrees to the right of vertical, we found that the last 2-3 degrees of missing rotation (our preparation is rotated 22.5 degrees left, which is more than the full available 20 degrees rotation of the Thorlabs mesoscope objective), along with several other factors, made this undesirable. First, it was very difficult to image auditory areas without the additional flexibility to rotate the objective more laterally. Second, it was difficult or impossible to attach the horizontal light shield and to establish a water meniscus with the objective fully rotated. One could use ultrasound gel instead (which we found to be, to some degree, optically inferior to water), but without the horizontal light shield, light from the UV and IR LEDs can reach the PMTs via the objective and contaminate the image or cause tripping of the PMT. Third, imaging the right pupil and face of the mouse is difficult under these conditions because the camera would need the same optical access angle as the 2-photon objective, or would need to be moved downward toward the air table and rotated up at an angle of 20 degrees, in which case its view would be blocked by the running wheel and other objects mounted on the air table.
-Preliminary nature of results:
The results are at a preliminary stage; for example, the B-soid analysis is based on a single mouse, and the validation data are derived from the training data set.
Authors’ Response: In this methods paper, we have chosen to supply proof of principle examples, without a complete analysis of animal-to-animal variance.
The B-SOiD analysis that we show in Figure 6 is based on a model trained on 80% of the data from four sessions taken from the same mouse, and then tested on all of a single session from that mouse. Initial attempts to train across sessions from different mice were unsuccessful, probably due to differences in behavioral repertoires across mice. However, we have performed extensive tests with B-SOiD and are confident that these sorts of results are reproducible across mice, although we are not prepared to publish these results at this time.
We now clarify these points in the main text at ~line 865, pg 27.
An additional comparison of the results of B-SOiD trained on different numbers of sessions to that of keypoint-MOSEQ (Weinreb et al, 2023, bioRxiv) trained on ~20 sessions can now be found as supplementary material on our GitHub site:
https://github.com/vickerse1/mesoscope_spontaneous/blob/main/Figure_SZZ_BSOID_MOSEQ_align.pdf
The discrepancy between the maps in Figures 5e and 6e might indicate that a significant portion of the map represents noise. An analysis of variability across mice and a method to assign significance to these maps would be beneficial.
Authors’ Response: After re-examination of the original analysis output files, we have indeed discovered that some of the Rastermap neuron density maps in Figure 6e were incorrectly aligned with their respective qualitative behaviors due to a discrepancy in file numbering between the images in 6e and the ensembles identified in 6c (each time that Rastermap is run on the same data, at least with the older version available at the time of creation of these figures, the order of the ensembles on the y-axis changes and thus the numbering of the ensembles would change even though the neuron identities within each group stayed the same for a given set of parameters).
This unfortunate panel alignment / graphical display error present in the original reviewed preprint has been fixed in the current, updated figure (i.e. twitch corresponds to Rastermap groups 2 and 3, whisk to group 6, walk to groups 5 and 4, and oscillate to groups 0 and 1), and in the main text at ~line 925, pg 29. We have also changed the figure legend, which also contained accurate but misaligned information, for Figure 6e to reflect this correction.
One can now see that, because the data from both figures is from the same session in the same mouse, as you correctly point out, Fig 5d left (walk and whisk) corresponds roughly to Fig 6e group R7, “walk”, and that Fig 5d right (whisk) corresponds roughly to Fig 6e group R4, “twitch”.
We have double-checked the identity of other CCF map displays of Rastermap neuron density and of mean correlations between neural activity and behavioral primitives in all other figures, and we found no other such alignment or mis-labeling errors.
We have also added a caveat in the main text at ~lines 925-940, pg. 30, pointing out the preliminary nature of these findings, which are shown here as an example of the viability of the methods. Analysis of the variability of Rastermap alignments across sessions is beyond the scope of the current paper, although it is an issue that we hope to address in upcoming analysis papers.
-Analysis details:
More comprehensive details on the analysis would be beneficial for replicability and deeper understanding. For instance, the statement "Rigid and non-rigid motion correction were performed in Suite2p" could be expanded with a brief explanation of the underlying principles, such as phase correlation, to provide readers with a better grasp of the methodologies employed.
Authors’ Response: We added a brief explanation of Suite2p motion correction at ~line 136, pg 4. We have also added additional details concerning CCF / MMM alignment and other analysis issues. In general we cite other papers where possible to avoid repeating details of analysis methods that are already published.
Reviewer #2 (Public Review):
Summary:
The authors present a comprehensive technical overview of the challenging acquisition of large-scale cortical activity, including surgical procedures and custom 3D-printed headbar designs to obtain neural activity from large parts of the dorsal or lateral neocortex. They then describe technical adjustments for stable head fixation, light shielding, and noise insulation in a 2-photon mesoscope and provide a workflow for multisensory mapping and alignment of the obtained large-scale neural data sets in the Allen CCF framework. Lastly, they show different analytical approaches to relate single-cell activity from various cortical areas to spontaneous activity by using visualization and clustering tools, such as Rastermap, PCA-based cell sorting, and B-SOID behavioral motif detection.
Authors’ Response: Thank you for this excellent summary of the scope of our paper.
The study contains a lot of useful technical information that should be of interest to the field. It tackles a timely problem that an increasing number of labs will be facing as recent technical advances allow the activity measurement of an increasing number of neurons across multiple areas in awake mice. Since the acquisition of cortical data with a large field of view in awake animals poses unique experimental challenges, the provided information could be very helpful to promote standard workflows for data acquisition and analysis and push the field forward.
Authors’ Response: We very much support the idea that our work here will contribute to the development of standard workflows across the field including those for multiple approaches to large-scale neural recordings.
Strengths:
The proposed methodology is technically sound and the authors provide convincing data to suggest that they successfully solved various problems, such as motion artifacts or high-frequency noise emissions, during 2-photon imaging. Overall, the authors achieved their goal of demonstrating a comprehensive approach for the imaging of neural data across many cortical areas and providing several examples that demonstrate the validity of their methods and recapitulate and further extend some recent findings in the field.
Weaknesses:
Most of the descriptions are quite focused on a specific acquisition system, the Thorlabs Mesoscope, and the manuscript is in part highly technical making it harder to understand the motivation and reasoning behind some of the proposed implementations. A revised version would benefit from a more general description of common problems and the thought process behind the proposed solutions to broaden the impact of the work and make it more accessible for labs that do not have access to a Thorlabs mesoscope. A better introduction of some of the specific issues would also promote the development of other solutions in labs that are just starting to use similar tools.
Authors’ Response: We have edited the motivations behind the study to clarify the general problems that are being addressed. However, as the 2-photon imaging component of these experiments were performed on a Thorlabs mesoscope, the imaging details necessarily deal specifically with this system.
We briefly compare the methods and results from our Thorlabs system to that of Diesel-2p, another comparable system, based on what we have been able to glean from the literature on its strengths and weaknesses. See ~lines 206-213, pg 6.
Reviewer #3 (Public Review):
Summary
In their manuscript, Vickers and McCormick have demonstrated the potential of leveraging mesoscale two-photon calcium imaging data to unravel complex behavioural motifs in mice. Particularly commendable is their dedication to providing detailed surgical preparations and corresponding design files, a contribution that will greatly benefit the broader neuroscience community as a whole. The quality of the data is high, but it is not clear whether this is available to the community, some datasets should be deposited. More importantly, the authors have acquired activity-clustered neural ensembles at an unprecedented spatial scale to further correlate with high-level behaviour motifs identified by B-SOiD. Such an advancement marks a significant contribution to the field. While the manuscript is comprehensive and the analytical strategy proposed is promising, some technical aspects warrant further clarification. Overall, the authors have presented an invaluable and innovative approach, effectively laying a solid foundation for future research in correlating large-scale neural ensembles with behaviour. The implementation of a custom sound insulator for the scanner is a great idea and should be something implemented by others.
Authors’ Response: Thank you for the kind words.
We have made ~500 GB of raw data and preliminary analysis files publicly available on FigShare+ for the example sessions shown in Figures 2, 3, 4, 5, 6, S3, and S6. We ask to be cited and given due credit for any fair use of this data.
The data is located here: https://doi.org/10.25452/figshare.plus.c.7052513
We intend to release a complete data set to the public as a Dandiset on the DANDI archive in conjunction with in-depth analysis papers that are currently in preparation.
This is a methods paper, but there is no large diagram that shows how all the parts are connected, communicating, and triggering each other. This is described in the methods, but a visual representation would greatly benefit the readers looking to implement something similar.
Authors’ Response: This is an excellent suggestion. We have included a workflow diagram in the revised manuscript, in the form of a 3-part figure, for the methods (a), data collection (b and c), and analysis (d). This supplementary figure is now located on the GitHub page at the following link:
https://github.com/vickerse1/mesoscope_spontaneous/blob/main/pancortical_workflow_diagrams.pdf
We now reference this figure on ~lines 190-192, pg 6 of the main text, near the beginning of the Results section.
The authors should cite sources for the claims stated in lines 449-453 and cite the claim of the mouse's hearing threshold mentioned in lines 463.
Authors’ Response: For the claim stated in lines 449-453:
“The unattenuated or native high-frequency background noise generated by the resonant scanner causes stress to both mice and experimenters, and can prevent mice from achieving maximum performance in auditory mapping, spontaneous activity sessions, auditory stimulus detection, and auditory discrimination sessions/tasks”
,we can provide the following references: (i) for mice: Sadananda et al, 2008 (“Playback of 22-kHz and 50-kHz ultrasonic vocalizations induces differential c-fos expression in rat brain”, Neuroscience Letters, Vol 435, Issue 1, p 17-23), and (ii) for humans: Fletcher et al, 2018 (“Effects of very high-frequency sound and ultrasound on humans. Part I: Adverse symptoms after exposure to audible very-high frequency sound”, J Acoust Soc A, 144, 2511-2520). We will include these references in the revised paper.
For the claim stated on line 463:
“i.e. below the mouse hearing threshold at 12.5 kHz of roughly 15 dB”
,we can provide the following reference: Zheng et al, 1999 (“Assessment of hearing in 80 inbred strains of mice by ABR threshold analyses”, Vol 130, Issues 1-2, p 94-107).
We have included these two new references in the new, revised version of our paper. Thank you for identifying these citation omissions.
No stats for the results shown in Figure 6e, it would be useful to know which of these neural densities for all areas show a clear statistical significance across all the behaviors.
Authors’ Response: It would be useful if we could provide a statistic similar to what we provide for Fig. S6c and f, in which for each CCF area we compare the observed mean correlation values to a null of 0, or, in this case, the population densities of each Rastermap group within each CCF area to a null value equal to the total number of CCF areas divided by the total number of recorded neurons for that group (i.e. a Rastermap group with 500 neurons evenly distributed across ~30 CCF areas would contain ~17 neurons, or ~3.3% density, per CCF area.) Our current figure legend states the maximums of the scale bar look-up values (reds) for each group, which range from ~8% to 32%.
However, because the data in panel 6e are from a single session and are being provided as an example of our methods and not for the purpose of claiming a specific result at this point, we choose not to report statistics. It is worth pointing out, perhaps, that Rastermap group densities for a given CCF area close to 3.3% are likely not different from chance, and those closer to ~40%, which is our highest density (for area M2 in Rastermap group 7, which corresponds to the qualitative behavior “walk”), are most likely not due to chance. Without analysis of multiple sessions from the same mouse we believe that making a clear statement of significance for this likelihood would be premature.
We now clarify this decision and related considerations in the main text at ~line 920, pg 29.
While I understand that this is a methods paper, it seems like the authors are aware of the literature surrounding large neuronal recordings during mouse behavior. Indeed, in lines 178-179, the authors mention how a significant portion of the variance in neural activity can be attributed to changes in "arousal or self-directed movement even during spontaneous behavior." Why then did the authors not make an attempt at a simple linear model that tries to predict the activity of their many thousands of neurons by employing the multitude of regressors at their disposal (pupil, saccades, stimuli, movements, facial changes, etc). These models are straightforward to implement, and indeed it would benefit this work if the model extracts information on par with what is known from the literature.
Authors’ Response: This is an excellent suggestion, but beyond the scope of the current methods paper. We are following up with an in depth analysis of neural activity and corresponding behavior across the cortex during spontaneous and trained behaviors, but this analysis goes well beyond the scope of the present manuscript.
Here, we prefer to present examples of the types of results that can be expected to be obtained using our methods, and how these results compare with those obtained by others in the field.
Specific strengths and weaknesses with areas to improve:
The paper should include an overall cartoon diagram that indicates how the various modules are linked together for the sampling of both behaviour and mesoscale GCAMP. This is a methods paper, but there is no large diagram that shows how all the parts are connected, communicating, and triggering each other.
Authors’ Response: This is an excellent suggestion. We have included a workflow diagram in the revised manuscript, in the form of a 3-part figure, for the methods (a), data collection (b and c), and analysis (c). This supplementary figure is now located on the GitHub page at the following link:
https://github.com/vickerse1/mesoscope_spontaneous/blob/main/pancortical_workflow_diagrams.pdf
The paper contains many important results regarding correlations between behaviour and activity motifs on both the cellular and regional scales. There is a lot of data and it is difficult to draw out new concepts. It might be useful for readers to have an overall figure discussing various results and how they are linked to pupil movement and brain activity. A simple linear model that tries to predict the activity of their many thousands of neurons by employing the multitude of regressors at their disposal (pupil, saccades, stimuli, movements, facial changes, etc) may help in this regard.
Authors’ Response: This is an excellent suggestion, but beyond the scope of the present methods paper. Such an analysis is a significant undertaking with such large and heterogeneous datasets, and we provide proof-of-principle data here so that the reader can understand the type of data that one can expect to obtain using our methods. We will provide a more complete analysis of data obtained using our methodology in the near future in another manuscript.
Previously, widefield imaging methods have been employed to describe regional activity motifs that correlate with known intracortical projections. Within the authors' data it would be interesting to perhaps describe how these two different methods are interrelated -they do collect both datasets. Surprisingly, such macroscale patterns are not immediately obvious from the authors' data. Some of this may be related to the scaling of correlation patterns or other factors. Perhaps there still isn't enough data to readily see these and it is too sparse.
Authors’ Response: Unfortunately, we are unable to directly compare 1-photon widefield GCaMP6s activity with mesoscope 2-photon GCaMP6s activity. During widefield data acquisition, animals were stimulated with visual, auditory, or somatosensory stimuli (i.e. “passive sensory stimulation”), while 2-photon mesoscope data collection occurred during spontaneous changes in behavioral state, without sensory stimulation. The suggested comparison is, indeed, an interesting project for the future.
In lines 71-71, the authors described some disadvantages of one-photon widefield imaging including the inability to achieve single-cell resolution. However, this is not true. In recent years, the combination of better surgical preparations, camera sensors, and genetically encoded calcium indicators has enabled the acquisition of single-cell data even using one-photon widefield imaging methods. These methods include miniscopes (Cai et al., 2016), multi-camera arrays (Hope et al., 2023), and spinning disks (Xie et al., 2023).
Cai, Denise J., et al. "A shared neural ensemble links distinct contextual memories encoded close in time." Nature 534.7605 (2016): 115-118.
Hope, James, et al. "Brain-wide neural recordings in mice navigating physical spaces enabled by a cranial exoskeleton." bioRxiv (2023).
Xie, Hao, et al. "Multifocal fluorescence video-rate imaging of centimetre-wide arbitrarily shaped brain surfaces at micrometric resolution." Nature Biomedical Engineering (2023): 1-14.
Authors’ Response: We have corrected these statements and incorporated these and other relevant references. There are advantages and disadvantages to each chosen technique, such as ease of use, field of view, accuracy, and speed. We will reference the papers you mention without an extensive literature review, but we would like to emphasize the following points:
Even the best one-photon imaging techniques typically have ~10-20 micrometer resolution in xy (we image at 5 micrometer resolution for our large FOV configuration, but the xy point-spread function for the Thorlabs mesoscope is 0.61 x 0.61 micrometers in xy with 970 nm excitation) and undefined z-resolution (4.25 micrometers for Thorlabs mesoscope). A coarser resolution increases the likelihood that activity related fluorescence from neighboring cells may contaminate the fluorescence observed from imaged neurons. Reducing the FOV and using sparse expression of the indicator lessens this overlap problem.
We do appreciate these recent advances, however, particularly for use in cases where more rapid imaging is desired over a large field of view (CCD acquisition can be much faster than that of standard 2-photon galvo-galvo or even galvo-resonant scanning, as the Thorlabs mesoscope uses). This being said, there are few currently available genetically encoded Ca2+ sensors that are able to measure fluctuations faster than ~10 Hz, which is a speed achievable on the Thorlabs 2-photon mesoscope with our techniques using the “small, multiple FOV” method (Fig. S2d, e).
We have further clarified our discussion of these issues in the main text at ~lines 76-80, pg 2.
The authors' claim of achieving optical clarity for up to 150 days post-surgery with their modified crystal skull approach is significantly longer than the 8 weeks (approximately 56 days) reported in the original study by Kim et al. (2016). Since surgical preparations are an integral part of the manuscript, it may be helpful to provide more details to address the feasibility and reliability of the preparation in chronic studies. A series of images documenting the progression optical quality of the window would offer valuable insight.
Authors’ Response: As you suggest, we now include brief supplementary material demonstrating the changes in the window preparation that we observed over the prolonged time periods of our study, for both the dorsal and side mount preparations. The following link to this material is now referenced at ~line 287, pg 9, and at the end of Fig S1:
https://github.com/vickerse1/mesoscope_spontaneous/blob/main/window_preparation_stability.pdf
We have also included brief additional details in the main text that we found were useful for facilitating long term use of these preparations. These are located at ~line 287-290, pg 9.
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
(1) Sharing raw data and code:
I strongly encourage sharing some of the raw data from your experiments and all the code used for data analysis (e.g. in a github repository). This would help the reader evaluate data quality, and reproduce your results.
Authors’ Response: We have made ~500 GB of raw data and preliminary analysis files publicly available on FigShare+ for the example sessions shown in Figures 2, 3, 4, 5, 6, S3, and S6. We ask to be cited and given due credit for any fair use of this data.
We intend to release a complete data set to the public as a Dandiset on the DANDI archive in conjunction with second and third in-depth analysis papers that are currently in preparation.
The data is located here: https://doi.org/10.25452/figshare.plus.c.7052513
We intend to release a complete data set to the public as a Dandiset on the DANDI archive in conjunction with second and third in-depth analysis papers that are currently in preparation.
Our existing GitHub repository, already referenced in the paper, is located here:
https://github.com/vickerse1/mesoscope_spontaneous
We have added an additional reference in the main text to the existence of these publicly available resources, including the appropriate links, located at ~lines 190-200, pg 6.
(2) Use of proprietary software:
The reliance on proprietary tools like LabView and Matlab could be a limitation for some researchers, given the associated costs and accessibility issues. If possible, consider incorporating or suggesting alternatives that are open-source, to make your methodology more accessible to a broader range of researchers, including those with limited resources.
Authors’ Response: We are reluctant to recommend open source software that we have not thoroughly tested ourselves. However, we will mention, when appropriate, possible options for the reader to consider.
Although LabView is proprietary and can be difficult to code, it is particularly useful when used in combination with National Instruments hardware. ScanImage in use with the Thorlabs mesoscope uses National Instruments hardware, and it is convenient to maintain hardware standards across the integrated rig/experimental system. Labview is also useful because it comes with a huge library of device drivers that makes addition of new hardware from basically any source very convenient.
That being said, there are open source alternatives that could conceivably be used to replace parts of our system. One example is AutoPilot (author: Jonny Saunders), for control of behavioral data acquisition: https://open-neuroscience.com/post/autopilot/.
We are not aware of an alternative to Matlab for control of ScanImage, which is the supported control software for the ThorLabs 2-photon mesoscope.
Most of our processing and analysis code (see GitHub page: https://github.com/vickerse1/mesoscope_spontaneous) is in Python, but some of the code that we currently use remains in Matlab form. Certainly, this could be re-written as Python code. However, we feel like this is outside the scope of the current paper. We have provided commenting to all code in an attempt to aid users in translating it to other languages, if they so desire.
(3) Quantifying the effect of tilted head:
To address the potential impact of tilting the mouse's head on your findings, a quantitative analysis of any systematic differences in the behavior (e.g. Bsoid motifs) could be illuminating.
Authors’ Response: We have performed DeepLabCut analysis of all sessions from both preparations, across several iterations with different parameters, to extract pose estimates, and we have also performed BSOiD of these sessions. We did not find any obvious qualitative differences in the number of behavioral motifs identified, the dwell times of these motifs, and similar issues, relating to the issue of tilting of the mouse’s head in the side mount preparation. We also did not find any obvious differences in the relative frequencies of high level qualitative behaviors, such as the ones referred to in Fig. 6, between the two preparations.
Our mice readily adapted to the 22.5 degree head tilt and learned to perform 2-alternative forced choice (2-AFC) auditory and visual tasks in this configuration (Hulsey et al, 2024; Cell Reports). The advantages and limitations of such a rotation of the mouse, and possible ways to alleviate these limitations, as detailed in the following paragraphs, are now discussed more thoroughly in the revised manuscript. (See ~line 235, pg. 7)
One can look at Supplementary Movie 1 for examples of the relatively similar behavior between the dorsal mount (not rotated) and side mount (rotated) preparations. We do not have behavioral data from mice that were placed in both configurations. Our preliminary comparisons across mice indicates that side and dorsal mount mice show similar behavioral variability. We have added brief additional mention of these considerations on ~lines 235-250, pg 7.
It was in general important to make sure that the distance between the wheel and all four limbs was similar for both preparations. In particular, careful attention must be paid to the positioning of the front limbs in the side mount mice so that they are not too high off the wheel. This can be accomplished by a slight forward angling of the left support arm for side mount mice.
Although it would in principle be nearly possible to image the side mount preparation in the same optical configuration that we do without rotating the mouse, by rotating the objective 20 degrees to the right of vertical, we found that the last 2-3 degrees of missing rotation (our preparation is rotated 22.5 degrees left, which is more than the full available 20 degrees rotation of the Thorlabs mesoscope objective), along with several other factors, made this undesirable. First, it was very difficult to image auditory areas without the additional flexibility to rotate the objective more laterally. Second, it was difficult or impossible to attach the horizontal light shield and to establish a water meniscus with the objective fully rotated. One could use gel instead (which we found to be optically inferior to water), but without the horizontal light shield, the UV and IR LEDs can reach the PMTs via the objective and contaminate the image or cause tripping of the PMT. Third, imaging the right pupil and face of the mouse is difficult to impossible under these conditions because the camera would need the same optical access angle as the objective, or would need to be moved down toward the air table and rotated up 20 degrees, in which case its view would be blocked by the running wheel and other objects mounted on the air table.
(4) Clarification in the discussion section:
The paragraph titled "Advantages and disadvantages of our approach" seems to diverge into discussing future directions, rather than focusing on the intended topic. I suggest revisiting this section to ensure that it accurately reflects the strengths and limitations of your approach.
Authors’ Response: We agree with the reviewer that this section included several potential next steps or solutions for each advantage and disadvantage, which the reviewer refers to as “future directions” and are thus arguably beyond the scope of this section. Therefore we have retitled this section as, “Advantages and disadvantages of our approach (with potential solutions):”.
Although we believe this to be a logical organization, and we already include a section focused purely on future directions in the Discussion section, we have refocused each paragraph of the advantages/disadvantages subsection to concentrate on the advantages and disadvantages per se. In addition, we have made minor changes to the “future directions” section to make it more succinct and practical. These changes can be found at lines ~1016-1077, pg 33-34.
Reviewer #2 (Recommendations For The Authors):
Below are some more detailed points that will hopefully help to further improve the quality and scope of the manuscript.
- While it is certainly favorable for many questions to measure large-scale activity from many brain regions, the introduction appears to suggest that this is a prerequisite to understanding multimodal decision-making. This is based on the argument that combining multiple recordings with movement indicators will 'necessarily obscure the true spatial correlation structures'. However, I don't understand why this is the case or what is meant by 'true spatial correlation structures'. Aren't there many earlier studies that provided important insights from individual cortical areas? It would be helpful to improve the writing to make this argument clearer.
Authors’ Response: The reviewer makes an excellent point and we have re-worded the manuscript appropriately, to reflect the following clarifications. These changes can be found at ~lines 58-71, pg. 2.
We believe you are referring to the following passage from the introduction:
“Furthermore, the arousal dependence of membrane potential across cortical areas has been shown to be diverse and predictable by a temporally filtered readout of pupil diameter and walking speed (Shimoaka et al, 2018). This makes simultaneous recording of multiple cortical areas essential for comparison of the dependence of their neural activity on arousal/movement, because combining multiple recording sessions with pupil dilations and walking bouts of different durations will necessarily obscure the true spatial correlation structures.”
Here, we do not mean to imply that earlier studies of individual cortical areas are of no value. This argument is provided as an example, of which there are others, of the idea that, for sequences or distributed encoding schemes that simultaneously span many cortical areas that are too far apart to be simultaneously imaged under conventional 2-photon imaging, or are too sparse to be discovered with 1-photon widefield imaging, there are some advantages of our new methods over conventional imaging methods that will allow for truly novel scientific analyses and insights.
The general idea of the present example, based on the findings of Shimoaka et al, 2018, is that it is not possible to directly combine and/or compare the correlations between behavior and neural activity across regions that were imaged in separate sessions, because the correlations between behavior and neural activity in each region appear to depend on the exact time since the behavior began (Shimoaka et al, 2018), in a manner that differs across regions. So, for example, if one were to record from visual cortex in one session with mostly brief walk bouts, and then from somatosensory cortex in a second session with mostly long walk bouts, any inferred difference between the encoding of walk speed in neural activity between the two areas would run the risk of being contaminated by the “temporal filtering” effect shown in Shimoaka et al, 2018. However, this would not be the case in our recordings, because the distribution of behavior durations corresponding to our recorded neural activity across areas will be exactly the same, because they were recorded simultaneously.
- The text describes different timescales of neural activity but is an imaging rate of 3 Hz fast enough to be seen as operating at the temporal dynamics of the behavior? It appears to me that the sampling rate will impose a hard limit on the speed of correlations that can be observed across regions. While this might be appropriate for relatively slow behaviors and spontaneous fluctuations in arousal, sensory processing and decision formation likely operate on faster time scales below 100ms which would even be problematic at 10 Hz which is proposed as the ideal imaging speed in the manuscript.
Authors’ Response: Imaging rate is always a concern and the limitations of this have been discussed in other manuscripts. We will remind the reader of these limitations, which must always be kept in mind when interpreting fluorescence based neural activity data.
Previous studies imaging on a comparable yet more limited spatial scale (Stringer et al, 2019) used an imaging speed of ~1 Hz. With this in view, our work represents an advance both in spatial extent of imaged cortex and in imaging speed. Specifically, we believe that ~1 Hz imaging may be sufficient to capture flip/flop type transitions between low and high arousal states that persist in general for seconds to tens of seconds, and that ~3-5 Hz imaging likely provides additional information about encoding of spontaneous movements and behavioral syllables/motifs.
Indeed, even 10 Hz imaging would not be fast enough to capture the detailed dynamics of sensory processing and decision formation, although these speeds are likely sufficient to capture “stable” encodings of sensory representations and decisions that must be maintained during a task, for example with delayed match-to-sample tasks.
In general we are further developing our preparations to allow us to perform simultaneous widefield imaging and Neuropixels recordings, and to perform simultaneous 1.2 x 1.2 mm 2-photon imaging and visually guided patch clamp recordings.
Both of these techniques will allow us to combine information across both the slow and fast timescales that you refer to in your question.
We have clarified these points in the Introduction and Discussion sections, at ~lines ~93-105, pg 3, and ~lines 979-983, pg 31 and ~lines 1039-1045, pg 33, respectively.
- The dorsal mount is very close to the crystal skull paper and it was ultimately not clear to me if there are still important differences aside from the headbar design that a reader should be aware of. If they exist, it would be helpful to make these distinctions a bit clearer. Also, the sea shell implants from Ghanbari et al in 2019 would be an important additional reference here.
Authors’ Response: We have added brief references to these issues in our revised manuscript at ~lines 89-97, pg 3:
Although our dorsal mount preparation is based on the “crystal skull paper” (Kim et al, 2016), which we reference, the addition of a novel 3-D printable titanium headpost, support arms, light shields, and modifications to the surgical protocols and CCF alignment represent significant advances that made this preparation useable for pan-cortical imaging using the Thorlabs mesoscope. In fact, we were in direct communication with Cris Niell, a UO professor and co-author on the original Kim et al, 2016 paper, during the initial development of our preparation, and he and members of his lab consulted with us in an ongoing manner to learn from our successful headpost and other hardware developments. Furthermore, all of our innovations for data acquisition, imaging, and analysis apply equally to both our dorsal mount and side mount preparations.
Thank you for mentioning the Ghanbari et al, 2019 paper on the transparent polymer skull method, “See Shells.” We were in fact not aware of this study. However, it should be noted that their preparation seems to, like the crystal skull preparation and our dorsal mount preparation, be limited to bilateral dorsal cortex and not to include, as does our cranial window side mount preparation and the through-the-skull widefield preparation of Esmaeili et al, 2021, a fuller range of lateral cortical areas, including primary auditory cortex.
- When using the lateral mount, rotating the objective, rather than the animal, appears to be preferable to reduce the stress on the animal. I also worry that the rather severe head tilt could be an issue when training animals in more complex behaviors and would introduce an asymmetry between the hemispheres due to the tilted body position. Is there a strong reason why the authors used water instead of an imaging gel to resolve the issue with the meniscus?
Authors’ Response: Our mice readily adapted to the 22.5 degree head tilt and learned to perform 2-alternative forced choice (2-AFC) auditory and visual tasks in this situation (Hulsey et al, 2024; Cell Reports). The advantages and limitations of such a rotation of the mouse, and possible ways to alleviate these limitations, as detailed in the following paragraphs, are now discussed more thoroughly in the revised manuscript. (See ~line 235, pg. 7)
One can look at Supplementary Movie 1 for examples of the relatively similar behavior between the dorsal mount (not rotated) and side mount (rotated) preparations. We do not have behavioral data from mice that were placed in both configurations. Our preliminary comparisons across mice indicates that side and dorsal mount mice show similar behavioral variability. We have added brief additional mention of these considerations on ~lines 235-250, pg 7.
It was in general important to make sure that the distance between the wheel and all four limbs was similar for both preparations. In particular, careful attention must be paid to the positioning of the front limbs in the side mount mice so that they are not too high off the wheel. This can be accomplished by a slight forward angling of the left support arm for side mount mice.
Although it would in principle be nearly possible to image the side mount preparation in the same optical configuration that we do without rotating the mouse, by rotating the objective 20 degrees to the right of vertical, we found that the last 2-3 degrees of missing rotation (our preparation is rotated 22.5 degrees left, which is more than the full available 20 degrees rotation of the objective), along with several other factors, made this undesirable. First, it was very difficult to image auditory areas without the additional flexibility to rotate the objective more laterally. Second, it was difficult or impossible to attach the horizontal light shield and to establish a water meniscus with the objective fully rotated. One could use gel instead (which we found to be optically inferior to water), but without the horizontal light shield, the UV and IR LEDs can reach the PMTs via the objective and contaminate the image or cause tripping of the PMT. Third, imaging the right pupil and face of the mouse is difficult to impossible under these conditions because the camera would need the same optical access angle as the objective, or would need to be moved down toward the air table and rotated up 20 degrees, in which case its view would be blocked by the running wheel and other objects mounted on the air table.
- In parts, the description of the methods is very specific to the Thorlabs mesoscope which makes it harder to understand the general design choices and challenges for readers that are unfamiliar with that system. Since the Mesoscope is very expensive and therefore unavailable to many labs in the field, I think it would increase the reach of the manuscript to adjust the writing to be less specific for that system but instead provide general guidance that could also be helpful for other systems. For example (but not exclusively) lines 231-234 or lines 371 and below are very Thorlabs-specific.
Authors’ Response: We have revised the manuscript so that it is more generally applicable to mesoscopic methods.
We will make revisions as you suggest where possible, although we have limited experience with the other imaging systems that we believe you are referring to. However, please note that we already mentioned at least one other comparable system in the original eLife reviewed pre-print (Diesel 2p, line 209; Yu and Smith, 2021).
Here are a couple of examples of how we have broadened our description:
(1) On lines ~231-234, pg 7, we write:
“However, if needed, the objective of the Thorlabs mesoscope may be rotated laterally up to +20 degrees for direct access to more ventral cortical areas, for example if one wants to use a smaller, flat cortical window that requires the objective to be positioned orthogonally to the target region.”
Here have modified this to indicate that one may in general rotate their objective lens if their system allows it. Some systems, such as the Thorlabs Bergamo microscope and the Sutter MOM system, allow more than 20 degrees of rotation.
(2) On line ~371, pg 11, we write:
“This technique required several modifications of the auxiliary light-paths of the Thorlabs mesoscope”
Here, we have changed the writing to be more general such as “may require…of one’s microscope.”
Thank you for these valuable suggestions.
- Lines 287-299: Could the authors quantify the variation in imaging depth, for example by quantifying to which extent the imaging depth has to be adjusted to obtain the position of the cortical surface across cortical areas? Given that curvature is a significant challenge in this preparation this would be useful information and could either show that this issue is largely resolved or to what extent it might still be a concern for the interpretation of the obtained results. How large were the required nominal corrections across imaging sites?
Authors’ Response: This information was provided previously (lines 297-299):
“In cases where we imaged multiple small ROIs, nominal imaging depth was adjusted in an attempt to maintain a constant relative cortical layer depth (i.e. depth below the pial surface; ~200 micrometer offset due to brain curvature over 2.5 mm of mediolateral distance, symmetric across the center axis of the window).”
This statement is based on a qualitative assessment of cortical depth based on neuron size and shape, the density of neurons in a given volume of cortex, the size and shape of blood vessels, and known cortical layer depths across regions. A ground-truth measurement of this depth error is beyond the scope of the present study. However, we do specify the type of glass, thickness, and curvature that we use, and the field curvature characterization of the Thorlabs mesoscope is given in Fig. 6 of the Sofroniew et al, 2016 eLife paper.
In addition, we have provided some documentation of online fast-z correction parameters on our GitHub page at:
https://github.com/vickerse1/mesoscope_spontaneous/tree/main/online_fast_z_correction
,and some additional relevant documentation can be found in our publicly available data repository on FigShare+ at: https://doi.org/10.25452/figshare.plus.c.7052513
- Given the size of the implant and the subsequent work attachments, I wonder to which extent the field of view of the animal is obstructed. Did the authors perform receptive field mapping or some other technique that can estimate the size of the animals' remaining field of view?
Authors’ Response: The left eye is pointed down ~22.5 degrees, but we position the mouse near the left edge of the wheel to minimize the degree to which this limits their field of view. One may view our Fig. 1 and Suppl Movies 1 and 6 to see that the eyes on the left and right sides are unobstructed by the headpost, light shields, and support arms. However, other components of the experimental setup, such as the speaker, cameras, etc. can restrict a few small portions of the visual field, depending on their exact positioning.
The facts that mice responded to left side visual stimuli in preliminary recordings during our multimodal 2-AFC task, and that the unobstructed left and right camera views, along with pupillometry recordings, showed that a significant portion of the mouse’s field of view, from either side, remains intact in our preparation.
We have clarified these points in the text at ~lines 344-346, pg. 11.
- Line 361: What does movie S7 show in this context? The movie seems to emphasize that the observed calcium dynamics are not driven by movement dynamics but it is not clear to me how this relates to the stimulation of PV neurons. The neural dynamics in the example cell are also not very clear. It would be helpful if this paragraph would contain some introduction/motivation for the optogenetic stimulation as it comes a bit out of the blue.
Authors’ Response: This result was presented for two reasons.
First, we showed it as a control for movement artifacts, since inhibition of neural activity enhances the relative prominence of non-activity dependent fluorescence that is used to examine the amplitude of movement-related changes in non-activity dependent fluorescence (e.g. movement artifacts). We have included a reference to this point at ~lines 587-588, pg 18.
Second, we showed it as a demonstration of how one may combine optogenetics with imaging in mesoscopic 2-P imaging. References to this point were already present in the original version of the manuscript (the eLife “ reviewed preprint”).
- Lines 362-370: This paragraph and some of the following text are quite technical and would benefit from a better description and motivation of the general workflow. I have trouble following what exactly is done here. Are the authors using an online method to identify the CCF location of the 2p imaging based on the vessel pattern? Why is it important to do this during the experiment? Wouldn't it be sufficient to identify the areas of interest based on the vessel pattern beforehand and then adjust the 2p acquisition accordingly? Why are they using a dial, shutter, and foot pedal and how does this relate to the working distance of the objective? Does the 'standardized cortical map' refer to the Allen common coordinate framework?
Authors’ Response: We have revised this section to make it more clear.
Currently, the general introduction to this section appears in lines 349-361. Starting in line 362, we currently present the technical considerations needed to implement the overall goals stated in that first paragraph of this section.
In general we use a post-hoc analysis step to confirm the location of neurons recorded with 2-photon imaging. We use “online” juxtaposition of the multimodal map image with overlaid CCF with the 2-photon image by opening these two images next to each other on the ScanImage computer and matching the vasculature patterns “by eye”. We have made this more clear in the text so that the interested reader can more readily implement our methods.
By use of the phrase “standardized cortical map” in this context, we meant to point out that we had not decided a priori to use the Allen CCF v3.0 when we started working on these issues.
- Does Fig. 2c show an example of the online alignment between widefield and 2p data? I was confused here since the use of suite2p suggests that this was done post-recording. I generally didn't understand why the user needed to switch back and forth between the two modes. Doesn't the 2p image show the vessels already? Also, why was an additional motorized dichroic to switch between widefield and 2p view needed? Isn't this the standard in most microscopes (including the Thorlabs scopes)?
Authors’ Response: We have explained this methodology more clearly in the revised manuscript, both at ~lines 485-500, pg 15-16, and ~lines 534-540, pg 17.
The motorized dichroic we used replaced the motorized mirror that comes with the Thorlabs mesoscope. We switched to a dichroic to allow for near-simultaneous optogenetic stimulation with 470 nm blue light and 2-photon imaging, so that we would not have to move the mirror back and forth during live data acquisition (it takes a few seconds and makes an audible noise that we wanted to avoid).
Figure 2c shows an overview of our two step “offline” alignment process. The image at the right in the bottom row labeled “2” is a map of recorded neurons from suite2p, determined post-hoc or after imaging. In Fig. 2d we show what the CCF map looks like when it’s overlaid on the neurons from a single suite2p session, using our alignment techniques. Indeed, this image is created post-hoc and not during imaging. In practice, “online” during imaging, we would have the image at left in the bottom row of Fig. 2c (i.e. the multimodal map image overlaid onto an image of the vasculature also acquired on the widefield rig, with the 22.5 degree rotated CCF map aligned to it based on the location of sensory responses) rotated 90 degrees to the left and flipped over a horizontal mirror plane so that its alignment matches that of the “online” 2-photon acquisition image and is zoomed to the same scale factor. Then, we would navigate based on vasculature patterns “by-eye” to the desired CCF areas, and confirm our successful 2-photon targeting of predetermined regions with our post-hoc analysis.
- Why is the widefield imaging done through the skull under anesthesia? Would it not be easier to image through the final window when mice have recovered? Is the mapping needed for accurate window placement?
Authors’ Response: The headpost and window surgeries are done 3-7 days apart to increase success rate and modularize the workflow. Multimodal mapping by widefield imaging is done through the skull between these two surgeries for two major reasons. First, to make efficient use of the time between surgeries. Second, to allow us to compare the multimodal maps to skull landmarks, such as bregma and lambda, for improved alignment to the CCF.
Anesthesia was applied to prevent state changes and movements of the mouse, which can produce large, undesired effects on neural responses in primary sensory cortices in the context of these mapping experiments. We sometimes re-imaged multimodal maps on the widefield microscope through the window, roughly every 30-60 days or whenever/if significant changes in vasculature pattern became apparent.
We have clarified these points in the main text at ~lines 510-522, pg 20-21, and we added a link to our new supplementary material documenting the changes observed in the window preparation over time:
https://github.com/vickerse1/mesoscope_spontaneous/blob/main/window_preparation_stability.pdf
Thank you for these questions.
- Lines 445 and below: Reducing the noise from resonant scanners is also very relevant for many other 2p experiments so it would be helpful to provide more general guidance on how to resolve this problem. Is the provided solution only applicable to the Thorlabs mesoscope? How hard would it be to adjust the authors' noise shield to other microscopes? I generally did not find many additional details on the Github repo and think readers would benefit from a more general explanation here.
Authors’ Response: Our revised Github repository has been modified to include more details, including both diagrams and text descriptions of the sound baffle, respectively:
However, we can not presently disclose our confidential provisional patent application. Complete design information will likely be available in early 2025 when our full utility patent application is filed.
With respect to your question, yes, this technique is adaptable to any resonant scanner, or, for that matter, any complicated 3D surface that emits sound. We first 3D scan the surface, and then we reverse engineer a solid that fully encapsulates the surface and can be easily assembled in parts with bolts and interior foam that allow for a tight fit, in order to nearly completely block all emitted sound.
It is this adaptability that has prompted us to apply for a full patent, as we believe this technique will be quite valuable as it may apply to a potentially large number of applications, starting with 2-photon resonant scanners but possibly moving on to other devices that emit unwanted sound.
- Does line 458 suggest that the authors had to perform a 3D scan of the components to create the noise reduction shield? If so, how was this done? I don't understand the connection between 3D scanning and printing that is mentioned in lines 464-466.
Authors’ Response: We do not want to release full details of the methodology until the full utility patent application has been submitted. However, we have now included a simplified text description of the process on our GitHub page and included a corresponding link in the main text:
We also clarified in the main text, at the location that you indicate, why the 3D scanning is a critical part of our novel 3D-design, printing, and assembly protocol.
- Lines 468 and below: Why is it important to align single-cell data to cortical areas 'directly on the 2-photon microscope'? Is this different from the alignment discussed in the paragraph above? Why not focus on data interpretation after data acquisition? I understand the need to align neural data to cortical areas in general, I'm just confused about the 'on the fly' aspect here and why it seems to be broken out into two separate paragraphs. It seems as if the text in line 485 and below could also be placed earlier in the text to improve clarity.
Authors’ Response: Here by “such mapping is not routinely possible directly on the 2-photon mesoscope” what we mean is that it is not possible to do multimodal mapping directly on the mesoscope - it needs to be done on the widefield imaging rig (a separate microscope). Then, the CCF is mapped onto the widefield multimodal map, which is overlaid on an image of the vasculature (and sometimes also the skull) that was also acquired on the widefield imaging rig, and the vasculature is used as a sort of Rosetta Stone to co-align the 2-photon image to the multimodal map and then, by a sort of commutative property of alignment, to the CCF, so that each individual neuron in the 2-photon image can be assigned a unique CCF area name and numerical identifier for subsequent analysis.
We have clarified this in the text, thank you.
The Python code for aligning the widefield and 2-photon vessel images would also be of great value for regular 2p users. It would strongly improve the impact of the paper if the repository were better documented and the code would be equally applicable for alignment of imaging data with smaller cranial windows.
Authors’ Response: All of the code for multimodal map, CCF, and 2-photon image alignment is, in fact, already present on the GitHub page. We have made some minor improvements to the documentation, and readers are more than welcome to contact us for additional help.
Specifically, the alignment you refer to starts in cell #32 of the meso_pre_proc_1.ipynb notebook. In general the notebooks are meant to be run sequentially, starting with cell #1 of meso_pre_proc_1, then going to the next cell etc…, then moving to meso_pre_proc_2, etc… The purpose of each cell is labeled at the top of the cell in a comment.
We now include a cleaned, abridged version of the meso_pre_proc_1.pynb notebook that contains only the steps needed for alignment, and included a direct link to this notebook in the main text:
Rotated CCF maps are in the CCF map rotation folder, in subfolders corresponding to the angle of rotation.
Multimodal map creation involves use of the SensoryMapping_Vickers_Jun2520.m script in the Matlab folder.
We updated the main text to clarify these points and included direct links to scripts relevant to each processing step.
- Figure 4a: I found it hard to see much of the structure in the Rastermap projection with the viridis colormap - perhaps also because of a red-green color vision impairment. Correspondingly, I had trouble seeing some of the structure that is described in the text or clearer differences between the neuron sortings to PC1 and PC2. Is the point of these panels to show that both PCs identify movement-aligned dynamics or is the argument that they isolate different movement-related response patterns? Using a grayscale colormap as used by Stringer et al might help to see more of the many fine details in the data.
Authors’ Response: In Fig. 4a the viridis color range is from blue to green to yellow, as indicated in the horizontal scale bar at bottom right. There is no red color in these Rastermap projections, or in any others in this paper. Furthermore, the expanded Rastermap insets in Figs. S4 and S5 provide additional detailed information that may not be clear in Fig 4a and Fig 5a.
We prefer, therefore, not to change these colormaps, which we use throughout the paper.
We have provided grayscale png versions of all figures on our GitHub page:
https://github.com/vickerse1/mesoscope_spontaneous/tree/main/grayscale_figures
In Fig 4a the point of showing both the PC1 and PC2 panels is to demonstrate that they appear to correspond to different aspects of movement (PC1 more to transient walking, both ON and OFF, and PC2 to whisking and sustained ON walk/whisk), and to exhibit differential ability to identify neurons with positive and negative correlations to arousal (PC1 finds both, both PC2 seems to find only the ON neurons).
We now clarify this in the text at ~lines 696-710, pg 22.
- I find panel 6a a bit too hard to read because the identification and interpretation of the different motifs in the different qualitative episodes is challenging. For example, the text mentions flickering into motif 13 during walk but the majority of that sequence appears to be shaped by what I believe to be motif 11. Motif 11 also occurs prominently in the oscillate state and the unnamed sequence on the left. Is this meaningful or is the emphasis here on times of change between behavioral motifs? The concept of motif flickering should be better explained here.
Authors’ Response: Here motif 13 corresponds to a syllable that might best be termed “symmetric and ready stance”. This tends to occur just before and after walking, but also during rhythmic wheel balancing movements that appear during the “oscillate” behavior.
The intent of Fig. 6a is to show that each qualitatively identified behavior (twitch, whisk, walk, and oscillate) corresponds to a period during which a subset of BSOiD motifs flicker back and forth, and that the identity of motifs in this subset differs across the identified qualitative behaviors. This is not to say that a particular motif occurs only during a single identified qualitative behavior. Admittedly, the identification of these qualitative behaviors is a bit arbitrary - future versions of BSOiD (e.g. ASOiD) in fact combine supervised (i.e. arbitrary, top down) and unsupervised (i.e. algorithmic, objective, bottom-up) methods of behavior segmentation in attempt to more reliably identify and label behaviors.
Flickering appears to be a property of motif transitions in raw BSOiD outputs that have not been temporally smoothed. If one watches the raw video, it seems that this may in fact be an accurate reflection of the manner in which behaviors unfold through time. Each behavior could be thought of, to use terminology from MOSEQ (B Datta), as a series of syllables strung together to make a phrase or sentence. Syllables can repeat over either fast or slow timescales, and may be shared across distinct words and sentences although the order and frequency of their recurrence will likely differ.
We have clarified these points in the main text at ~lines 917-923, pg 29, and we added motif 13 to the list of motifs for the qualitative behavior labeled “oscillate” in Fig. 6a.
- Lines 997-998: I don't understand this argument. Why does the existence of different temporal dynamics make imaging multiple areas 'one of the keys to potentially understanding the nature of their neuronal activity'?
Authors’ Response: We believe this may be an important point, that comparisons of neurobehavioral alignment across cortical areas cannot be performed by pooling sessions that contain different distributions of dwell times for different behaviors, if in fact that dependence of neural activity on behavior depends on the exact elapsed time since the beginning of the current behavioral “bout”. Again, other reasons that imaging many areas simultaneously would provide a unique advantage over imaging smaller areas one at a time and attempting to pool data across sessions would include the identification of sequences or neural ensembles that span many areas across large distances, or the understanding of distributed coding of behavior (an issue we explore in an upcoming paper).
We have clarified these points at the location in the Discussion that you have identified. Thank you for your questions and suggestions.
Minor
Line 41: What is the difference between decision, choice, and response periods?
Authors’ Response: This now reads “...temporal separation of periods during which cortical activity is dominated by activity related to stimulus representation, choice/decision, maintenance of choice, and response or implementation of that choice.”
Line 202: What does ambulatory mean in this context?
Authors’ Response: Here we mean that the mice are able to walk freely on the wheel. In fact they do not actually move through space, so we have changed this to read “able to walk freely on a wheel, as shown in Figs. 1a and 1b”.
Is there a reason why 4 mounting posts were used for the dorsal mount but only 1 post was sufficient for the lateral mount?
Authors’ Response: Here, we assume you mean 2 posts for the side mount and 4 posts for the dorsal mount.
In general our idea was to use as many posts as possible to provide maximum stability of the preparations and minimize movement artifacts during 2-photon imaging. However, the design of the side mount headpost precluded the straight-forward or easy addition of a right oriented, second arm to its lateral/ventral rim - this would have blocked access of both the 2-photon objective and the right face camera. In the dorsal mount, the symmetrical headpost arms are positioned further back (i.e. posterior), so that the left and right face cameras are not obscured.
When we created the side mount preparation, we discovered that the 2 vertical 1” support posts were sufficient to provide adequate stability of the preparation and minimize 2-photon imaging movement artifacts. The side mount used two attachment screws on the left side of the headpost, instead of the one screw per side used in the dorsal mount preparation.
We have included these points/clarifications in the main text at ~lines 217-230, pg 7.
Figure S1g appears to be mislabeled.
Authors’ Response: Yes, on the figure itself that panel was mislabeled as “f” in the original eLife reviewed preprint. We have changed this to read “g”.
Line 349 and below: Why is the method called pseudo-widefield imaging?
Authors’ Response: On the mesoscope, broad spectrum fluorescent light is passed through a series of excitation and emission filters that, based on a series of tests that we performed, allow both reflected blue light and epifluorescence emitted (i.e. Stokes-shifted) green light to reach the CCD camera for detection. Furthermore, the CCD camera (Thorlabs) has a much smaller detector chip than that of the other widefield cameras that we use (RedShirt Imaging and PCO), and we use it to image at an acquisition speed of around 10 Hz maximum, instead of ~30-50 Hz, which is our normal widefield imaging acquisition speed (it also has a slower readout than what we would consider to be a standard or “real” 1-photon widefield imaging camera).
For these 3 reasons we refer to this as “pseudo-widefield” imaging. We would not use this for sensory activity mapping on the mesoscope - we primarily use it for mapping cortical vasculature and navigating based on our multimodal map to CCF alignment, although it is actually “contaminated” with some GCaMP6s activity during these uses.
We have briefly clarified this in the text.
Figures 4d & e: Do the colors show mean correlations per area? Please add labels and units to the colorbars as done in panel 4a.
Authors’ Response: For both Figs 4 and 5, we have added the requested labels and units to each scale bar, and have relabeled panels d to say “Rastermap CCF area cell densities”, and panels e to say “mean CCF area corrs w/ neural activity.”
Thank you for catching these omissions/mislabelings.
Line 715: what is superneuron averaging?
Authors’ Response: This refers to the fact that when Rastermap displays more than ~1000 neurons it averages the activity of each group of adjacent 50 neurons in the sorting to create a single display row, to avoid exceeding the pixel limitations of the display. Each single row representing the average activity of 50 neurons is called a “superneuron” (Stringer et al, 2023; bioRxiv).
We have modified the text to clarify this point.
Line 740: it would be good to mention what exactly the CCF density distribution quantifies.
Authors’ Response: In each CCF area, a certain percentage of neurons belongs to each Rastermap group. The CCF density distribution is the set of these percentages, or densities, across all CCF areas in the dorsal or side mount preparation being imaged in a particular session. We have clarified this in the text.
Line 745: what does 'within each CCF' mean? Does this refer to different areas?
Authors’ Response: The corrected version of this sentence now reads: “Next, we compared, across all CCF areas, the proportion of neurons within each CCF area that exhibited large positive correlations with walking speed and whisker motion energy.”
How were different Rastermap groups identified? Were they selected by hand?
Authors’ Response: Yes, in Figs. 4, 5, and 6, we selected the identified Rastermap groups “by hand”, based on qualitative similarity of their activity patterns. At the time, there was no available algorithmic or principled means by which to split the Rastermap sort. The current, newer version of Rastermap (Stringer et al, 2023) seems to allow for algorithmic discretization of embedding groups (we have not tested this yet), but it was not available at the time that we performed these preliminary analyses.
In terms of “correctness” of such discretization or group identification, we intend to address this issue in a more principled manner in upcoming publications. For the purposes of this first paper, we decided that manual identification of groups was sufficient to display the capabilities and outcomes of our methods.
We clarify this point briefly at several locations in the revised manuscript, throughout the latter part of the Results section.
Reviewer #3 (Recommendations For The Authors):
In "supplementary figures, protocols, methods, and materials", Figure S1 g is mislabeled as Figure f.
Authors’ Response: Yes, on the figure itself this panel was mislabeled as “f” in the original reviewed preprint. We have changed this to read “g”.
In S1 g, the success rate of the surgical procedure seems quite low. Less than 50% of the mice could be imaged under two-photon. Can the authors elaborate on the criteria and difficulties related to their preparations?
Authors’ Response: We will elaborate on the difficulties that sometimes hinder success in our preparations in the revised manuscript.
The success rate indicated to the point of “Spontaneous 2-P imaging (window) reads 13/20, which is 65%, not 50%. The drop to 9/20 by the time one gets to the left edge of “Behavioral Training” indicates that some mice do not master the task.
Protocol I contains details of the different ways in which mice either die or become unsuitable or “unsuccessful” at each step. These surgeries are rather challenging - they require proper instruction and experience. With the current protocol, our survival rate for the window surgery alone is as high as 75-100%. Some mice can be lost at headpost implantation, in particular if they are low weight or if too much muscle is removed over the auditory areas. Finally, some mice survive windowing but the imageable area of the window might be too small to perform the desired experiment.
We have added a paragraph detailing this issue in the main text at ~lines 287-320, pg 9.
In both Suppl_Movie_S1_dorsal_mount and Suppl_Movie_S1_side_mount provided (Movie S1), the behaviour video quality seems to be unoptimized which will impact the precision of Deeplabcut. As evident, there were multiple instances of mislabeled key points (paws are switched, large jumps of key points, etc) in the videos.
Many tracked points are in areas of the image that are over-exposed.
Despite using a high-speed camera, motion blur is obvious.
Occlusions of one paw by the other paws moving out of frame.
As Deeplabcut accuracy is key to higher-level motifs generated by BSOi-D, can the authors provide an example of tracking by exclusion/ smoothing of mislabeled points (possibly by the median filtering provided by Deeplabcut), this may help readers address such errors.
Authors’ Response: We agree that we would want to carefully rerun and carefully curate the outputs of DeepLabCut before making any strong claims about behavioral identification. As the aim of this paper was to establish our methods, we did not feel that this degree of rigor was required at this point.
It is inevitable that there will be some motion blur and small areas of over-exposure, respectively, when imaging whiskers, which can contain movement components up to ~150 Hz, and when imaging a large area of the mouse, which has planes facing various aspects. For example, perfect orthogonal illumination of both the center of the eye and the surface of the whisker pad on the snout would require two separate infrared light sources. In this case, use of a single LED results in overexposure of areas orthogonal to the direction of the light and underexposure of other aspects, while use of multiple LEDs would partially fix this problem, but still lead to variability in summated light intensity at different locations on the face. We have done our best to deal with these limitations.
We now briefly point out these limitations in the methods text at ~lines 155-160, pg 5.
In addition, we have provided additional raw and processed movies and data related to DeepLabCut and BSOiD behavioral analysis in our FigShare+ repository, which is located at:
https://doi.org/10.25452/figshare.plus.c.7052513
In lines 153-154, the authors mentioned that the Deeplabcut model was trained for 650k iterations. In our experience (100-400k), this seems excessive and may result in the model overfitting, yielding incorrect results in unseen data. Echoing point 4, can the authors show the accuracy of their Deeplabut model (training set, validation set, errors, etc).
Authors’ Response: Our behavioral analysis is preliminary and is included here as an example of our methods, and not to make claims about any specific result. Therefore we believe that the level of detail that you request in our DeepLabCut analysis is beyond the scope of the current paper. However, we would like to point out that we performed many iterations of DeepLabCut runs, across many mice in both preparations, before converging on these preliminary results. We believe that these results are stable and robust.
We believe that 650k iterations is within the reasonable range suggested by DLC, and that 1 million iterations is given as a reasonable upper bound. This seems to be supported by the literature for example, see Willmore et al, 2022 (“Behavioral and dopaminergic signatures of resilience”, Nature, 124:611, 124-132). Here, in a paper focused squarely on behavioral analysis, DLC training was run with 1.3 million iterations with default parameters.
We now note, on ~lines 153-154, pg 5, that we used 650K iterations, a number significantly less than the default of 1.03 million, to avoid overfitting.
In lines 140-141, the authors mentioned the use of slicing to downsample their data. Have any precautions, such as a low pass filter, been taken to avoid aliasing?
Authors’ Response: Most of the 2-photon data we present was acquired at ~3 Hz and upsampled to 10 Hz. Most of the behavioral data was downsampled from 5000 Hz to 10 Hz by slicing, as stated. We did not apply any low-pass filter to the behavioral data before sampling. The behavioral variables have heterogeneous real sampling/measurement rates - for example, pupil diameter and whisker motion energy are sampled at 30 Hz, and walk speed is sampled at 100 Hz. In addition, the 2-photon acquisition rate varied across sessions.
These facts made principled, standardized low-pass filtering difficult to implement. We chose rather to use a common resampling rate of 10 Hz in an unbiased manner. This downsampled 10 Hz rate is also used by B-SOiD to find transitions between behavioral motifs (Hsu and Yttri, 2021).
We do not think that aliasing is a major factor because the real rate of change of our Ca2+ indicator fluorescence and behavioral variables was, with the possible exception of whisker motion energy, likely at or below 10 Hz.
We now include a brief statement to this effect in the methods text at ~lines 142-146, pg. 4.
Line 288-299, the authors have made considerable effort to compensate for the curvature of the brain which is particularly important when imaging the whole dorsal cortex. Can the authors provide performance metrics and related details on how well the combination of online curvature field correction (ScanImage) and fast-z "sawtooth"/"step" (Sofroniew, 2016)?
Authors’ Response: We did not perform additional “ground-truth” experiments that would allow us to make definitive statements concerning field curvature, as was done in the initial eLife Thorlabs mesoscope paper (Sofroniew et al, 2016).
We estimate that we experience ~200 micrometers of depth offset across 2.5 mm - for example, if the objective is orthogonal to our 10 mm radius bend window and centered at the apex of its convexity, a small ROI located at the lateral edge of the side mount preparation would need to be positioned around 200 micrometers below that of an equivalent ROI placed near the apex in order to image neurons at the same cortical layer/depth, and would be at close to the same depth as an ROI placed at or near the midline, at the medial edge of the window. We determined this by examining the geometry of our cranial windows, and by comparing z-depth information from adjacent sessions in the same mouse, the first of which used a large FOV and the second of which used multiple small FOVs optimized so that they sampled from the same cortical layers across areas.
We have included this brief explanation in the main text at ~lines 300-311, pg 9.
In lines 513-515, the authors mentioned that the vasculature pattern can change over the course of the experiment which then requires to re-perform the realignment procedure. How stable is the vasculature pattern? Would laser speckle contrast yield more reliable results?
Authors’ Response: In general the changes in vasculature we observed were minimal but involved the following: i) sometimes a vessel was displaced or moved during the window surgery, ii) sometimes a vessel, in particular the sagittal sinus, enlarged or increased its apparent diameter over time if it is not properly pressured by the cranial window, and iii) sometimes an area experiencing window pressure that is too low could, over time, show outgrowth of fine vascular endings. The most common of these was (i), and (iii) was perhaps the least common. In general the vasculature was quite stable.
We have added this brief discussion of potential vasculature changes after cranial window surgery to the main text at ~lines 286-293, pg 9.
We already mentioned, in the main text of the original eLife reviewed preprint, that we re-imaged the multimodal map (MMM) every 30-60 days or whenever changes in vasculature are observed, in order to maintain a high accuracy of CCF alignment over time. See ~lines 507-511, pg 16.
We are not very familiar with laser speckle contrast, and it seems like a technique that could conceivably improve the fine-grained accuracy of our MMM-CCF alignment in some instances. We will try this in the future, but for now it seems like our alignments are largely constrained by several large blood vessels present in any given FOV, and so it is unclear how we would incorporate such fine-grained modifications without applying local non-rigid manipulations of our images.
In lines 588-598, the authors mentioned that the occasional use of online fast-z corrections yielded no difference. However, it seems that the combination of the online fast-z correction yielded "cleaner" raster maps (Figure S3)?
Authors’ Response: The Rastermaps in Fig S3a and b are qualitatively similar. We do not believe that any systematic difference exists between their clustering or alignments, and we did not observe any such differences in other sessions that either used or didn’t use online fast-z motion correction.
We now provide raw data and analysis files corresponding to the sessions shown in Fig S3 (and other data-containing figures) on FigShare+ at:
https://doi.org/10.25452/figshare.plus.c.7052513
Ideally, the datasets contained in the paper should be available on an open repository for others to examine. I could not find a clear statement about data availability. Please include a linked repo or state why this is not possible.
Authors’ Response: We have made ~500 GB of raw data and preliminary analysis files publicly available on FigShare+ for the example sessions shown in Figures 2, 3, 4, 5, 6, S3, and S6. We ask to be cited and given due credit for any fair use of this data.
The data is located here:
Vickers, Evan; A. McCormick, David (2024). Pan-cortical 2-photon mesoscopic imaging and neurobehavioral alignment in awake, behaving mice. Figshare+. Collection:
https://doi.org/10.25452/figshare.plus.c.7052513
We intend to release a complete data set to the public as a Dandiset on the DANDI archive in conjunction with second and third in-depth analysis papers that are currently in preparation.
-
-
fromthemachine.org fromthemachine.orgDEN -1
-
SON Ye R O C K O F . . . S A G E S ? H E A R D E R O R I T R E A L L Y D O E S M E A N "FREEDOM" B R E A D I S L I F E Tying up loose eadds, in a similar vain to the connection between the Burning Bush and universal voting now etched by-stone, there exists a similar missing Link connecting the phrase "it's not a a gam" to Mary Magdeline to a pattern that shows us that the Holy Trinity and our timelines are narrated by a series of names of video game systems and their manufacturers from "Nintendo" to Genesis and the rock of SEGA. Through a "kiss" and the falling of a wallthe words bread and read are tied up and twisted with the story of this Revelation and the heart of the word Creation, "be the reason it's A.D." It's a strong connection between the idea that virtual reality and Heaven are linked by more than simply "technology" but that this message that shows us that these tools for understanding have fallen from the sky in order to help us understand why it is so important, why I call it a moral mandate, that we use this information to follow the map delivered to us in the New Testament and literally end world hunger, and literally heal the sick; because of the change in circumstance revealed to us. These simple things, these few small details that might seem like nothing, or maybe appear to be "changing everything" they are not difficult things to do, in light of Creation, and few would doubt that once we do see them implementied here... the difference between Heaven and Hell will be ever so clear. A while ago, in a place called Kentucky... this story began with a sort of twisted sci-fi experience that explained a kind of "God machine" that could manipulate time and reality, and in that story, in that very detailed and interesting story that I lived through, this machine was keyed to my DNA, in something like the "Ancient technology" of Stargate SG-1 and Atlantis mythology. My kind brother Seth made a few appearances in the story, not actually in person but in fairly decent true to life holograms that I saw and spoke to every once in awhile. He looked a little different, he had long hair; but that's neither here nor there, and he hasn't really had long hair since I was a little boy. He happens to be a genetic engineer, and I happen to be a computer person (although he's that too, now; just nowhere near as good as me... with computers) so the story talked a little bit about how I would probably not have used DNA as a key, since I'm not a retard, and he probably wouldn't either, because works in that field (cyclone, huracan, tornado). So then the key we imagined was something ... well, Who cares what the key is, right? o back to the task at hand, not so long ago, in a place called Plantation I was struck by lightning, literally (well not literally) the answer to a question that nobody knew was implanted in my mind, and it all came from asking a single simple question. I was looking for more chemistry elements in the names of the books of the Holy Bible, after seeing Xenon at the "sort of beginning" of Exodus, where it screams "let there be light" in Linux and chemistry (and I've told you that a hundred times by now). So it didn't take long to follow the light of that word and read Genesis backwards, and see, at the very beginning of that book, Silicon... in reverse. So, what about God's DNA, anyway? What's he really made of? SIM MON S WILD ER ROD DEN BERRY o after seeing Silicon, and connecting that to the numerous attempts I've made to show a message connecting The Matrix to the Fifth Element (as Silicon) describing what it is that God believes we should do with this knowledge--and see that it is narrated as the miracles of Jesus Christ in the New Testament... these names came to me in quick succession, an answer to the question. I suppose any Gene will do, these three though, have a very important tie to the message that connects Joshua's Promised Land of flowing Milk and Honies to ... a kiss that begins the new day (I hope) ... and a message about exactly how we might go about doing magical things like ending world hunger and healing the sick using technology described ... in Star Trek and Stargate. A "religion of the Stars" is being born. That's great... it starts with an earthquake. R.E.M. and a band ... 311. Oooh, I can see it coming down... The Petty Reckless. An evening's love starts with a kiss. Dave Matthews Band. I wanna rock and roll all night and party every day. Adam. I mean Kiss. Are you starting to see a pattern form? Birds, snakes, and aeroplanes? It's that, it's the end of the world as we know it, and I feel fine. In that song we see clues that more than just the Revelation of Christ is narrated by John on an island called Patmos. There yet another Trinity, starting with "Pa" and hearting Taylor Momsen's initials... most likely for a reason... and the Revelation ends with a transition that I hope others will agree with me turns "original sin" into something closer to "obviously salvation" when we finally understand the character that is behind the message of da i of Ra... and begin to see the same design in the names of Asmodai and in this Revelation focusing on freedom and truth that really does suggest Taylor can't talk to me in any way other than "letting freedom sing" in this narrative of kismet and fate and free will and ... then we see that narrative continue in the names of bands, just like the 3/11/11 earthquake is narrated in not just R.E.M.'s song but in the name 311. Just like the 9/11 attack is narrated not just in that same song (released in 1987) and "Inside Job" (released in 2000) but also in "Fucked up world." Dear all of you walking dumb and blind, this same quake is narrated in Taylor's Zombie; waiting for the day to shake, all very similar to Cairo and XP, perhaps a "fad" of doublethink in the minds of the authors singing about a clear prophesy in the Bible; this connection between the day, 3/11 though, and the name of a band and the day of an arrest and the verse Matthew that tells you clearly you have now been baptized in water and fire... it shows us the design of a story whose intent and purpose is to ensure that we no longer allow for things like hurricanes and earthquakes and murder and rape to be "simulated" that we build a better system, that doesn't allow for 'force majeure" to take lives for no reason at all. Not just in band names, but in the angels names too, in all of our names; we see this narration continue. The Holy Water that is central to the baptism of Christ is etched into Taylor's name, between "sen" and "mom" the key to the two Mary's whose names contain the Spanish for "sea" in a sort of enlightenment hidden in plain sight. In "Simmons" the key connection between today, this Biblical Monday, and the word "simulation" that ties to Simpsons and simians and keep it simple stupid, and in Simmons the missing "s" of Kismet, finally completing the question. It's a song and dance that started a long time ago, as you can see from the ancient Hebrew word for "fate" and in more recent years a connection to the ballroom of Atlantis in the Doors 5 to 1 and Dave sang about it in Rapunzel and then Taylor shook a tambourine on the beach only minutes away from me--but never said "hi." The battle of the bands continues tying some door knocking to a juxtaposition between "Sweet Things" and "Knocking on Heavens door" all the way to a Gossip Girl episode where little J asked a question that I can't be sure she knew was related, she said... "who's that, at the door?" What it really all amounts to, though, is the whole world witnessing the Creation of Adam and Eve from a little girl stuttering out "the the" at the sight of the Grinch himself, and then later not even able to get those words off her lips... about seeing how Creation and modern art are inextricably tied to religion, to heaven, and to freedom. The bottom line here, hopefully obvious now, is that you can't keep this message "simple" it's a Matrix woven between more points of light than I can count, and many more that I'm sure you will find. It's a key to seeing how God speaks to me, and to you; and how we are, we really are that voice. Tay, if you don't do something just because God called it "fate" you are significantly more enslaved than if you do--and you wanted to. "Now I see that you and me, were never meant, never meant to be..." she sang before I mentioned her, and before she ever saw me... in a song she calls "Nothing Left to Lose" and I see is not really just another word for freedom. We have plenty to lose by not starting the fire, not the least of which is Heaven itself. Understand what "force majeure" really means to you and I. Ha, by the way. IN CASE YOU FORGOT YESTERDAY'S MESSAGE "DADDY, I WANT IT NOW." VERUKA SALT. whose name means "to see (if) you are the Body of Christ" whined, in the story of Will Why Won Ka, about nothing more or less than Heaven on Hearth, than seeing an end to needless torture and pain. To see if you are the "Salt of the Earth" warming the road to Heaven; honestly to see if you can break through this inane lie of "I don't understand" and realize that breaking this story and talking about what is being presented not just by me and you but by history and God himself is the key to the car that drives us home. To see how Cupid you really are. STOP NODDING, TURN AROUND AND CALL A REPORTER. The story of Willy Wonka ties directly to the Promised Land of Flowing Milk and Honey to me; by showing us a river of chocolate and a the everlasting God starter, (er is it guardian of B stopper) that opens the doors of perception about exactly what kinds of mistake may have been made in the past in this transition to Heaven that we are well on the way of beginning. Here, in the Land of Nod, that is also Eden and also the Heart of the Ark we see warnings about "flowing milk and honey" being akin to losing our stable ecosystem, to losing the stuff of life itself, biology and evolution, and if we don't understand--this is probably exactly the mistake that was made and the cause of the story of Cain and Abel. So here we are talking about genetic engineering and mind uploading and living forever, and hopefully seeing that while all things are possible with God--losing the wisdom of the message of religion is akin to losing life in the Universe and with that any hope of eternal longevity. With some insight into religion, you can connect the idea that without bees our stable ecosystem might collapse, to the birds and the bees, and a message about stability and having more than one way to pollinate the flowers and trees and get some. Janet and Nanna, by the way, both have pretty brown eyes, but that probably comes as no surprise to you. Miss Everything, on the other hand (I hear, does not have brown eyes), leads us to glimpse how this message about the transition of our society might continue on in the New Testament, and suggest that we do need to eat, and have dinner conversation, and that a Last Supper might be a little bit more detrimental to our future than anyone had ever thought, over and over and over again. To see how religion really does make clear that this is what the message is about, to replace the flowing milk we have a "Golden Cow" that epitomizes nothing less than "not listening to Adam" and we have a place that believes the Hammer of Judah Maccabee should be ... extinct. You are wrong. Of course the vibrating light here ties this Gene to another musical piece disclosing something... "Wild Thing" I make your heart sing. You can believe the Guitar Man is here to steal the show and deliver bread for the hungry and for the wise. Here's some, it's not just Imagine Dragons telling you to listen to the radio but Jefferson Starshiptoo, and Live. When you wake up, you can hear God "singing" to you on the radio every single day; many of us already do. He's telling you to listen to me, and I do not understand why you do not. You don't look very Cupid, if you ask me. WHAT DO YOU THINK YOU ARE, DAN RE Y NO LDS? I think we all know what the Rod of Jesus Christ is by now. ​ It is a large glowing testament to freedom and truth, and a statement about blindness and evil that is unmistakable. To say that seeing it is the gateway to Heaven would be an understatement of it's worth, of the implication that not seeing it is obvious Hell when it is linked to everything from nearly every story of the Holy Bible from Isaac to Isaiah to "behold he is to coming" and if you weren't sure if the Hand of God were in action here--it's very clear that it is; that linking Tricky Dick and Watergate to Seagate ... really delivering crystal clear understanding that the foundation of Heaven is freedom and that you have none today because you refuse to see the truth. It is the doorway to seeing that what has been going on in this place hasn't been designed to hide me, but to hide a prosperous future from you--to hide the truth about our existence and the purpose of Creation--that all told, you are standing at the doorstep of Heaven and stammering your feet, closing your eyes, and saying "you don't want to help anyone." If delivering freedom, truth, and equality to you does not a den make, well, you can all suck it ... from God, to you. Between Stargate and Star Trek it's pretty easy to see a roadmap to very quickly and easily be able to end world hunger and heal the sick without drastically changing the way our society works, it's about as simple as a microwave, or a new kind of medicine--except it's not so easy to see why it is that you are so reluctant to talk about the truth that makes these things so easy to do. You see, your lack of regard for anyone anywhere has placed you in a position of weakness, and if you do nothing today, you will not be OK tomorrow. It's pretty easy to see how Roddenberry's name shows that this message comes from God, that he's created this map that starts with an Iron Rod throughout our history proving Creation, whose heart is a Den of Family who care about the truth, and about freedom, and about helping each other--not what you are--you are not that today. Today you are sick, and I'd like you to look at the mirror he's made for you, and be eshamden (or asham). Realize, realize... what you are. What you've become, just as I have... the devil in a sweet, sweet kiss. -Dave J. Matthews .WHSOISKEYAV { border-width: 1px; border-style: dashed; border-color: rgb(15,5,254); padding: 5px; width: 503px; text-align: center; display: inline-block; align: center; p { align: center; } /* THE SCORE IS LOVE FIVE ONE SAFETY ONE FIELD GOAL XIVDAQ: TENNIS OR TINNES? TONNES AND TUPLE(s) */ } <style type="text/css"> code { white-space: pre; } google_ad_client = "ca-pub-9608809622006883"; google_ad_slot = "4355365452"; google_ad_width = 728; google_ad_height = 90; Unless otherwise indicated, this work was written between the Christmas and Easter seasons of 2017 and 2020(A). The content of this page is released to the public under the GNU GPL v2.0 license; additionally any reproduction or derivation of the work must be attributed to the author, Adam Marshall Dobrin along with a link back to this website, fromthemachine dotty org. That's a "." not "dotty" ... it's to stop SPAMmers. :/ This document is "living" and I don't just mean in the Jeffersonian sense. It's more alive in the "Mayflower's and June Doors ..." living Ethereum contract sense [and literally just as close to the Depp/Caster/Paglen (and honorably PK] 'D-hath Transundancesense of the ... new meaning; as it is now published on Rinkeby, in "living contract" form. It is subject to change; without notice anywhere but here--and there--in the original spirit of the GPL 2.0. We are "one step closer to God" ... and do see that in that I mean ... it is a very real fusion of this document and the "spirit of my life" as well as the Spirit's of Kerouac's America and Vonnegut's Martian Mars and my Venutian Hotel ... and *my fusion* of Guy-A and GAIA; and the Spirit of the Earth .. and of course the God given and signed liberties in the Constitution of the United States of America. It is by and through my hand that this document and our X Commandments link to the Bill or Rights, and this story about an Exodus from slavery that literally begins here, in the post-apocalyptic American hartland. Written ... this day ... April 14, 2020 (hey, is this HADAD DAY?) ... in Margate FL, USA. For "official used-to-v TAX day" tomorrow, I'm going to add the "immultible incarnite pen" ... if added to the living "doc/app"--see is the DAO, the way--will initi8 the special secret "hidden level" .. we've all been looking for. Nor do just mean this website or the totality of my written works; nor do I only mean ... this particular derivation of the GPL 2.0+ modifications I continually source ... must be "from this website." I also mean *the thing* that is built from ... bits and piece of blocks of sand-toys; from Ethereum and from Rust and from our hands and eyes working together ... from this place, this cornerstone of the message that is ... written from brick and mortar words and events and people that have come before this poit of the "sealed W" that is this specific page and this time. It's 3:28; just five minutes--or is it four, too layne. This work is not to be redistributed according to the GPL unless all linked media on Youtube and related sites are intact--and historical references to the actual documented history of the art pieces (as I experience/d them) are also available for linking. Wikipedia references must be available for viewing, as well as the exact version of those pages at the time these pieces were written. All references to the Holy Bible must be "linked" (as they are or via ... impromptu in-transit re-linking) to the exact verses and versions of the Bible that I reference. These requirements, as well as the caveat and informational re-introduction to God's DAO above ... should be seen as material modifications to the original GPL2.0 that are retroactively applied to all works distributed under license via this site and all previous e-mails and sites. /s/ wso If you wanna talk to me get me on facebook, with PGP via FlowCrypt or adam at from the machine dotty org -----BEGIN PGP PUBLIC KEY BLOCK-----
this was written sometime i think around 2016. it's hard to recall the exact date; but if you check in the original gitlog there is one that has an original commit.
SONYe
R O C K O F . . . S A G E S ?
**\ **
I T R E A L L Y D O E S M E A N "FREEDOM" B R E A D I S L I F E
Tying up loose eadds, in a similar vain to the connection between the Burning Bush and universal voting now etched by-stone, there exists a similar missing Link connecting the phrase "it's not a a gam" to Mary Magdeline to a pattern that shows us that the Holy Trinity and our timelines are narrated by a series of names of video game systems and their manufacturers from "Nintendo" to Genesis and the rock of SEGA. Through a "kiss" and the falling of a wallthe words bread and read are tied up and twisted with the story of this Revelation and the heart of the word Creation, "be the reason it's A.D." It's a strong connection between the idea that virtual reality and Heaven are linked by more than simply "technology" but that this message that shows us that these tools for understanding have fallen from the sky in order to help us understand why it is so important, why I call it a moral mandate, that we use this information to follow the map delivered to us in the New Testament and literally end world hunger, and literally heal the sick; because of the change in circumstance revealed to us. These simple things, these few small details that might seem like nothing, or maybe appear to be "changing everything" they are not difficult things to do, in light of Creation, and few would doubt that once we do see them implementied here... the difference between Heaven and Hell will be ever so clear.
A while ago, in a place called Kentucky... this story began with a sort of twisted sci-fi experience that explained a kind of "God machine" that could manipulate time and reality, and in that story, in that very detailed and interesting story that I lived through, this machine was keyed to my DNA, in something like the "Ancient technology" of Stargate SG-1 and Atlantis mythology. My kind brother Seth made a few appearances in the story, not actually in person but in fairly decent true to life holograms that I saw and spoke to every once in awhile. He looked a little different, he had long hair; but that's neither here nor there, and he hasn't really had long hair since I was a little boy. He happens to be a genetic engineer, and I happen to be a computer person (although he's that too, now; just nowhere near as good as me... with computers) so the story talked a little bit about how I would probably not have used DNA as a key, since I'm not a retard, and he probably wouldn't either, because works in that field (cyclone, huracan, tornado). So then the key we imagined was something ... well, Who cares what the key is, right?
**\ **
o back to the task at hand, not so long ago, in a place called Plantation I was struck by lightning, literally (well not literally) the answer to a question that nobody knew was implanted in my mind, and it all came from asking a single simple question. I was looking for more chemistry elements in the names of the books of the Holy Bible, after seeing Xenon at the "sort of beginning" of Exodus, where it screams "let there be light" in Linux and chemistry (and I've told you that a hundred times by now). So it didn't take long to follow the light of that word and read Genesis backwards, and see, at the very beginning of that book, Silicon... in reverse.
*\ *
So, what about God's DNA, anyway*? *
What's he really made of?
SIM MON S WILD ER ROD DEN BERRY
o after seeing Silicon, and connecting that to the numerous attempts I've made to show a message connecting The Matrix to the Fifth Element (as Silicon) describing what it is that God believes we should do with this knowledge--and see that it is narrated as the miracles of Jesus Christ in the New Testament... these names came to me in quick succession, an answer to the question. I suppose any Gene will do, these three though, have a very important tie to the message that connects Joshua's Promised Land of flowing Milk and Honies to ... a kiss that begins the new day (I hope) ... and a message about exactly how we might go about doing magical things like ending world hunger and healing the sick using technology described ... in Star Trek and Stargate. A "religion of the Stars" is being born.
That's great... it starts with an earthquake. R.E.M. and a band ... 311. Oooh, I can see it coming down... The Petty Reckless. An evening's love starts with a kiss. Dave Matthews Band. I wanna rock and roll all night and party every day. Adam. I mean Kiss. Are you starting to see a pattern form? Birds, snakes, and aeroplanes? It's that, it's the end of the world as we know it, and I feel fine.
*\ *
*\ *
*\ *
In that song we see clues that more than just the Revelation of Christ is narrated by John on an island called Patmos. There yet another Trinity, starting with "Pa" and hearting Taylor Momsen's initials... most likely for a reason... and the Revelation ends with a transition that I hope others will agree with me turns "original sin" into something closer to "obviously salvation" when we finally understand the character that is behind the message of da i of Ra... and begin to see the same design in the names of Asmodai and in this Revelation focusing on freedom and truth that really does suggest Taylor can't talk to me in any way other than "letting freedom sing" in this narrative of kismet and fate and free will and ... then we see that narrative continue in the names of bands, just like the 3/11/11 earthquake is narrated in not just R.E.M.'s song but in the name 311. Just like the 9/11 attack is narrated not just in that same song (released in 1987) and "Inside Job" (released in 2000) but also in "Fucked up world."
Dear all of you walking dumb and blind, this same quake is narrated in Taylor's Zombie; waiting for the day to shake, all very similar to Cairo and XP, perhaps a "fad" of doublethink in the minds of the authors singing about a clear prophesy in the Bible; this connection between the day, 3/11 though, and the name of a band and the day of an arrest and the verse Matthew that tells you clearly you have now been baptized in water and fire... it shows us the design of a story whose intent and purpose is to ensure that we no longer allow for things like hurricanes and earthquakes and murder and rape to be "simulated" that we build a better system, that doesn't allow for 'force majeure" to take lives for no reason at all.
Not just in band names, but in the angels names too, in all of our names; we see this narration continue. The Holy Water that is central to the baptism of Christ is etched into Taylor's name, between "sen" and "mom" the key to the two Mary's whose names contain the Spanish for "sea" in a sort of enlightenment hidden in plain sight. In "Simmons" the key connection between today, this Biblical Monday, and the word "simulation" that ties to Simpsons and simians and keep it simple stupid*, and in Simmons the missing "s" of Kismet, finally completing the question.***
***\
*\
*\ *
It's a song and dance that started a long time ago, as you can see from the ancient Hebrew word for "fate" and in more recent years a connection to the ballroom of Atlantis in the Doors 5 to 1 and Dave sang about it in Rapunzel and then Taylor shook a tambourine on the beach only minutes away from me--but never said "hi." The battle of the bands continues tying some door knocking to a juxtaposition between "Sweet Things" and "Knocking on Heavens door" all the way to a Gossip Girl episode where little J asked a question that I can't be sure she knew was related, she said... "who's that, at the door?"
*\ *
What it really all amounts to, though, is the whole world witnessing the Creation of Adam and Eve from a little girl stuttering out "the the" at the sight of the Grinch himself, and then later not even able to get those words off her lips... about seeing how Creation and modern art are inextricably tied to religion, to heaven, and to freedom.
*\ *
*\ *
*\ *
The bottom line here, hopefully obvious now, is that you can't keep this message "simple" it's a Matrix woven between more points of light than I can count, and many more that I'm sure you will find. It's a key to seeing how God speaks to me, and to you; and how we are, we really are that voice. Tay, if you don't do something just because God called it "fate" you are significantly more enslaved than if you do--and you wanted to. "Now I see that you and me, were never meant, never meant to be..." she sang before I mentioned her, and before she ever saw me... in a song she calls "Nothing Left to Lose" and I see is not really just another word for freedom.
We have plenty to lose by not starting the fire, not the least of which is Heaven itself. Understand what "force majeure" really means to you and I. Ha, by the way.
IN CASE YOU FORGOT YESTERDAY'S MESSAGE
**\ **
*\ *
*\ *
"DADDY, I WANT IT NOW."
VERUKA SALT. whose name means "to see (if) you are the Body of Christ" whined, in the story of Will Why Won Ka, about nothing more or less than Heaven on Hearth, than seeing an end to needless torture and pain. To see if you are the "Salt of the Earth" warming the road to Heaven; honestly to see if you can break through this inane lie of "I don't understand" and realize that breaking this story and talking about what is being presented not just by me and you but by history and God himself is the key to the car that drives us home. To see how Cupid you really are.
STOP NODDING, TURN AROUND AND CALL A REPORTER.
The story of Willy Wonka ties directly to the Promised Land of Flowing Milk and Honey to me; by showing us a river of chocolate and a the everlasting God starter, (er is it guardian of B stopper) that opens the doors of perception about exactly what kinds of mistake may have been made in the past in this transition to Heaven that we are well on the way of beginning. Here, in the Land of Nod, that is also Eden and also the Heart of the Ark we see warnings about "flowing milk and honey" being akin to losing our stable ecosystem, to losing the stuff of life itself, biology and evolution, and if we don't understand--this is probably exactly the mistake that was made and the cause of the story of Cain and Abel. So here we are talking about genetic engineering and mind uploading and living forever, and hopefully seeing that while all things are possible with God--losing the wisdom of the message of religion is akin to losing life in the Universe and with that any hope of eternal longevity.\ With some insight into religion, you can connect the idea that without bees our stable ecosystem might collapse, to the birds and the bees, and a message about stability and having more than one way to pollinate the flowers and trees and get some. Janet and Nanna, by the way, both have pretty brown eyes, but that probably comes as no surprise to you.\ Miss Everything, on the other hand (I hear, does not have brown eyes), leads us to glimpse how this message about the transition of our society might continue on in the New Testament, and suggest that we do need to eat, and have dinner conversation, and that a Last Supper might be a little bit more detrimental to our future than anyone had ever thought, over and over and over again. To see how religion really does make clear that this is what the message is about, to replace the flowing milk we have a "Golden Cow" that epitomizes nothing less than "not listening to Adam" and we have a place that believes the Hammer of Judah Maccabee should be ... extinct. You are wrong.
*\ *
*\ *
Of course the vibrating light here ties this Gene to another musical piece disclosing something... "Wild Thing" I make your heart sing. You can believe the Guitar Man is here to steal the show and deliver bread for the hungry and for the wise. Here's some, it's not just Imagine Dragons telling you to listen to the radio but Jefferson Starship*too, and Live. *
*\ *
When you wake up, you can hear God "singing" to you on the radio every single day; many of us already do. He's telling you to listen to me, and I do not understand why you do not. You don't look very Cupid, if you ask me.**
***\
WHAT DO YOU THINK YOU ARE,
DAN RE Y NO LDS?
**\ **
I think we all know what the Rod of Jesus Christ is by now.
​
It is a large glowing testament to freedom and truth, and a statement about blindness and evil that is unmistakable. To say that seeing it is the gateway to Heaven would be an understatement of it's worth, of the implication that not seeing it is obvious Hell when it is linked to everything from nearly every story of the Holy Bible from Isaac to Isaiah to "behold he is to coming" and if you weren't sure if the Hand of God were in action here--it's very clear that it is; that linking Tricky Dick and Watergate to Seagate ... really delivering crystal clear understanding that the foundation of Heaven is freedom and that you have none today because you refuse to see the truth.
It is the doorway to seeing that what has been going on in this place hasn't been designed to hide me, but to hide a prosperous future from you--to hide the truth about our existence and the purpose of Creation--that all told, you are standing at the doorstep of Heaven and stammering your feet, closing your eyes, and saying "you don't want to help anyone."
If delivering freedom, truth, and equality to you does not a den make,
well, you can all suck it
**\ **

Between Stargate and Star Trek it's pretty easy to see a roadmap to very quickly and easily be able to end world hunger and heal the sick without drastically changing the way our society works, it's about as simple as a microwave, or a new kind of medicine--except it's not so easy to see why it is that you are so reluctant to talk about the truth that makes these things so easy to do. You see, your lack of regard for anyone anywhere has placed you in a position of weakness, and if you do nothing today, you will not be OK tomorrow.\ It's pretty easy to see how Roddenberry's name shows that this message comes from God, that he's created this map that starts with an Iron Rod throughout our history proving Creation, whose heart is a Den of Family who care about the truth, and about freedom, and about helping each other--not what you are--you are not that today. Today you are sick, and I'd like you to look at the mirror he's made for you, and ***be eshamden (or asham). ***

Realize, realize... what you are. What you've become, just as I have... the devil in a sweet, sweet kiss.**
***\
-Dave J. Matthews



Unless otherwise indicated, this work was written between the Christmas and Easter seasons of 2017 and 2020(A). The content of this page is released to the public under the GNU GPL v2.0 license; additionally any reproduction or derivation of the work must be attributed to the author, Adam Marshall Dobrin along with a link back to this website, fromthemachine dotty org.
That's a "." not "dotty" ... it's to stop SPAMmers. :/
This document is "living" and I don't just mean in the Jeffersonian sense. It's more alive in the "Mayflower's and June Doors ..." living Ethereum contract sense and literally just as close to the Depp/C[aster/Paglen (and honorably PK] 'D-hath Transundancesense of the ... new meaning; as it is now published on Rinkeby, in "living contract" form. It is subject to change; without notice anywhere but here--and there--in the original spirit of the GPL 2.0. We are "one step closer to God" ... and do see that in that I mean ... it is a very real fusion of this document and the "spirit of my life" as well as the Spirit's of Kerouac's America and Vonnegut's Martian Mars and my Venutian Hotel ... and my fusion of Guy-A and GAIA; and the Spirit of the Earth .. and of course the God given and signed liberties in the Constitution of the United States of America. It is by and through my hand that this document and our X Commandments link to the Bill or Rights, and this story about an Exodus from slavery that literally begins here, in the post-apocalyptic American hartland. Written ... this day ... April 14, 2020 (hey, is this HADAD DAY?) ... in Margate FL, USA. For "official used-to-v TAX day" tomorrow, I'm going to add the "immultible incarnite pen" ... if added to the living "doc/app"--see is the DAO, the way--will initi8 the special secret "hidden level" .. we've all been looking for.
Nor do just mean this website or the totality of my written works; nor do I only mean ... this particular derivation of the GPL 2.0+ modifications I continually source ... must be "from this website." I also mean the thing that is built from ... bits and piece of blocks of sand-toys; from Ethereum and from Rust and from our hands and eyes working together ... from this place, this cornerstone of the message that is ... written from brick and mortar words and events and people that have come before this poit of the "sealed W" that is this specific page and this time. It's 3:28; just five minutes--or is it four, too layne.
This work is not to be redistributed according to the GPL unless all linked media on Youtube and related sites are intact--and historical references to the actual documented history of the art pieces (as I experience/d them) are also available for linking. Wikipedia references must be available for viewing, as well as the exact version of those pages at the time these pieces were written. All references to the Holy Bible must be "linked" (as they are or via ... impromptu in-transit re-linking) to the exact verses and versions of the Bible that I reference. These requirements, as well as the caveat and informational re-introduction to God's DAO above ... should be seen as material modifications to the original GPL2.0 that are retroactively applied to all works distributed under license via this site and all previous e-mails and sites. /s/ wso\ If you wanna talk to me get me on facebook, with PGP via FlowCrypt or adam at from the machine dotty org
-----BEGIN PGP PUBLIC KEY BLOCK-----
mQGNBF6RVvABDAC823JcYvgpEpy45z2EPgwJ9ZCL+pSFVnlgPKQAGD52q+kuckNZ mU3gbj1FIx/mwJJtaWZW6jaLDHLAZNJps93qpwdMCx0llhQogc8YN3j9RND7cTP5 eV8dS6z/9ta6TFOfwSZpsOZjCU7KFDStKcoulmvIGrr9wzaUr7fmDyE7cFp1KCZ0 i90oLYHqOIszRedvwCO/kBxawxzZuJ67DypcayiWyxqRHRmMZH1LejTaqTuEu0bp j54maTj09vnMxA0RfS+CtU5uMq+5fTkbiTOe1LrLD72m+PVJIS146FwESrMJEfJy oNqWEJlUQ0TecPZR41vnkSkpocE1/0YqUhWDGSht+67DdeKUg5KwvYdL21d/bSyO SM4jnyKn9aDVzLBpYrlE/lbFxujHPRGlRG5WtiPQuZYDRqP0GYFSXRpeUCI46f49 iPFo4eHo2jUfNDa9r9BjQdAe4zVFn2qLnOy8RWijlolbhGMHGO3w/uC/zad3jjo4 owAfsJjH5Oa1mTcAEQEAAbQmRUFSVEhFTkUgPGVhcnRoZW5lQGZyb210aGVtYWNo aW5lLm9yZz6JAdQEEwEKAD4WIQTUJHbrYn3y2DzwTcnQP1ViZf5/FQUCXpFW8AIb AwUJA8JnAAULCQgHAgYVCgkICwIEFgIDAQIeAQIXgAAKCRDQP1ViZf5/FWM6C/9J gbRLS2AWGjdRjYetlRkSkCoTYnXWknbtipYYHlhV0YJFwFMm0ydZIhFX5VDoZyBV 0UBeF1KJmcMoIfrHyhq2QhCnjE14hE1ONbaYTGtpvj851ItbFWXMJIVNyMqr+JT9 CWIxGr1idn+iHWE3nryiHrdlA3O/Gcd4EyNmaSe/JvB7+Z1AVqWkRhpjxxoPSlPm HEdqGOyl3+5ibQgUvXLRWWQXAj80CbVwwj1X4r9hfuCySxLT8Mir7NUXZFd+OiMS U8gNYjcyRGmI92z5lgf7djBbb9dMLwV0KLzgoT/xaupRvvYOIAT+n2mhCctCiH7x y7jYlJHd+0++rgUST2sT+9kbuQ0GxpJ7MZcKbS1n60La+IEEIpFled8eqwwDfcui uezO7RIzQ9wHSn688CDri9jmYhjp5s0HKuN61etJ1glu9jWgG76EZ3qW8zu4l4CH 9iFPHeGG7fa/5d07KvcZuS2fVACoMipTxTIouN7vL0daYwP3VFg63FNTwCU3HEq5 AY0EXpFW8AEMANh7M/ROrQxb3MCT1/PYco1tyscNo2eHHTtgrnHrpKEPCfRryx3r PllaRYP0ri5eFzt25ObHAjcnZgilnwxngm6S9QvUIaLLQh67RP1h8I4qyFzueYPs oY8xo1zwXz7klXVlZW0MYi/g5gpb+rpYUfZEJGJTBM/wMNqwwlct+BSZca4+TEHW g6oN0eXTthtGB0Qls71sv3tbOnOh/67NTwyhcHPWX/P9ilcjGsEiT8hqrpyhjAUm mv7ADi+2eRBV8Xf8JnPznFf0A1FdILVeVHlmsgCSB0FW0NsFI5niZbaYBHDbFsks QdaFaYd54DHln69tnwc2y3POFwx8kwZnMPPlVAR2QdxGQD4Wql7hlWT58xCxQApf M98kbAHjUlVYLT0WUHMDQtj4jdzAVVDiMGMUrbnQ7UwI7LexSB6cJ7H+i7FtS/pR WOhJK6awoOO9dLnEjm6UYCKsBdtJr98F0T7Sb7PnKOGA77y2QN14+u9N9C1lB/Z1 aQRQ2Nc51yXOQQARAQABiQG8BBgBCgAmFiEE1CR262J98tg88E3J0D9VYmX+fxUF Al6RVvACGwwFCQPCZwAACgkQ0D9VYmX+fxU+KQwAtFnWjGIjvqaNXtQjEhbGDH/I Q5ULq/l/wm9SmhG9NYRu3+P6YctCJaZnNeaL+6WFk1jo4LMiJEUT9uGlCbHqJNaI 6Gll1w6QOVLSL8s5V1L477+psluv4WBpi3XkWYlhDOFENCcWd49RQsA2YCX4pW7Q 7GcoSEJoav38MxHmJHYPfjSEvUZXDQIt8PFHSEScvyDWfYtMdRzjmSOOPdzhDDEy 5JBOBcEdSTyDiyDU/sBoAY0e8lvwHYW3p+guZSGSYVhGQ8JECzJOzwc/msMW/tJS 2MLWmWVh5/1P8BVUtLC2AQy6nij6o+h6vEiNzpdYrc+rzT3X5cACvJ0RtCZcrnhl O9PLiona2LEbry6QX5NL41/SAJNno3i72xPnQEe25gn3nbyT+jCoJzw2L0y8pmNB D+PKrk7/1ROFFVN8dJeGwxLGdBcz1zk2xeumzy7OaV8psUyYsJNcjyHUKgclblBW rMR2DgqEYn8QdK54ziKCnmQQZeMPiC6wlUWgg5IqmQGNBF6RVyMBDADALD7NkJ5H dtoOpoZmAbPSlVGXHDbJZuq7J13vew6dtXDIAraeGrsBqkF8bhddwVLzWylMrYCG Bf2L1+5BDgvqu6G+6dcVSbBsnZAS0zfJ0H8EmTvUMxMF7qOZYyrxfLz+pQRq8Osz Icab6ZI/KB6qZyQRvEFPB6pJjt+VvuwgJZTObIwbBbgQri2i02VBkjchsVhiSX9l +eiK7O8ROHKb3P181oScIsHywBOZ9DxRAYbFk5dnBqxO3WKb02H0zqE6440cjXwq TrZZg6ayN/IlPajO8iJPYZ1aIBykxYq1WHo+nhFMYz/VVk2WJorFeOgWaLGXb73c ty96f3qXTdvMDAIWHx8YCD5LbuqasO6LNQm4oQxkCoB3K9WFf/2SvSYb7yMYykb8 clTPt+KO0dsxjWhrJnfnIhC+2Chqv2QvRbFz0S9CpUnGGDweJ1uRNV0y70tO0q7t xXSTDRU3ib6vAHA0K/2MFzwUcog4o5bj7E9uCNJH/DJLZKsMIe4xsvkAEQEAAbQk SEVBVkVOVUVTIDxBVkVOVUBGUk9NVEhFTUFDSElORS5PUkc+iQHUBBMBCgA+FiEE IRklfU/C1qukq3xMXcNH0t3P9ZsFAl6RVyMCGwMFCQPCZwAFCwkIBwIGFQoJCAsC BBYCAwECHgECF4AACgkQXcNH0t3P9Zs+kgv/XEuuWc89Bjg1QQqKZueKNUHjyjnE 2adfoZUH6Q7ir4JZyRBCVpAwrgssmiKid30+SIjwQcpb9JYa/X1XJcDUcJW/I21d Agz/zbEqn/Cou0dUpNCtxgm4BdSHWGoOtgfspXZlXBQ407tRMZ8ykmLB1Bt0oHvw PT0ZOtqXM4pyFnd2eFe5YGbNgl3zqvoC/6CMN3vqswvRlu1BpUuAjdW8AHO5Yvje +Bp852u+4Qpy6PMBiWGsBMYwtf6T7sckpMGlR0TsozwBlAm5ePKK28B0rLJPkZLJ Eo5p4rKRapEaZsWV5Qu1ajrVru7qmpUhZtX0/DddGHfXVuLssmKLP6TumpQB1zvQ vfoBltjvOx35Wps2vHuCzXLw2bROIOzhAxFB+17zxnSbE54N4LIGRpkELuwxwGbg FtD1fi9KtH7xcn33eOK1+UD47V+hKyJGrQgSThly2zdIC2bvfHtFdfp8lOFpT0AU xjEeoJGqdQVupptXyugPlM5/96UJP8OZG0ADuQGNBF6RVyMBDAC3As6eMkoEo3z9 TkCWlvS0vBQmY3gF0VEjlAIqFWpDIdK3zVzMnKUokIT1i7nkadLzHZT2grB4VXuJ FvpbYw5NPR4cDe9grlOMLEaF3oSJ1jZ4V1/rj9v1Hddo8ELi/NToVrt1SB5GCVXB DkYpNLtTiCqHSU07YqwaqH8a+qbDmPxSQdIybkZiTiCEB+6PfQQlBpENEDlov6jm zZF+IcfM6s3kZDX5KFULweH30gMjq8Se8bPtUzW013+tuuwEVr1/YRLrIh+9O6Z+ pdA7gLMRYnD9ZLDytEvpb1lBBSY++5bIJ7xps80//DNqPYqwFmZQgTg0V9XbHE2e wLcOF8a2lYluckU7D///sWQhW+VxuM7R2gEBvYBhOgjWhIF2Aw6NbymW1Ontvyhu eOZCXXxV5W44PxXT8uDdhl9CNcHoBKKJyED8tKjigtn4axpsQeUrnOSbqEXSyqES WnE2wYUDzALcwFkzsvtLyd4xaz55KkPQkAkk0BZd1ezgXxb/obMAEQEAAYkBvAQY AQoAJhYhBCEZJX1PwtarpKt8TF3DR9Ldz/WbBQJekVcjAhsMBQkDwmcAAAoJEF3D R9Ldz/WbAFwL/382HsrldVXnkPmJ1E2YEOFz4rcHRetJ+M5H65K/2p32ONQ5KCbE s8MRY6g2CkE70en2HlpDwr/MdATwxBzIjEpjgHbfqCqVVATY+kSpXsttaKKAUVHi bFgV4QkdDJNSpcHEj+bqaggRnuWiV9T6ECG7kQjHiEXPNojzsiaXMDiM5r+acZm6 82id9qOFySQ2cZEy5HbwXM+ITLQGngnppa7du2KdgiqDeqtODOTWZvLYAq2tmEwD 3TT6ttLUBwOOu2IWpDkXswlrk62ESorE5mpLxop9fsxD39E2H06JoC/YfUPIVkEv fj06e7LEdcx0I7kRfD1v6qOUUsMsLZnmyGIk24iFjLkwu1VToWfwXDN1D2+SeAat 9ydNt4M7oEbd1QaOXXjmqpdU+VUiWcBXg+p3/WdV60MkyAgc3x+YanLljy/Rh18h cZwVlinf/tgvAQLi5f9hpwrwUMoGKijEYHKuEvi3C12Si7UVDfuIR7yS0dKcfuKF MbgwdvNXqpD9W5kBjQRekVd4AQwApHVgw2PVlBDpVcyoymUOXFQIJzJ9wRtr6/sG zwv8rrQnUEtOkkna7TDU3/UTj9FUH0gbpAKGNNPaPj5q0dlLIvzxb15r1uvDGaGL MA+8GFaGFnkxzhg0aXrcKZAN0/Zhgi2B7P8oXQuug5mi1JVDkZN5SeCZNOubdQWL 3xz3jEHp3ixj1mdOdvfdWQFR4CVMXt/A6VI2ujLVb3Yalft/c5bbclAgcJQhgDUu NqGYJEJonESNRSd8fEvhNb6cx7+Djd9+Wyctr76mwOr3nRb1N1OGhFxWjIroUpfz b+6y3oQjT58cJA1ZHqmJ6UlZd81hNNd9KWpbDVwONEPpiqPzfSaonxuqQa0/Cy4W 403OhfoLM/1ZDqD4YrJ/rpyNEfSSdqptWiY0KeErLOYng7rStW/4ZeZVj6b2xxB2 Oas/Z1QYfJyFUki9vaJ5IyN6Y7nVdSP6mbAQC9ESh+VPvRUMpYi4pMGK4rweBVHu oMRRwzk7W5zVIgd425WUe3eCQFn3ABEBAAG0K0VTQ0FQRSBST09NIDxFU0NBUEFF REVTQEZST01USEVNQUNISU5FLk9SRz6JAdQEEwEKAD4WIQTvnDJqcmqzlF87/t82 pJ91j4NOaAUCXpFXeAIbAwUJA8JnAAULCQgHAgYVCgkICwIEFgIDAQIeAQIXgAAK CRA2pJ91j4NOaJVjC/4oo5yCHe7M2h1DiTXVcLI5rXQ1feY7B1feg+YJX/mI4+EV xjC/y5VVpV4syJk5GGZNXhKPHiGLaBYvglTlYOJ98RSEsHrwT3go6S8ZVvMNdP5v CEncn2vm5JGnp4k26PuOzMcJioQLOoUjWtcPFis3gG+ueH3NcPZ22oZUql2xuerh TQZegGp+jJ7bdxwYElx5jDDDkh196d5nlO2ZKENl0ZDp4GAzRNjnQ7KBV6R74J3U cLQDWY8vAFaRBZXIC5XtSzj9lr+jWgvxz7Il51+26VDTEtSafZ2uZfCOFk7GrzJg
sneak preview
now linking to the next page ... in the discussion:
https://fromthemachine.org/2017/08/waiting-for-that-green-light.html
-
-
fromthemachine.org fromthemachine.orgJOHN7171
-
Unless otherwise indicated, this work was written between the Christmas and Easter seasons of 2017 and 2020(A). The content of this page is released to the public under the GNU GPL v2.0 license; additionally any reproduction or derivation of the work must be attributed to the author, Adam Marshall Dobrin along with a link back to this website, fromthemachine dotty org. That's a "." not "dotty" ... it's to stop SPAMmers. :/ This document is "living" and I don't just mean in the Jeffersonian sense. It's more alive in the "Mayflower's and June Doors ..." living Ethereum contract sense [and literally just as close to the Depp/Caster/Paglen (and honorably PK] 'D-hath Transundancesense of the ... new meaning; as it is now published on Rinkeby, in "living contract" form. It is subject to change; without notice anywhere but here--and there--in the original spirit of the GPL 2.0. We are "one step closer to God" ... and do see that in that I mean ... it is a very real fusion of this document and the "spirit of my life" as well as the Spirit's of Kerouac's America and Vonnegut's Martian Mars and my Venutian Hotel ... and *my fusion* of Guy-A and GAIA; and the Spirit of the Earth .. and of course the God given and signed liberties in the Constitution of the United States of America. It is by and through my hand that this document and our X Commandments link to the Bill or Rights, and this story about an Exodus from slavery that literally begins here, in the post-apocalyptic American hartland. Written ... this day ... April 14, 2020 (hey, is this HADAD DAY?) ... in Margate FL, USA. For "official used-to-v TAX day" tomorrow, I'm going to add the "immultible incarnite pen" ... if added to the living "doc/app"--see is the DAO, the way--will initi8 the special secret "hidden level" .. we've all been looking for. Nor do just mean this website or the totality of my written works; nor do I only mean ... this particular derivation of the GPL 2.0+ modifications I continually source ... must be "from this website." I also mean *the thing* that is built from ... bits and piece of blocks of sand-toys; from Ethereum and from Rust and from our hands and eyes working together ... from this place, this cornerstone of the message that is ... written from brick and mortar words and events and people that have come before this poit of the "sealed W" that is this specific page and this time. It's 3:28; just five minutes--or is it four, too layne. This work is not to be redistributed according to the GPL unless all linked media on Youtube and related sites are intact--and historical references to the actual documented history of the art pieces (as I experience/d them) are also available for linking. Wikipedia references must be available for viewing, as well as the exact version of those pages at the time these pieces were written. All references to the Holy Bible must be "linked" (as they are or via ... impromptu in-transit re-linking) to the exact verses and versions of the Bible that I reference. These requirements, as well as the caveat and informational re-introduction to God's DAO above ... should be seen as material modifications to the original GPL2.0 that are retroactively applied to all works distributed under license via this site and all previous e-mails and sites. /s/ wso
and now,
ladies and gentlemen, aesir and cherubim ...
whatever that means; we will continue to look and search for what is the coonection between the GNU 2.0 "the new two point owe" GPL and of course messages from Google and Government regarding Roe v. Wade and "good luck, e"
here we are;
FOSSwire The differences between the GPL, LGPL and the BSD April 6, 2007 Avatar for peter Peter Upfold There are a lot of different open source licences out there, and it can sometimes be a bit confusing if you're not intimate with the details of each one. So here's a quick roundup of three of the most popular licenses and the difference between them.
Just a quick disclaimer - I'm not a lawyer, so don't depend on my explanations on the licences here. All the usual disclaimers apply.
GNU General Public Licence The GNU General Public Licence, or GPL as it's often called, is the most popular free software licence and it's used by many different projects, including the Linux kernel, the GNU tools and literally hundreds of others.
You can find the legal text for the GPL here, but here's a quick summary of what it means.
Basically, you're allowed to use, redistribute and change the software, but any changes you make must also be licensed under the GPL. So that means you have to give everyone else the same rights as you got. Fair's fair, right?
There are also other restrictions and there's quite a nice human-readable version at the Creative Commons site.
The GNU Lesser General Public Licence The LGPL is similar to the GPL, but is more designed for software libraries where you want to allow non-GPL applications to link to your library and utilise it. If you modify the software, you still have to give back the source code, but you are allowed to link it with proprietary stuff without giving the source code to all of that back.
Again, there's a nice friendly look at this on the Creative Commons site.
The BSD Licence In contrast to the GNU licences, the BSD licence is very permissive. Used originally by the BSD operating system, it covers a fair amount of software.
The BSD basically says "here's the source code, do whatever you want with it, but if you have problems, it's your problem". That means you can take BSD'ed code and turn it into a proprietary application if you so wish - there's nothing saying you have to give the code back (although it is nice to do so).
The BSD licence is very small because it is so simple, and often looks like this:
Redistribution and use in source and binary forms, with or without modification, are permitted provided that the following conditions are met: * Redistributions of source code must retain the above copyright notice, this list of conditions and the following disclaimer. * Redistributions in binary form must reproduce the above copyright notice, this list of conditions and the following disclaimer in the documentation and/or other materials provided with the distribution. * Neither the name of the [[whoever]] nor the names of contributors may be used to endorse or promote products derived from this software without specific prior written permission.
THIS SOFTWARE IS PROVIDED BY THE COPYRIGHT HOLDERS AND CONTRIBUTORS "AS IS" AND ANY EXPRESS OR IMPLIED WARRANTIES, INCLUDING, BUT NOT LIMITED TO, THE IMPLIED WARRANTIES OF MERCHANTABILITY AND FITNESS FOR A PARTICULAR PURPOSE ARE DISCLAIMED. IN NO EVENT SHALL THE COPYRIGHT OWNER OR CONTRIBUTORS BE LIABLE FOR ANY DIRECT, INDIRECT, INCIDENTAL, SPECIAL, EXEMPLARY, OR CONSEQUENTIAL DAMAGES (INCLUDING, BUT NOT LIMITED TO, PROCUREMENT OF SUBSTITUTE GOODS OR SERVICES; LOSS OF USE, DATA, OR PROFITS; OR BUSINESS INTERRUPTION) HOWEVER CAUSED AND ON ANY THEORY OF LIABILITY, WHETHER IN CONTRACT, STRICT LIABILITY, OR TORT (INCLUDING NEGLIGENCE OR OTHERWISE) ARISING IN ANY WAY OUT OF THE USE OF THIS SOFTWARE, EVEN IF ADVISED OF THE POSSIBILITY OF SUCH DAMAGE.
There are also several other licences (the MIT for example) that are similar in spirit to the BSD.
Obviously, that's not all the licences - there are plenty, and developers choose them for different circumstances. Some are restrictive, but preserve the free-ness of the code like the GPL, and some are much more permissive.
The Free Software Foundation call the GPL-style restrictions (you must release any modifications under the same licence) 'copyleft'. Mr Stallman himself has an essay about this and other issues on the GNU site. Bear in mind though, this article does push Stallman's personal views on software licences quite heavily. Take with a pinch of salt if necessary.
Avatar for peterPeter Upfold Tips & TutorialsLicensing/LegalFundamentals HOME » ARTICLES » FOSSwire All articles
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
eLife assessment
In this study, the authors develop a useful strategy for fluorophore-tagging endogenous proteins in human induced pluripotent stem cells (iPSCs) using a split mNeonGreen approach. Experimentally, the methods are solid, and the data presented support the author's conclusions. Overall, these methodologies should be useful to a wide audience of cell biologists who want to study protein localization and dynamics at endogenous levels in iPSCs.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
In this manuscript, the authors have applied an asymmetric split mNeonGreen2 (mNG2) system to human iPSCs. Integrating a constitutively expressed long fragment of mNG2 at the AAVS1 locus, allows other proteins to be tagged through the use of available ssODN donors. This removes the need to generate long AAV donors for tagging, thus greatly facilitating high-throughput tagging efforts. The authors then demonstrate the feasibility of the method by successfully tagging 9 markers expressed in iPSC at various, and one expressed upon endoderm differentiation. Several additional differentiation markers were also successfully tagged but not subsequently tested for expression/visibility. As one might expect for high-throughput tagging, a few proteins, while successfully tagged at the genomic level, failed to be visible. Finally, to demonstrate the utility of the tagged cells, the authors isolated clones with genes relevant to cytokinesis tagged, and together with an AI to enhance signal-to-noise ratios, monitored their localization over cell division.
Strengths:
Characterization of the mNG2 tagged parental iPSC line was well and carefully done including validation of a single integration, the presence of markers for continued pluripotency, selected offtarget analysis, and G-banding-based structural rearrangement detection.
The ability to tag proteins with simple ssODNs in iPSC capable of multi-lineage differentiation will undoubtedly be useful for localization tracking and reporter line generation.
Validation of clone genotypes was carefully performed and highlights the continued need for caution with regard to editing outcomes.
Weaknesses:
IF and flow cytometry figures lack quantification and information on replication. How consistent is the brightness and localization of the markers? How representative are the specific images? Stability is mentioned in the text but data on the stability of expression/brightness is not shown.
To address this comment, we have quantified the mean fluorescence intensity of the tagged cell populations in Fig. S3B-T. This data correlates well with the expected expression levels of each gene relative to the others (Fig. S3A), apart from CDH1 and RACGAP1, which are described in the discussion.
The images in Fig. 2 show tagged populations enriched by FACS so they are non-clonal and are representative of the diversity of the population of tagged cells.
The images shown in Fig. 3 are representative of the clonal tagged populations. The stability of the tag was not quantified directly. However, the fluorescence intensity was very stable across cells in clonal populations. Since these populations were recovered from a single cell and grown for several weeks, this low variability across cells in a population suggests that these tags are stable.
The localization of markers, while consistent with expectations, is not validated by a second technique such as antibody staining, and in many cases not even with Hoechst to show nuclear vs cytoplasmic.
We find that the localization of each protein is distinct and consistent with previous studies. To address this comment, we have added an overlay of the green fluorescence images with brightfield images to better show the location of the tagged protein relative to the nuclei and cytoplasm. We have also added references to other studies that showed the same localization patterns for these proteins in iPSCs and other relevant cell lines.
For the multi-germ layer differentiation validation, NCAM is also expressed by ectoderm, so isn't a good solo marker for mesoderm as it was used. Indeed, the kit used for the differentiation suggests Brachyury combined with either NCAM or CXCR4, not NCAM alone.
Since Brachyury is the most common mesodermal marker, we first tested differentiation using anti-Brachyury antibodies, but they did not work well for flow cytometry. We then switched to anti-NCAM antibodies. Since we used a kit for directed differentiation of iPSCs into the mesodermal lineage, NCAM staining should still report for successful differentiation. In the context of mixed differentiation experiments (embryoid body formation or teratoma assay), NCAM would not differentiate between ectoderm and mesoderm. The parental cells (201B7) have also been edited at the AAVS1 locus in multiple other studies, with no effect on their differentiation potential.
Only a single female parental line has been generated and characterized. It would have been useful to have several lines and both male and female to allow sex differences to be explored.
We agree that it would be interesting (and important) to study differences in protein localization between female and male cell types, and from different individuals with different genetic backgrounds. We see our tool as opening a door for cell biology to move away from randomly collected, transformed, differentiated cell types to more directed comparative studies of distinct normal cell types. Since few studies of cell biological processes have been done in normal cells, a first step is to understand how processes compare in an isogenic background, then future studies can reveal how they compare with other individuals and sexes. We hope that either our group or others will continue to build similar lines so that these studies can be done.
The AI-based signal-to-noise enhancement needs more details and testing. Such models can introduce strong assumptions and thus artefacts into the resolved data. Was the model trained on all markers or were multiple models trained on a single marker each? For example, if trained to enhance a single marker (or co-localized group of markers), it could introduce artefacts where it forces signal localization to those areas even for others. What happens if you feed in images with scrambled pixel locations, does it still say the structures are where the training data says they should be? What about markers with different localization from the training set? If you feed those in, does it force them to the location expected by the training data or does it retain their differential true localization and simply enhance the signal?
The image restoration neural network was used as in Weigert et al., 2018. The model was trained independently for each marker. Each trained model was used only on the corresponding marker and with the same imaging conditions as the training images. From visual inspection, the fluorescent signal in the restored images was consistent with the signal in the raw images, both for interphase and mitotic cells. We found very few artefacts of the restoration (small bright or dark areas) that were discarded. We did not try to restore scrambled images or images of mismatched markers.
Reviewer #2 (Public Review):
Summary:
The authors have generated human iPSC cells constitutively expressing the mNG21-10 and tested them by endogenous tagging multiple genes with mNG211 (several tagged iPS cell lines clones were isolated). With this tool, they have explored several weakly expressed cytokinesis genes and gained insights into how cytokinesis occurs.
Strengths:
Human iPSC cells are used.
Weaknesses:
i) The manuscript is extremely incremental, no improvements are present in the split-fluorescent (split-FP) protein variant used nor in the approach for endogenous tagging with split-FPs (both of them are already very well established and used in literature as well as in different cell types).
Although split fluorescent proteins and the endogenous tagging methodology had been developed previously, their use in human stem cells has not been explored. We argue that human iPSCs are a valuable model for cell biologists to study cellular processes in differentiating cells in an isogenic context for proper comparison. Many normal human cell types have not been studied at the cellular/subcellular level, and this tool will enable those studies. Importantly, other existing cell lines required transformation to persist in culture and represent a single, differentiated cell type that is not normal. Moreover, the protocols that we developed along with this methodology (e.g. workflows for iPSC clonal isolation that include automated colony screening and Nanopore sequencing) will be useful to other groups undertaking gene editing in human cells. Therefore, we argue that our work opens new doors for future cell biology studies.
ii) The fluorescence intensity of the split mNeonGreen appears rather low, for example in Figure 2C the H2BC11, ANLN, SOX2, and TUBB3 signals are very noisy (differences between the structures observed are almost absent). For low-expression targets, this is an important limitation. This is also stated by the authors but image restoration could not be the best solution since a lot of biologically relevant information will be lost anyway.
The split mNeonGreen tag is one of the brighter fluorescent proteins that is available. The low expression that the reviewer refers to for H2BC11, ANLN, TUBB3 and SOX2 is expected based on their predicted expression levels. Further, these images were taken with cells in dishes using lower resolution imaging and were not intended to be used for quantification. As shown in the images in Figures 3H, when using a different microscope with different optical settings and higher magnification, the localization is very clear and quantifiable without needing to use restoration (e.g., compare H2BC11 and ANLN). Using microscopes with high NA objectives, lasers and EMCCD or sCMOS cameras with high sensitivity can sufficiently detect levels of very weakly expressing proteins that can be quantified above background and compared across cells. It is worth noting that each tag may be studied in very different contexts. For example, ANLN will be useful for studies of cytokinesis, while the loss of SOX2 expression and gain of TUBB3 expression may be used to screen for differentiation rather than for localization per se. The reason for endogenous tagging is to study proteins at their native levels rather than using over-expression or fixation with antibodies where artefacts can be introduced. Endogenous tags tag will also enable studies of dynamic changes in localization during differentiation in an isogenic background as described previously.
Importantly, image restoration is not required to image any of these probes! We use it to demonstrate how a researcher can increase the temporal resolution of imaging weakly-expressed proteins for extended periods of time. This data can be used to compare patterns of localization and reveal how patterns change with time and during differentiation. Imaging with fewer timepoints and altered optical settings will still permit researchers to extract quantifiable information from the raw data without requiring image restoration.
iii) There is no comparison with other existing split-FP variants, methods, or imaging and it is unclear what the advantages of the system are.
We are not sure what the reviewer means by this comment. In the future, we plan to incorporate an additional split-FP variant (e.g., split sfCherry) in this iPSC line to enable the imaging of more than one protein in the same cell. However, the split mNeonGreen system is still amenable for use with dyes with different fluorescence spectra that can mark other cellular components, especially for imaging over shorter timespans. In addition to tagging efficiency, the main advantage of split FPs is its scale, as demonstrated by the OpenCell project by tagging 1,310 proteins endogenously (Cho et al., 2022). We developed protocols that facilitate the identification of edited cell lines with high throughput. We also used multiple imaging methods throughout the study that relied on the use of different microscopes and flow cytometry, demonstrating the flexibility of this tagging system. Even for more weakly expressing proteins, the probe could be sufficiently visualized by multiple systems. Such endogenous tags can be used for everything from simply knowing when cells have differentiated (e.g., loss of SOX2 expression, gain of differentiation markers), to studying biological processes over a range of timescales.
Reviewer #3 (Public Review):
The authors report on the engineering of an induced Pluripotent Stem Cell (iPSC) line that harbours a single copy of a split mNeonGreen, mNG2(1-10). This cell line is subsequently used to take endogenous protein with a smaller part of mNeonGreen, mNG2(11), enabling the complementation of mNG into a fluorescent protein that is then used to visualize the protein. The parental cell is validated and used to construct several iPSC lines with endogenously tagged proteins. These are used to visualize and quantify endogenous protein localisation during mitosis.
I see the advantage of tagging endogenous loci with small fragments, but the complementation strategy has disadvantages that deserve some attention. One potential issue is the level of the mNG2(1-10). Is it clear that the current level is saturating? Based on the data in Figure S3, the expression levels and fluorescence intensity levels show a similar dose-dependency which is reassuring, but not definitive proof that all the mNG2(11)-tagged protein is detected.
We have not quantified the levels of mNG21-10 expression directly. However, the increase in fluorescence observed with highly expressed proteins (e.g., ACTB) supports that mNG21-10 levels must be sufficiently high to permit differences among endogenous proteins with vastly different expression levels. To ensure high expression, we used a previously validated expression system comprised of the CAG promoter integrated at the AAVS1 locus, which has previously been used to provide high and stable transgene expression (e.g. Oceguera-Yanez et al., 2016). We acknowledge that it is difficult to confirm that all of the endogenous mNG211-tagged protein is ‘detectable’.
Do the authors see a difference in fluorescence intensity for homo- and heterozygous cell lines that have the same protein tagged with mNG2(11)? One would expect two-fold differences, or not?
To answer this question, we measured the fluorescence intensity of homozygous and heterozygous clones carrying smNG2-anillin and smNG2-RhoA. We found homozygous clones that were approximately twice as bright as the corresponding heterozygous clones (Fig. S4H and I). This suggests that the complementation between mNG21-10 and mNG211 occurs efficiently over a range of mNG211 expression, since anillin is expressed weakly and RhoA is expressed more strongly in iPSCs. However, we also observed some homozygous clones that were not brighter than the corresponding heterozygous clones, which could be due to undetected byproducts of CRISPR or clonal variation in protein expression.
Related to this, would it be favourable to have a homozygous line for expressing mNG2(1-10)?
Our heterozygous cell line leaves the other AAVS1 allele available for integrations of other transgenes for future experiments. While a homozygous line could express more mNG2(1-10), it does not seem to be rate-limiting even with a highly-expressed protein like beta-actin, and we are not sure that it is necessary. The value gained by having the free allele could outweigh the difference in mNG2(1-10) levels.
The complementation seems to work well for the proteins that are tested. Would this also work for secreted (or other organelle-resident) proteins, for which the mNG2(11) tag is localised in a membrane-enclosed compartment?
The interaction between the 1-10 and 11 fragments is strong and should be retained when proteins are secreted. It was recently shown that secreted proteins tagged with GFP11 can be detected when interacting with GFP1-10 in the extracellular space, albeit using over-expression (Minegishi et al., 2023). However, in our work, the mNG21-10 fragment is cytosolic and we have only explored proteins localized to the nucleus or the cytoplasm similar to Cho et al., (2022). By GO annotation, 75% of human proteins are present in the cytoplasm and/or nucleus, which still covers a wide range of proteins of interest. Future versions of our line could include incorporating organelle-targeting peptides to drive the large fragment to specific, non-cytosolic locations.
The authors present a technological advance and it would be great if others could benefit from this as well by having access to the cell lines.
As discussed below, some of the resources are already available, and we are working to make the mNG21-10 cell line available for distribution.
Recommendations for the authors:
Reviewer #2 (Recommendations For The Authors):
The manuscript is methodological, the main achievement is the generation of a stable iPSC with the split Neon system available for the scientific community. Although it is technically solid, the judgement of this reviewer is that the manuscript should be considered for a more specialised/methodological/resource-based journal.
Indeed, we have submitted this article under the “tools and resources” category of eLife, which publishes methodology-centered papers of high technical quality. We felt this was a good venue for the audience that it can reach compared to more specialized journals that may be more limited in scope. For example, our system will be a useful resource for cell biologists and they are more likely to see it in eLife compared to more specialized journals.
Reviewer #3 (Recommendations For The Authors):
(1) The authors present a technological advance and it would be great if others can benefit from this as well. Therefore access to the materials (and data) would be valuable (the authors do a great job by listing all the repair templates and primers).
We have added several pieces of data and information to the supplementary materials, as described below.
For instance:
What is the (complete/plasmid) sequence of the AAVS1-mNG2(1-10) repair plasmid? Will it be deposited at Addgene?
The plasmids used in this paper are now available on Addgene, along with their sequences [ID 206042 for pAAVS1-Puro-CAG-mNG2(1-10) and 206043 for pH2B-mNG2(11)].
The ImageJ code for the detection of colonies is interesting and potentially valuable. Will the code be shared (e.g. at Github, or as supplemental text)?
The ImageJ macro has been uploaded to the CMCI Github page (https://github.com/CMCI/colony_screening). The parameters are optimized to perform segmentation on images obtained using a Cytation5 microscope with our specific settings, but they can be tweaked for any other sets of images. The following text has been added to the methods section: “The code for this macro is available on Github (https://github.com/CMCI/colony_screening)”.
The cell line with the mNG2(1-10) as well as other cell lines can be of interest to others. Will the cell lines be made available? If so, can the authors indicate how?
We are in the process of depositing our cell line in a public repository. This process may take some time for quality control. For now, the cells can be made available by requesting them from the corresponding authors.
(2) How well does the ImageJ macro for detection of the colonies in the well work? Is there any comparison of analysis by a human vs. the macro?
In our most recent experiment, the colony screening macro correctly identified 99.5% of wells compared to manual annotation (83/84 positive wells and 108/108 negative wells). For each 96-well plate, imaging takes 25 minutes, and it takes 7 minutes for analysis. Despite a few false negatives, we expect this macro to be useful for large-scale experiments where multiple 96-well plates need to be screened, which would take hours manually.
(3) The CDH labeling was not readily detected by FACS, but was visible by microscopy. Is the labeling potentially disturbed by the procedure (low extracellular calcium + trypsin?) to prepare the cell for FACS?
It is not clear why the CDH labelling was not detected by FACS. As the reviewer suggests, there could be several reasons: E-cadherin could be broken down by the dissociation reagent (Accutase), or recycled into the cell following the loss of adhesion and the low extracellular calcium in PBS. However, the C-terminal intracellular tail of E-cadherin was tagged, which should not be affected by Accutase. Moreover, recycling into the cell should still result in a detectable fluorescent signal. Notably, the flow cytometry experiments were done as quickly as possible after dissociation to minimize the time that E-cadherin could be degraded or recycled. We also resuspended the cells in MTeSR Plus media instead of PBS, and compared cells grown on iMatrix511 to those grown on Matrigel in case differences in the extracellular matrix affected Ecadherin expression. Another possibility is that the microscopy used for detection of E-cadherin in cells involved using a sweptfield livescan confocal microscope with high NA objective, 100mW 488nm laser and an EMCCD camera with high sensitivity, and perhaps this combination permitted detection better than the detector on the BD FACSMelody used for FACs.
(4) The authors write that the "Tubulin was cytosolic during interphase" which is surprising (and see also figure 3H), as I was expecting it to be incorporated in microtubules. May this be an issue of insufficient resolution (if I'm right this was imaged with 20x, NA=0.35 and so the resolution could be improved by imaging at higher NA)?
Indeed, as the reviewer points out, our terminology (cytosol vs. microtubule) reflects the low resolution of the imaging for the cell populations in dishes and the individual alpha-tubulin monomers being labelled with the mNG211 tag, which are present as cytoplasmic monomers as well as polymers on microtubules. However, even in this image (Fig. 2C), the mitotic spindle microtubules are visible as they are so robust compared to the interphase microtubules. Notably, when we imaged cells from the cloned tagged cell line using a microscope designed for live imaging with a higher NA objective (see above), endogenous tagged TUBA1B was even more clearly visible in spindle microtubules, and was weakly observed in some microtubules in interphase cells, although they are slightly out of focus (Fig. 3H). If we had focused on a lower focal plane where the interphase cells are located and altered the optical settings, we would see more microtubules.
(5) It would be nice to have access to the Timelapse data as supplemental movies (.e.g from the experiments shown in Figure 4).
We have added the movies corresponding to the timeplase images as supplementary movies (Movies S1-6), with the raw and restored movies shown side-by-side.
(6) In Figure 3B, the order of the colors in the bar is reversed relative to the order of the legend. Would it be possible to use the same order? That makes it easier for me (as a colorblind person) to match the colors in the figure with that of the legend.
We have modified the legend in Fig 2B and 3B to be in the same order as the bars.
-
-
Local file Local file
-
This has led some women to commit to formingtheir own cooperatives. But while the Lutonde case provides valuableinspiration, it is still the only women's mining cooperative in 3TG. Itnevertheless epitomizes the motivation and desire of women in easternDRC, specifically their drive to take advantage of new opportunities inASM and their willingness to position themselves to derive benefitsfrom activities throughout the supply chain
new mining code -new opportunities for women
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
Response to reviewers
We wish to thank the reviewers for the time taken to appraise the manuscript and the helpful feedback to improve it. We have taken onboard the suggested feedback and incorporated it into the revision. The findings of the revised manuscript are unchanged. Below is a point-by-point response to specific comments.
Public reviews
Reviewer 1
Thank you to reviewer 1 for the thorough and insightful review of our manuscript. We are pleased that the strengths of our research, particularly the use of whole-genome bisulfite sequencing, the combination of animal and human data, and the investigation of a potential dietary intervention were recognized. We are confident that these aspects contribute significantly to the value and originality of our work.
We acknowledge the concerns regarding the statistical rigor of the study, particularly the sample size and data analysis methods. We would like to address these points in more detail:
Sample size: While we agree that a larger sample size would be ideal, the chosen sample size (n=4 per group) is consistent with other murine whole-genome bisulfite sequencing experiments in the field. We have carefully considered the cost-benefit trade-off in selecting this approach. In the revision we discuss the potential limitations of this sample size.
Data analysis: We acknowledge the inconsistencies in the study reporting and have committed to improving the clarity in the revision. We carefully reviewed the concerns regarding the use of causal language and the interpretation of differences in our results. In some cases, the use of causal language is justified by the intervention study design. We also believe other explanations like stochastic variation affecting the same genomic regions in different tissues, are exceedingly unlikely from a statistical viewpoint. In the revision we have adopted a balanced approach to the language.
Confounders: We acknowledge the importance of accounting for potential confounders such as birthweight, alcohol exposure and sex. The pups selected for genome analysis were matched for sex and on litter size as a proxy for in utero alcohol exposure. This careful selection of mice for genome analysis was intentionally guided to mitigate potential confounding.
Statistical rigour: We acknowledge the importance of multiple testing correction in the genome-wide analysis. We used the DSS method of Feng et al (PMID: 2456180) which employs a two-step procedure for assessing significance of a region. Instead of a single p-value for the whole DMR, DSS uses the area statistic to rank candidate regions and control the false discovery rate through shrinkage estimation methods. This approach reduces the risk of reporting false positives due to multiple testing across numerous CpG sites. It is similar in respects to employing local FDR correction at 0.05 level, with an additional minimum effect size threshold applied, and particularly suited to experiments where the number of replicates is low. In the revision we have committed to improving the clarity of the reporting of statistical methods.
Reviewer 2
Thank you to reviewer 2 for the comprehensive and valuable feedback on our manuscript. We take your concerns about the generalizability of our findings and the interpretation of certain results seriously. We would like to address your specific criticisms in detail:
Generalizability and Human Data: We agree that the generalizability of mouse models to human conditions has limitations. However, our study focused on understanding the early molecular alterations caused by moderate PAE, which can be more effectively modelled in a controlled environment like mice. To clarify this, we have strengthened the manuscript by emphasizing the focus on moderate PAE in the title and throughout the paper.
Transcriptome Analysis: We recognize the importance of investigating the functional consequences of PAE-induced DMRs and agree that transcriptome analysis would be highly valuable. We are currently planning to conduct future transcriptomic studies to understand the link between DMRs and gene expression.
Species-Specificity and DMR Enrichment: We acknowledge the likelihood of species-specific PAE effects. Our finding of enrichment of DMRs in non-coding regions was consistent with observations from the Lussier study of FASD. We agree there is further work to do and now highlight this in the discussion.
Tissue Sample Locations: Due to technical restrictions of processing newborn mouse tissue, we are unable to enhance the manuscript with specific tissue regions sampled.
Interpretation of Shared Genomic Regions: We appreciate your point about the alternative explanation for the shared genomic regions between brain and liver. Our interpretation is that regions identified in the alcohol group only affected equally in both tissues are likely established stochastically (as a result of the exposure) in the early embryo and then maintained in the germ layers. We have revised to suggest this is the most likely explanation and we acknowledge a more detailed examination in more tissues would be warranted for proof.
Additional Feedback
Reviewer 1
Introduction
- Line 65 - alcohol consumption is not always preventable and these statements further increase the stigma associated with FASD. A better way to say this would be "a leading cause of neurodevelopmental impairments".
We have implemented this suggestion in revised manuscript.
- The studies cited in lines 87-89 are somewhat outdated, as several more recent studies with better sample sizes have been published in recent years. I would recommend citing more recent publications in addition to these studies. Similarly, the authors should also cite Portales-Casamar et al., 2016 (Epigenetic & Chromatin) for the validation in humans, as it was the original study for those data.
We have added a citation for the study mentioned by Portales-Casamar et al. (2016) in the revised manuscript.
- Lines 95-95 - the authors should elaborate further on the "encouraging results" from choline supplementation studies, as these details may help interpret the findings from their own study.
In the revised manuscript, we replaced “encouraging results” with “results suggesting a high methyl donor diet (HMD) could at least partially mitigate the adverse effects of PAE on various behavioural outcomes”.
- Minor point: DNA methylation is preferable to "methylation" alone when not referring to specific CpGs or sites, as methylation can also refer to protein or RNA methylation.
“Methylation” has been replaced with “DNA methylation” in revised manuscript
Results
- Line 118 - HMD should be defined here.
HMD defined in revised manuscript
- The figures in the main manuscript and supplemental materials are not in the same order as they are presented in the text.
We apologise for this and thank the reviwer for their attendtion to detail. In the revision we have corrected the order of figures to match the text.
- It is concerning that the H20-HMD group had lower baseline weights, which could impact the findings from these analyses. Please discuss how these differences were accounted for in the study design and analyses.
We appreciate the reviewer's concern about the lower baseline weight in the H20-HMD group. We agree that this difference could potentially affect our findings. However, we want to emphasize that total weight gain during pregnancy was statistically similar across all groups by linear mixed effect model. Additionally, all dams were within the healthy weight range for their strain. While we cannot completely rule out any potential influence of baseline weight, we believe the similarity in weight gain and the healthy range of all dams suggest that the in-utero experience of pups regarding weight-related factors was likely comparable across groups.
- I have some concerns regarding the cutoffs used to identify the DMRs, particularly given the small N and number of tests. The authors should report the number of DMRs that meet a multiple testing threshold; if none, they should use a more stringent threshold than p<0.05, as one would expect 950,000 CpGs to meet that threshold by chance (19,000,000 CpGs x 0.05). The authors should also report the number of DMRs tested, as this will be a more appropriate benchmark for their analyses than the number of CpGs (they should also report the specific number here).
We appreciate the reviewer's concerns regarding the DMR cut-offs. We agree that clarifying the methods and justifying our choices is crucial. Our implementation of the DSS method for defining DMRs employs a local FDR p<0.05 cut-off, with additional delta beta threshold of 5%. We have clarified this in the methods section of the revised manuscript . We want to emphasize that the local FDR approach effectively mitigates the concern of chance findings by adjusting for multiple comparisons across the genome. Line 414-420 in the revised methods contains the following amended text
“Differentially methylated regions (DMRs) were identified within each tissue using a Bayesian hierarchical model comparing average DNA methylation ratios in each CpG site between PAE and non-PAE mice using the Wald test with smoothing, implemented in the R package DSS (46). False-discovery rate control was achieved through shrinkage estimation methods. We declared DMRs as those with a local FDR P-value < 0.05 based on the p-values of each individual CpG site in the DMR, and minimum mean effect size (delta) of 5%”
- I also have concerns about the delta cutoff for their DMRs. First, it is not clear if this cutoff is set for a single CpG or across the DMR (even then, it is not clear if this is a mean, median, max, min, etc.) Second, since the authors analyzed CpGs with 10X coverage, they can only reliably detect a delta of 0.1 (1/10 reads).
Thank you for raising this important point. In the revision we have clarified the effect size cutoff reflects the mean effect across CpGs within the DMR as follows (line 418)
“We declared DMRs as those with a local FDR P-value < 0.05 based on the p-values of each individual CpG site in the DMR, and minimum mean effect size (delta) of 5%”
We chose the mean as it provides a comprehensive representation of the overall methylation change within the region, while ensuring all individual CpGs used in the analysis had at least 10x coverage. It is not true that we can only detect a delta of 1/10 reads, the mean effect is the relative difference in means between groups and is not dependent on the underlying sequencing depth.
- Prenatal alcohol exposure is known to impact cell type proportions in the brain, which could lead to differences in DNAm patterns. The authors should address this possibility in the discussion, as well as examine their list of DMRs to determine if they are associated with specific brain cell types. The possibility of cell type differences in the liver should also be discussed.
We agree with the reviewer that PAE-induced alterations in cell type proportions can influence DNA methylation patterns. While isolating specific cell types in our current study's brain and liver samples was not achievable due to tissue limitations, we acknowledge this as a limitation and recognize the need for further investigations incorporating single-cell or cell type-specific approaches in the discussion.
- It is interesting, but maybe not surprising, that more DMRs were identified in the liver compared to the brain. This finding would warrant some additional interpretation in the discussion.
We appreciate and agree that this finding indeed warrants further interpretation. We have added the following sentence into the discussion section of the revised manuscript that provides some potential factors behind this observation.
Lines 263 “Indeed, most of the observed effects were tissue-specific, with more perturbations to the epigenome observable in liver tissue, which may reflect the liver’s specific role in metabolic detoxification of alcohol. Alternatively, cell type composition differences between brain and liver might explain differential sensitivity to alcohols effects”.
- Lines 148-149 - I disagree about the enrichment of decreased DNAm in brain DMRs, as 52.6% is essentially random chance. The authors should also include a statistical test here, such as a chi-squared test, to support this statement.
We agree that a revised interpretation is warranted. The updated manuscript has been amended as follows: “Lower DNA methylation with early moderate PAE in NC mice was more frequently observed in liver DMRs (93.5% of liver DMRs), while brain DMRs were almost equally divided between lower and higher DNA methylation with early moderate PAE (52.6% of brain DMRs had lower DNA methylation with early moderate PAE).”
- Similarly, I would recommend the authors use increased/decreased DNAm, rather than hypermethylated/hypomethylation, as the latter terms are better suited to DNAm values near 100% or 0%.
The use of hyper/hypo methylation is still considered common and well understood even for moderate changes. We agree the use of increased/decreased is more inclusive for a broader audience, so we have amended all references accordingly in the main text.
- Lines 153-155 - please report the statistics to support these enrichment results. A permutation test would be well suited to this analysis.
The reporting of statistics related to the enrichment test has now been amended to read “Overlap permutation tests showed liver DMRs were enriched in inter-CpG regions and non-coding intergenic regions (p < 0.05), while being depleted in all CpG regions and genic regions except 1to5kb, 3UTR and 5UTR regions, where there was no significant difference (Figure 2f).”
- Line 156 - "overwhelming enrichment" is a very strong statement considering the numbers themselves.
Omitted “overwhelming” in revised manuscript. Revised manuscript states: “Using open chromatin assay and histone modification datasets from the ENCODE project, we found enrichment (p < 0.05) of DMRs in open chromatin regions (ATAC-seq), enhancer regions (H3K4me1), and active gene promoter regions (H3K27ac), in mouse fetal forebrain tissue and fetal liver (Table 2).”
- Lines 165-167 - Please describe the analyses and metrics used to determine if the DNAm differences were mitigated in the HMD groups. As it stands, it is not clear if they are simply not significant, or if the delta was decreased. In terms of a figure, a scatter plot of the deltas for these DMRs would be better suited to visualizing these changes.
To determine whether DMRs were mitigated we simply applied the same statistical testing procedure on the subset of PAE DMRs in the group of mice exposed to the HM diet. The sample size is the same, and the burden on multiple testing is reduced as we did not test the entire genome. We believe our interpretation stands although we have urged caution in the discussion as follows (line 319)
“Another key finding from this study was that HMD mitigated some of the effects of PAE on DNA methylation. Although a plausible alternative explanation is that some of the PAE regions were not reproduced in the set of mice given the folate diet, our data are consistent with preclinical studies of choline supplementation in rodent models (34, 35) (36). Moreover, a subset of PAE regions were statistically replicated in subjects with FASD, suggestive or robust associations. Although our findings should be interpreted with caution, they collectively support the notion that alcohol induced perturbation of epigenetic regulation may occur, at least in part, through disruption of the one-carbon metabolism.”
- Given the lenient threshold to identify DMRs, it is possible that PAE-associated DMRs are simply false positives and do not "replicate" in a different subset of animals. One way to check this would be to determine whether there are any differences between mitigated/unmitigated DMRs and the strength of their initial associations. Should the mitigated DMRs skew towards higher p-values and lower deltas, one might consider that these findings could be false positives.
We appreciate the reviewer's concern about potential false positives due to the chosen DMR identification threshold. We reiterate the DMR calling thresholds were adjusted for local FDR; however, we acknowledge the need for further validation. We haven't observed this trend of mitigated DMRs having higher p-values and lower deltas, but we have replicated some PAE DMRs in independent human datasets and found support for their biological plausibility in the context of PAE.
- Related to the HMD analyses, I am concerned that the EtOH-HMD group consumed less alcohol, which could manifest in the PAE-induced DMRs disappearing, unrelated to the HMD exposure. The authors should comment on whether the pups were matched for ethanol exposure and include sensitivity analyses that include ethanol level as a covariate to confirm that their results are not simply due to decreased alcohol exposure.
We appreciate the reviewer's concern regarding the lower alcohol consumption by Dams in the EtOH-HMD group and its potential impact on DMRs. We agree that consistent in utero exposure is crucial for reliable results. Our pup selection for genomic analysis involved matching litter size as a proxy for in utero exposure, so even through the average alcohol consumption was lower for the EtOH-HMD group, we matched pups across treatment groups based on litter size as a proxy for alcohol intake levels, excluding pups with significantly different exposure levels. We agree more robust methods including direct measurement of blood alcohol content would improve the study. We have now incorporated this into the discussion of the revised manuscript on lines 351: “Additionally, we employed an ad-libitum alcohol exposure model rather than direct dosing of dams. Although the trajectories of alcohol consumption were not statistically different between groups, this introduces more variability into alcohol exposure patterns, and might might impact offspring methylation data”
- Lines 172 - please be more specific about the neurocognitive domains tested.
In the revision we have included more detail about the neurocognitive domains tested (originally mentioned in the results) in the methods as follows:
“These tests included the open field test (locomotor activity, anxiety) (38), object recognition test (locomotor activity, spatial recognition) (39), object in place test (locomotor activity, spatial recognition) (40), elevated plus maze test (locomotor activity, anxiety) (41), and two trials of the rotarod test (motor coordination, balance) (42)”
- Line 191 - please report the tissue type used in the human study, as well as the method used to estimate cell type proportions.
We stated in the results section that buccal swabs were used in both human cohorts.
We added to the revised manuscript that cell type proportions were estimated using the EpiDISH R package.
- Related to validation, it is unclear whether the human-identified DMRs were also validated in mice, or if the authors are showing their own DMRs. Please also discuss why DMRs might not have been replicated in AQUA.
We used human data sets to validate observations from our murine model, focusing on regions identified in our early moderate PAE model. This is now explicitly state on line 209 of the revision:
“We undertook validation studies by examining PAE sensitive regions identified in our murine model using existing DNA methylation data from human cohorts to address the generalizability of our findings.”
“In the section entitled ‘Candidate Gene Analysis..’ we used our murine data sets to reproduce previously published associations that included regions identified in both animal and human studies. We posit the lack of replication of our early moderate PAE regions in AQUA is explained in part by species-specific differences and considering the striking differences in effect size seen in regions that did replicate in FASD subjects, the exposure may need to be of sufficient magnitude and duration for the effects seen in brain and liver to survive reprogramming in the blood. The AQUA cohort is largely enriched for low to moderate patterns of alcohol consumption.
- Line 197 - please provide a citation for the ethanol-sensitive regions. There are also several existing DNAm analyses in brain tissues from animal models that should be included as part of these analyses, as several have shown brain-region and sex-specific DMRs related to prenatal alcohol exposure. These contrasts might help the authors further delineate the effects of prenatal alcohol in their model and expand on current literature to explain the deficits caused by alcohol exposure.
Our candidate gene/region selection was informed by a systematic review of previously published human and animal studies reporting associations between in utero exposure to PAE and offspring DNA methylation. We synthesized evidence across several models, tissues and methylation platforms to arrive at a core set of reproducible associations. Line 481 of the methods now includes a citation to our systematic review which details our selection criteria.
Discussion
- Line 211 - This is a strong statement for one hypothesis. It is also possible that different cell types have similar responses to prenatal alcohol exposure. In this scenario, perturbations need not arise before germ layer separation. The authors should soften this causal statement.
We appreciate this point although given the genome size relative to the size of the DMRs we have detected, the chance that different cell types would respond similarly in exactly the same regions seems exceedingly rare. We posit a more likely explanation is early perturbations in the embryo are established stochastically as a result of the exposure (supported by the interventional design) and maintained in the differentiating tissues. We agree further work is needed to prove this, specifically in a wider set of tissues from multiple germ layers so we have amended the discussion as follows:
“These perturbations may have been established stochastically because of alcohol exposure in the early embryo and maintained in the differentiating tissue. Further analysis in different germ layer tissues is required to formally establish this.”
- Lines 222-224 - I completely agree with this statement. However, the authors had the opportunity to examine dosage effects in their model as they measured alcohol-levels from the dams. At the very least, I would recommend sensitivity analyses in their DMRs to assess whether alcohol level/dosage influences their results.
Although a great suggestion to improve the manuscript, we did not have opportunity to examine dosages by design as we selected mice for genome analysis with matched exposure patterns. It would be fascinating to conduct a sensitivity analysis.
Methods:
- Please include the lysis protocol.
Thank you for picking up this error in our reporting. We have now included the following details in the methods which improve the reproducibility of this study: “Ten milligrams of tissue were collected from each liver and brain and lysed in Chemagic RNA Tissue10 Kit special H96 extraction buffer”.
- Please include the total reads for each sample and details of the QC pipeline, including filtering flags, quality metrics, and genome build.
Thank you for suggesting improvements to our reporting which improve the reproducibility of this study. We have included a new supplementary tableTab of sequencing statistics and details of the quality metrics. Please note the genome build is explicitly stated in the methods already.
- Please make your code publicly available to ensure that these analyses can be replicated.
Thank you for this suggestion. A data availability statement has now been included in the revision and code will be made available upon request
- Why were Y chromosome reads included in the dataset?
Y chromosomal reads were not included in the DMR analysis. Amended “We filtered the X chromosomal reads” to “We filtered the sex chromosomal reads” in revised manuscript.
- Please provide the number of total CpGs available for analysis.
Added sentence into results section of revised manuscript: “A total of 21,842,961 CpG sites were initially available for analysis.” We also clarified that the ~19,000,000 CpGs were analysed following coverage filtering.
- Please provide the parameters for the DMR analysis and report how the p-values and deltas were calculated.
We have addressed this in previous comments
- The supplemental materials for the human data are missing.
Thank you for picking up this oversight. The revision now includes an additional data supplement which details the analysis of the human data sets for interested readers.
Tables and figures
- Table 1. It is not clear how the DMRs for this table were selected. The exact p-values and FDR should also be reported in this table. The number of CpGs in these DMRS should also be reported.
Table 1 includes select DMRs that were consistently detected in both brain and liver tissue. These are particularly of interest as they represent regions highly sensitive to alcohol exposure. We agree that exact reporting of p-values would be ideal. Instead of a single p-value for the whole DMR, DSS uses the area statistic to rank candidate regions and control the false discovery rate (FDR) through shrinkage estimation methods. In the revision we have now included region size and number of CpGs in table 1.
- Table 3. Please include p-values for the DMR analyses.
As above we report the area-statistic which is an equivalent measure to assess evidence for differential methylation.
- Figure 2 (Figure 4 in revised manuscript). Please report the N for these analyses. It also seems that the pairwise t-tests were only compared to the H20-NC, which does not provide much insight into the PAE group. The relevance of the sexP analysis to the present manuscript is also unclear.
Figure 2 is now Figure 4 in the revision and the sample size has been included in figure legend. We compared all groups to the control group (H20-NC) as we aimed to determine any differences in intervention groups from the control.
We apologies for lack of clarity around the ‘sex P’ terminology. This refers to the p-value for the main effect of sex on the behavioural outcome. We agree it lacks relevance since the regression models were adjusted for sex. In the revision we have updated the methods as follows (line426) and removed references to sex P
“To examine the effect of alcohol exposure on behavioural outcomes we used linear regression with alcohol group (binary) as the main predictor adjusted for diet and sex.”
- Figure 3ef (Figure 2ef in revised manuscript). It is unclear how the regions random regions were generated. A permutation test would be relevant to determine whether there are any actual enrichment differences.
As stated in methods section: “DMRs were then tested for enrichment within specific genic and CpG regions of the mouse genome, compared to a randomly generated set of regions in the mouse genome generated with resampleRegions in regioneR, with equivalent means and standard deviations.”
- Figure 5. Please include the gene names for these DMRs, as well as their genomic locations. It would also be relevant to annotate these plots with the max, min, and mean delta between groups.
Thank you, we considered this however the DMRs are not in genes so we cannot apply a gene label. The locations are reported on the x-axis and the statistics are shown in Table 3.
- Figure S1b and S2c- It is quite worrisome that the PAE-HMD group drank less throughout pregnancy than their PAE counterparts. Please discuss how this was addressed in the analyses.
We appreciate the reviewer's concern regarding the lower alcohol consumption in the PAE-HMD group and its potential impact on DMRs. We agree that consistent in-utero exposure is crucial for reliable results. Although the total amount of liquid consumed over pregnancy was lower in this group, they started with a lower baseline and the trajectory was not statistically different compared to other groups.
We have now incorporated this into the discussion section of the revised manuscript on lines 336: “Additionally, we employed an ad-libitum alcohol exposure model rather than direct dosing of dams. Although the trajectories of alcohol consumption were not statistically different between groups, this introduces more variability into alcohol exposure patterns, and might might impact offspring methylation data.”
- Figure S1cd. See my comments about Figure 2.
Suggested changes have been incorporated.
- Figure S2d. it is not clear to what the statistics presented in this panel refer. Please clarify and discuss the implications of dietary intake differences on your findings.
Added sentence to caption in revised manuscript: “Statistical analysis involved linear mixed-effects regression comparing trajectories of treatment groups to H2O-NC baseline control group.”
- Figure S3. See my comments about Figure 2.
Suggested changes have been incorporated
- Figure S4. I am confused by the color legend, as it seems both colors are PAE. I also do not see how any regions show increased or decreased DNAm in PAE based on this plot (also no statistics are presented to support these conclusions).
The plot is intended to show there are no gross changes in methylation when averaged across all CpGs within different regulatory genomic contexts. Statistics are not included as it is intuitive from the plot that the means are the same. We have updated the figure legend which now reads
“Figure S4. No evidence for global disruption of methylation by PAE. The figure shows methylation levels averaged across CpGs in different regulatory genomic contexts. Neither brain tissue (A & B), nor liver tissue (C & D) were grossly affected by PAE exposure (blue bars). Bars represent means and standard deviation.”
-
-
learn-us-east-1-prod-fleet01-xythos.content.blackboardcdn.com learn-us-east-1-prod-fleet01-xythos.content.blackboardcdn.com
-
What year were you born?Please respond in YYYY format____________3. What are the first four letters of the name of your hometown?Please respond in nnn formatDo not capitalize the first letter____________4. What is the zip code of your home address?Please respond in ZZZZZ format____________End Group: Unique User ID5. What is your gender?Select one.o Femaleo Maleo Non-binary/ third gendero Prefer not to answero Prefer to self-describe: ________________6. Do you currently live in New York or consider New York your primary state of residence?Select one.o Yes -----> Continue to number 7o No -----> Skip to number 8
Starts with the demographic questions since they already previously stated they would ask them in the consent form. Typically demographic questions are last however since they already informed the reader about them, there will be no surprise to seeing them. Also, the demographic questions are very simple and broad. The researcher is only collecting the information needed and not asking in-depth questions. This is so they do not scare away the participant with too personal of questions.
-
You will be asked to provide your birth year, zip code of residence, and the first 4 letters of the name ofyour hometown to generate a unique user code associated with your responses. This information will beused to count the number of participants and to estimate the average number of surveys participantscomplete. Providing this information is completely voluntary and will not be available to anyone exceptthe primary investigators. Your name will not be associated with the responses you provide, or the datagenerated from them. You will also be asked to provide the coordinates of the location where youcompleted the survey. Coordinate information is important
This section relates to the demographic questions. Since people are hesitant to answer personal information, explaining why it is needed and that it is optional puts the participant at ease and makes them more likely to answer. Also allows the participant not to be surprised when asked about it in the survey.
-
-
docs.google.com docs.google.com
-
(Texas Mexicans and California Mexicans are very different from each other, like the Scottish and the Irish-fundamentally the same genetic code, but completely different in accent and habits.)
true, nice analogy
-
-
engl201.opened.ca engl201.opened.ca
-
This may mean, “supplementing ‘the humanities’ own methodologicaltoolkits’ with theoretical insights from software, critical code and platform studies”
This could be speaking about any tools, but I'm going to mention AI, and how everyone makes it out to be the worst thing in the world when in reality, (she had a hidden agenda! she put my tender, heart in a blender, and STILL I surrendered!) It's quite a useful tool.
-
-
apnews.com apnews.com
-
frostbite
frostbite: the superficial or deep freezing of the tissues of some part of the body
The WHO's code of practice for fishery products indicates that shrimp should be stored and processed at temperatures below 4°C.
-
-
ipfs.indy0.net ipfs.indy0.netDeno 1.01
-
Deno is (and always will be) a single executable file. Like a web browser, it knows how to fetch external code.
knows how to fetch external code
-
-
naturalistic-data.org naturalistic-data.org
-
masker.fit_transform
AttributeError: 'Brain_Data' object has no attribute 'fit_transform'
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
Response to Reviewers’ Public Comments
We are grateful for the reviewers’ comments. We have modified the manuscript accordingly and detail our responses to their major comments below.
(1) Reviewer 2 was concerned that transformation of continuous functional data into categorical form could reduce precision in estimating the genetic architecture.
We agree that transforming continuous data into categories may reduce resolution, but it also improves accuracy when the continuous data are affected by measurement noise. In our dataset, many genotypes are at the lower bound of measurement, and the variation in measured fluorescence among these genotypes is largely or entirely caused by measurement noise. By transforming to categorical data, we dramatically reduced the effect of this noise on the estimation of genetic effects. We modified the results and discussion sections to address this point.
(2) Reviewer 2 asked about generalizability of our findings.
Because our paper is the first use of reference-free analysis of a 20-state combinatorial dataset, generalizability is at this point unknown. However, a recent manuscript from our group confirms the generality of the simplicity of genetic architecture: using reference-free methods to analyze 20 published combinatorial deep mutational scans, several of which involve 20-state libraries, we found that main and pairwise effects account for virtually all of the genetic variance across a wide variety of protein families and types of biochemical functions (Park Y, Metzger BPH, Thornton JW. 2023. The simplicity of protein sequence-function relationships. BioRxiv, 2023.09.02.556057). Concerning the facilitating effect of epistasis on the evolution of new functions, we speculate that this result is likely to be general: we have no reason to think that the underlying cause of this observation – epistasis brings genotypes with different functions closer in sequence space to each other and expands the total number of functional sequences – arises from some peculiarity of the mechanisms of steroid receptor DBD folding or DNA binding. However, we acknowledge that our data involve sequence variation at those sites in the protein that directly mediate specific protein-DNA contact; it is plausible that sites far from the “active site” may have weaker epistatic interactions and therefore have weaker effects on navigability of the landscape. We have addressed these issues in the discussion.
(3) Reviewer 3 asked “in which situation would the authors expect that pairwise epistasis does not play a crucial role for mutational steps, trajectories, or space connectedness, if it is dominant in the genotype-phenotype landscape?”
The question addressed in our paper is not whether epistasis shapes steps, trajectories or connectedness in sequence space but how it does so and what its particular effects are on the evolution of new functions. The dominant view in the field has been that the primary role of epistasis is to block evolutionary paths. We show, however, that in multi-state sequence space, epistasis facilitates rather than impedes the evolution of new functions. It does this by increasing the number of functional genotypes and bringing genotypes with different functions closer together in sequence space. This finding was possible because of the difference in approach between our paper and prior work: most prior work considered only direct paths in a binary sequence space between two particular starting points – and typically only considering optimization of a single function – whereas we studied the evolution of new functions in a multi-state amino acid space, under empirically relevant epistasis informed by complete combinatorial experiments. The result is a clear demonstration that the net effect of real-world levels of epistasis on navigability of the multidimensional sequence landscape is to make the evolution of new functions easier, not harder.
(4) Reviewer 3 asked for “an explanation of how much new biological results this paper delivers as compared with the paper in which the data were originally published.”
Starr 2017 did not use their data to characterize the underlying genetic architecture of function by estimating main and epistatic effects of amino acid states and combinations; it also did not evaluate the importance of epistasis in generating functional variants, determining the transcription factor’s specificity, or shaping evolutionary navigability on the landscape.
(5) Reviewer 3 requested an explanation of how the results would have been (potentially) different if a reference-based approach were used, and how reference-based analysis compares with other reference-free approaches to estimating epistasis.
This topic has been covered in detail in a recent manuscript from our group (Park et al. Biorxiv 2023.09.02.556057). Briefly, reference-free approaches provide the most efficient explanation of an entire genotype-phenotype map, explaining the maximum amount of genetic variance and reducing sensitivity to experimental noise and missing genotypes compared to reference-based approaches. Reference-based approaches tend to infer much more epistasis, especially higher-order epistasis, because measurement error and local idiosyncrasy near the wild-type sequence propagate into spurious high-order terms. Reference-based analyses are appropriate for characterizing only the immediate sequence neighborhood of a particular “wild-type” protein of interest. Reference-free approaches are therefore best suited to understanding genotype-phenotype landscapes as a whole. We have clarified these issues in the revised discussion.
(6) Reviewer 3 suggested that the comparison between the full and main-effects-only model should involve a re-estimation of main effects in the latter case.
This is indeed what we did in our analysis. We have clarified the description in the results and methods sections to make this clear.
(7) Reviewer 3 asked about the applicability of the approach to data beyond those analyzed in the present study and requirements to use it.
Our approach could be used for any combinatorial DMS dataset in which the phenotypic data are categorical (or can be converted to categorical form). Complete sampling is not required: a virtue of reference-free analysis is that by averaging the estimated effects of states and combinations over all variants that contain them, reference-free analysis is highly robust to missing data (except at the highest possible order of epistasis, where only a single variant represents a high-order effect) as long as variant sampling is unbiased with respect to phenotype. All the required code are publicly available at the github link provided in this manuscript. We have also described a general form of reference-free analysis for continuous data and applied it to 20 protein datasets in a recent publication (Park et al. Biorxiv 2023.09.02.556057).
(8)Reviewer 3 suggested that the text could be shortened and made less dense.
We agree and have done a careful edit to streamline the narrative.
Response to Reviewers’ Non-Public Recommendations
(1) Reviewer 1 noted that specific epistatic effects might in some cases produce global nonlinearities in the genotype-phenotype relationship. They then asked how our results might change if we did not impose a nonlinear transformation as part of the genotype-phenotype model. The reviewer’s underlying concern was that the non-specific transformation might capture high-order specific epistatic effects and thus reducing their importance.
Because our data are categorical, we required a model that characterizes the effect of particular amino acid states and combinations on the probability that a variant is in a null, weak, or strong activation class. A logistic model is the classic approach to this kind of analysis. The model structure assumes that amino acid states and combinations have additive effects on the log-odds of being in one functional class versus the lower functional class(es); the only nonlinear transformation is that which arises mathematically when log-odds are transformed into probability through the logistic link function. Thinking through the reviewer’s comment, we have concluded that our model does not make any explicit transformation to account for nonlinearity in the relationship between the effects of specific sequence states/combinations and the measured phenotype (activation class). If additional global nonlinearities are present in the genotype-phenotype relationship – such as could be imposed by limited dynamic range in the production of the fluorescence phenotype or the assay used to measure it – it is possible that the sigmoid shape of the logistic link function may also accommodate these nonlinearities. We have noted this part in the revised manuscript.
(2) Reviewer 1 observed that our model seems to prefer sets of several pairwise interactions among states across sites rather than fewer high-order interactions among those same states.
This finding arises because the pattern of phenotypic variation across genotypes in our dataset is consistent with that which would be produced by pairwise interactions rather than by high-order interactions. In a reference-free framework, these patterns are distinct from each other: a group of second-order terms cannot fit the patterns produced by high-order epistasis, and high-order terms cannot fit the pattern produced by pairwise interactions. Similarly, main-effect terms cannot fit the pattern of phenotypes produced by a pairwise interaction, and a pairwise epistatic term cannot fit the pattern produced by main effects of states at two sites. For example, third-order terms are required when the genotypes possessing a particular triplet of states deviate from that expected given all the main and second-order effects of those states; this deviation cannot be explained by any combination of first- and second-order effects.
We explain this point in detail in our recent manuscript (Park Y, Metzger BPH, Thornton JW. 2023. The simplicity of protein sequence-function relationships. BioRxiv, 2023.09.02.556057) and we summarize it here. Consider the simple example of two sites with two possible states (genotypes 00, 01, 10, and 11). If there are no main effects and no pairwise effects, this architecture will generate the same phenotype for all four variants – the global average (or zero-order effect). If there are pairwise effects but no main effects, this architecture will generate a set of phenotypes on which the average phenotype of genotypes with a 0 at the first site (00 and 01) equals the global average – as does the average of those with 0 at the second site (00 and 10). The epistatic effect causes the individual genotypes to deviate from the global average. This pattern can be fit only by a pairwise epistatic term, not by first-order terms. Conversely, if there are main effects but no pairwise effects, then the average phenotype of genotypes 00 and 01 will deviate from the global average (by an amount equal to the first-order effect), as will the average of (00 and 10): the phenotype of each genotype will be equal to the sum of the relevant first-order effects for the state it contains. This pattern cannot be fit by second-order model terms. The same logic extends to higher orders: a cluster of second-order terms cannot explain variation generated by third-order epistasis, because third-order variation is by definition is the deviation from the best second-order model.
(3) Reviewer 1 suggested several places in the text where citations to prior work would be appropriate.
We appreciate these suggestions and have modified the manuscript to refer to most of these works.
(4) Reviewer 1 pointed to the paper of Gong et al eLife 2013 and asked whether it is known how robust the proteins in our study are to changes in conformation/stability compared to other proteins, and whether this might impact the likelihood of observing higher-order epistasis in this system.
The DBDs that we study here are very stable, and previous work shows that mutations affect DNA specificity primarily by modifying the DBD’s affinity rather than its stability (McKeown et al., Cell 2014). Additionally, Gong et al.’s findings pertain to a globally nonlinear relationship between stability and function, which arises from the Boltzmann relationship between the energy of folding and occupancy of the folded state. Because our data are categorical – based on rank-order of measured phenotype rather than fluorescence as a continuous phenotype – the kind of global nonlinearity observed in Gong’s study are not expected to produce spurious estimates of epistasis in our work. We have modified the discussion to discuss the point.
(5) Reviewer 1 asked a) why the epistatic models produce landscapes on which variants have fewer neighbors on average than main-effects only models and b) why the average distance from all ERE-specific nodes to all SRE-specific nodes is greater with epistasis (but the average distance from ERE to nearest SRE is lower with epistasis).
In the main effects-only landscape, the functional genotypes are relatively similar to each other, because each must contain several of the states that contribute the most to a positive genetic score. Moreover, ERE-specific nodes are similar to each other, and SRE-specific nodes are similar to each other, because each must contain one or more of a relatively small number of specificity-determining states. When epistasis is added to the genetic architecture, two things happen: 1) more genotypes become functional because there are more combinations that can exceed the threshold score to produce a functional activator and 2) these additional functional variants are more different from each other – in general, and within the classes of ERE- or SRE-specific variants – because there are now more diverse combinations of states that can yield either phenotype. As a result, a broader span of sequence space is occupied, but ERE- and SRE-specific variants are more interspersed with each other. This means that the average distance between all pairs of nodes is greater, and this applies to all ERE-SRE pairs, as well. However, the interspersing means that the closest single SRE to any particular ERE is closer than it was without epistasis. We have added this explanation to the main text.
(6) Reviewer 2 asked us to explain why average path length increases with pairwise epistasis as the strength of selection for specificity increases.
This behavior occurs because of the existence of a local peak in the pairwise model. Genotypes on this peak contained few connections to other genotypes, all of which were less SRE specific. Thus, with strong selection, i.e. high population size, the simulations became stuck on the local peak, cycling among the genotypes many times before leaving, resulting in a large increase in the mean step number. As shown in the rest of the figure, when the longest set of paths are removed, there are still differences in the average number of steps with and without epistasis. This issue is described in the methods section.
(7) Reviewers made several suggestions for clarity in the text and figures.
We have modified the paper to address all of these comments.
(8) Reviewer 3 stated that the code should be available.
The code is available at https://github.com/JoeThorntonLab/DBD.GeneticArchitecture.
-
-
www.education.gouv.fr www.education.gouv.fr
-
recours effectif à l'ensemble du panel des sanctions réglementaires fixé à l'article R. 511-13 du code de l'éducation et reproduit dans le règlement intérieur
-
-
www.biorxiv.org www.biorxiv.org
-
Reviewer #1 (Public Review):
Summary:
Semenova et al. have studied a large cross-sectional cohort of people living with HIV on suppressive ART, N=115, and performed high dimensional flow cytometry to then search for associations between immunological and clinical parameters and intact/total HIV DNA levels.
A number of interesting data science/ML approaches were explored on the data and the project seems a serious undertaking. However, like many other studies that have looked for these kinds of associations, there was not a very strong signal. Of course, the goal of unsupervised learning is to find new hypotheses that aren't obvious to human eyes, but I felt in that context, there were (1) results slightly oversold, (2) some questions about methodology in terms mostly of reservoir levels, and (3) results were not sufficiently translated back into meaning in terms of clinical outcomes.
Strengths:
The study is evidently a large and impressive undertaking and combines many cutting-edge statistical techniques with a comprehensive experimental cohort of people living with HIV, notably inclusive of populations underrepresented in HIV science. A number of intriguing hypotheses are put forward that could be explored further. Sharing the data could create a useful repository for more specific analyses.
Weaknesses:
Despite the detailed experiments and methods, there was not a very strong signal for the variable(s) predicting HIV reservoir size. The Spearman coefficients are ~0.3, (somewhat weak, and acknowledged as such) and predictive models reach 70-80% prediction levels, though sometimes categorical variables are challenging to interpret.
There are some questions about methodology, as well as some conclusions that are not completely supported by results, or at minimum not sufficiently contextualized in terms of clinical significance.
On associations: the false discovery rate correction was set at 5%, but data appear underdetermined with fewer observations than variables (144vars > 115ppts), and it isn't always clear if/when variables are related (e.g inverses of one another, for instance, %CD4 and %CD8).
The modeling of reservoir size was unusual, typically intact and defective HIV DNA are analyzed on a log10 scale (both for decays and predicting rebound). Also sometimes in this analysis levels are normalized (presumably to max/min?, e.g. S5), and given the large within-host variation of level we see in other works, it is not trivial to predict any downstream impact of normalization across population vs within-person.
Also, the qualitative characterization of low/high reservoir is not standard and naturally will split by early/later ART if done as above/below median. Given the continuous nature of these data, it seems throughout that predicting above/below median is a little hard to translate into clinical meaning.
Lastly, the work is comprehensive and appears solid, but the code was not shared to see how calculations were performed.
-
-
dl.acm.org dl.acm.org
-
Computational thinking is thinking recursively. Itis parallel processing. It is interpreting code as dataand data as code. It is type checking as the general-ization of dimensional analysis. It is recognizingboth the virtues and the dangers of aliasing, or giv-ing someone or something more than one name. Itis recognizing both the cost and power of indirectaddressing and procedure call. It is judging a pro-gram not just for correctness and efficiency but foraesthetics, and a system’s design for simplicity andelegance
I agree that it involves thinking recursively, parallel processing, and interpreting code as data and vice versa. It requires us to delve into the intricacies of type checking as a generalization of dimensional analysis and understanding the concept of aliasing, which can have both benefits and risks.
-
-
-
Reviewer #2 (Public Review):
Summary:
In this manuscript, L&S investigates the important general question of how humans achieve invariant behavior over stimuli belonging to one category given the widely varying input representation of those stimuli and more specifically, how they do that in arbitrary abstract domains. The authors start with the hypothesis that this is achieved by invariance transformations that observers use for interpreting different entries and furthermore, that these transformations in an arbitrary domain emerge with the help of the transformations (e. g. translation, rotation) within the spatial domain by using those as "scaffolding" during transformation learning. To provide the missing evidence for this hypothesis, L&S used behavioral category learning studies within and across the spatial, auditory and visual domains, where rotated and translated 4-element token sequences had to be learned to categorize and then the learned transformation had to applied in new feature dimensions within the given domain. Through single- and multiple-day supervised training and unsupervised tests, L&S demonstrated by standard computational analyses that in such setups, space and spatial transformations can, indeed, help with developing and using appropriate rotational mapping whereas the visual domain cannot fulfill such a scaffolding role.
Strengths:
The overall problem definition and the context of spatial mapping-driven solution to the problem is timely. The general design of testing the scaffolding effect across different domains is more advanced than any previous attempts clarifying the relevance of spatial coding to any other type of representational codes. Once the formulation of the general problem in a specific scientific framework is done, the following steps are clearly and logically defined and executed. The obtained results are well interpretable, and they could serve as a good steppingstone for deeper investigations. The analytical tools used for the interpretations are adequate. The paper is relatively clearly written.
Weaknesses:
Some additional effort to clarify the exact contribution of the paper, the link between analyses and the claims of the paper and its link to previous proposals would be necessary to better assess the significance of the results and the true nature of the proposed mechanism of abstract generalization.
(1) Insufficient conceptual setup: The original theoretical proposal (the Tolman-Eichenbaum-Machine, Whittington et al., Cell 2020) that L&S relate their work proposes that just as in the case of memory for spatial navigation, humans and animal create their flexible relational memory system of any abstract representation by a conjunction code that combines on the one hand, sensory representation and on the other hand, a general structural representation or relational transformation. The TEM also suggest that the structural representation could contain any graph-interpretable spatial relations, albeit in their demonstration 2D neighbor relations were used. The goal of L&S's paper is to provide behavioral evidence for this suggestion by showing that humans use representational codes that are invariant to relational transformations of non-spatial abstract stimuli and moreover, that humans obtain these invariances by developing invariance transformers with the help of available spatial transformers. To obtain such evidence, L&S use the rotational transformation. However, the actual procedure they used actually solved an alternative task: instead of interrogating how humans develop generalizations in abstract spaces, they demonstrated that if one defines rotation in an abstract feature space embedded in visual or auditory modality that is similar to the 2D space (i.e. has two independent dimensions that are clearly segregable and continuous), humans cannot learn to apply rotation of 4-piece temporal sequences in those spaces while they can do it in 2D space, and with co-associating a one-to-one mapping between locations in those feature spaces with locations in the 2D space an appropriate shaping mapping training will lead to successful application of rotation in the given task (and in some other feature spaces in the given domain). While this is an interesting and challenging demonstration, it does not shed light on how humans learn and generalize only that humans CAN do learning and generalization in this, highly constrained scenario. This result is a demonstration of how a stepwise learning regiment can make use of one structure for mapping a complex input into a desired output. The results neither clarify how generalizations would develop in abstract spaces nor the question if this generalization uses transformations developed in the abstract space. The specific training procedure ensures success in the presented experiments but the availability and feasibility of an equivalent procedure in natural setting is a crucial part of validating the original claim and that has not been done in the paper.
(2) Missing controls: The asymptotic performance in Exp 1 after training in the three tasks was quite different in the three tasks (intercepts 2.9, 1.9, 1.6 for spatial, visual and auditory, respectively; p. 5. para. 1, Fig 2BFJ). It seems that the statement "However, or main question was how participants would generalise learning to novel, rotated exemplars of the same concept." assumes that learning and generalization are independent. Wouldn't it be possible, though, that the level of generalization depends on the level of acquiring a good representation of the "concept" and after obtaining an adequate level of this knowledge, generalization would kick in without scaffolding? If so, a missing control is to equate the levels of asymptotic learning and see whether there is a significant difference in generalization. A related issue is that we have no information what kind of learning in the three different domains were performed, albeit we probably suspect that in space the 2D representation was dominant while in the auditory and visual domains not so much. Thus, a second missing piece of evidence is the model fitting results of the ⦰ condition that would show which way the original sequences were encoded (similar to Fig 2 CGK and DHL). If the reason for lower performance is not individual stimulus difficulty but the natural tendency to encode the given stimulus type by a combo of random + 1D strategy that would clarify that the result of the cross-training is, indeed, transferring the 2D-mapping strategy.
Tags
Annotators
URL
-
-
-
Converts Word doc to HTML code
doc to html
-
-
stackoverflow.com stackoverflow.com
-
The reason it works when using one statement is that local swallows the return type of the right hand side (e.g. local foo=$(false) actually returns the zero status code); that's one of bash's many pitfalls.
-
-
omnidatacleaningguide.netlify.app omnidatacleaningguide.netlify.app
-
omni_table
testing on a code chunk
-
-
-
Here are some of the main reasons to use multiple methods in your programs: Reducing Complexity: Divide a problem into subproblems to solve it a piece at a time. Reusing Code: Avoid repetition of code. Maintainability and Debugging: Smaller methods are easier to debug and understand.
Reasons to use methods
-
-
aclanthology.org aclanthology.org
-
We firstly pre-process dialog act A into a se-quence of control codes using the following format:A′ = [ I ( s1 = v1 , · · · sP = vP ) ] (4)Meanwhile, the output sequence x′ is pre-processed via appending x with a special start to-ken [BOS] and an end token [EOS].
Đầu tiên, hành động hội thoại A được tiền xử lý thành một chuỗi code với format như sau: A' = [I (s1 = v1, …, sp = vp)] Trong khi đó, chuỗi đầu ra x' được tiền xử lý bằng việc thêm vào x 2 token đặc biệt là [BOS] và [EOS].
-
-
www.biorxiv.org www.biorxiv.org
-
Note: This response was posted by the corresponding author to Review Commons. The content has not been altered except for formatting.
Learn more at Review Commons
Reply to the reviewers
Reviewer #1
In this paper, authors report that radiation, acidic pH, hypoxia, and drugs that interfere with lipid synthesis, all of which affect lipid droplets (LD), also affect the production of small extracellular vesicles (sEVs). In addition, they also report that LD content and sEV secretion are also modulated in CR-CSCs. Authors conclude that sEV formation and secretion is directly linked to LDs, and that their studies may open the way to new clinical perspectives. However, some important issues need to be addressed before the paper can be considered for publication.
My _main concern is that the notion that LDs and sEVs are linked remains vague. Do cells contain more LDs and secrete more sEVs because these two pathways are selectively up-regulated via some mechanism____s_ that controls both pathways in a concerted manner? Or do cells with more LDs and more sEVs also contain more of everything, perhaps as a result of metabolic activity? __
We appreciate the Reviewer's observations. Indeed, this comment represents the main pillar of the entire manuscript. We have attempted to uncover the molecular mechanism behind this novel and intriguing organelle connection. First of all, we have adapted the manuscript emphasizing that the LD – sEV connection might be direct or indirect. Our omic data suggested that some proteins belonging to the RAB family, mainly Rab18, Rab7a and Rab5c, could play a pivotal role in the LDs-sEVs axis. To strengthen those results, we have performed additional experiments by silencing the expression of the three candidate Rabs. Rab5c seems to be a good candidate to modulate the LD-sEV connection. We believe that Rab5c is not the only contributor to the LD-sEV connection but is part of a whole set of different elements that regulate this axis. However, it is quite challenging to rule out other molecular candidates as co-contributors to this phenomenon, especially when considering cellular metabolic pathways.
We recognize that external stimuli, such as radiation, pH, and lipid-interfering drugs, may exert their effects on other cellular organelles, even though we have strived to analyze each individual phenomenon rigorously. We are confident that our work lays the foundation for further research in the field.
__A direct corollary of this issue is whether increased sEV secretion reflects more endosomes and lysosomes (e.g. LysoTracker-positive compartments) or whether sEV secretion is selectively up-regulated. __
Thanks to the Reviewer’ suggestion, we have analyzed both the lysosome and endosome contents in our experimental cell systems. These data are now included in the manuscript in Figure S8. We have observed that it is unlikely that lysosomes are directly involved in the LD – sEV connection. However, the expression of Rab7a, a regulator of the late endosomal pathway, correlated with the LD content of the cells and their sEV release. Therefore, the endosomal pathway might be a good candidate to contribute to this LD – sEV connection.
__At one point, authors argue that cells that secrete more sEVs also contain more MVBs, but this issue remains elusive. To what extent is the increase in LDs and sEVS correlated in particular with an increase in endosome-lysosomes, and ER-Golgi (LDs originate from the ER)? __
We thank the Reviewer for this comment. We agree that the analyses of sEVs secreted in the media might not reflect the MVB content in the cells. However, two experiments, one on Panc01 cells and another one on MCF7 cells, showed that the number of MVBs, assessed by confocal microscopy using CD63 staining (MCF7) or CD63 and Alix plasmids (PANC-01), was directly correlated with the number of released sEVs in the media (Figure Fig S3C and 4J).
In addition, we included additional experiments assessing the lysosome content in HT29 LDHigh and LDLowcells. Hereby, we confirmed that HT29 LDHigh cells showed a higher LD content than HT29 LDLow cells. Inversely, by studying the lysotracker area per cell, we showed that HT29 LDLow population has a higher lysosomal content as compared to their counterpart, HT29 LDHigh cells (test = Wilcoxon rank sum test with continuity correction_ W = 85127, p-value = 7.255e-07 for LDs and W = 49321, p-value = 1.14e-11 for Lysotracker). However, we could not demonstrate a clear correlation between the number of LDs in the cell and the lysotracker signal.
Finally, we have also studied the expression of GM130, a Golgi-shaping protein (Ref. 1) and Rab7, a late-endocytic protein (Fig S8C). While the expression of Rab7 (endosome) seemed to correlate with the LD and sEV contents, the expression of GM130 (Golgi) gave back no coherent results. Indeed, it was inversely correlated to the LD and sEV amount, in accordance with what was already reported elsewhere (Ref 2 and 3)
- Nakamura N. Emerging new roles of GM130, a cis-Golgi matrix protein, in higher order cell functions. J Pharmacol Sci. (2010) 112:255–64. Doi: 10.1254/jphs.09R03CR
- Lydia-Ann L.S. Harris, James R. Skinner, Trevor M. Shew, Nada A. Abumrad, Nathan E. Wolins. Monoacylglycerol disrupts Golgi structure and perilipin 2 association with lipid droplets.Doi.org/10.1101/2021.07.09.451829
- Alvin Kamili, Nuruliza Roslan, Sarah Frost, Laurence C. Cantrill, Dongwei Wang, Austin Della-Franca, Robert K. Bright, Guy E. Groblewski, Beate K. Straub, Andrew J. Hoy, Yuyan Chen, Jennifer A. Byrne; TPD52 expression increases neutral lipid storage within cultured cells. J Cell Sci 1 September 2015; 128 (17): 3223–3238. Doi: 10.1242/jcs.167692
Authors conclude that the data with lipid inhibitors strengthen the connection between LDs and sEVs (Fig 2 and S2). However, is this regulation selective, or does it merely reflect the general effect of these inhibitors on membrane-related processes? The same comment applies to the role of iron metabolism after knockdown of ferritin heavy chain (Fig 3 and S3), acidic pH and X-ray radiation (Fig 4 and S4)____.
We thank the Reviewer for the interesting observation. As previously mentioned, we cannot rule out other potential contributors to the LDs-sEVs connection upon lipid inhibitor treatments and/or the others external stimuli applied to our cell systems.
The data presented in this manuscript merely represent a novel and unexplored (at least so far) organelle connection, direct or indirect, with a broad clinical implication. As the membrane-related processes (such as Endosomes, Golgi apparatus, Exosome (sEV) pathway, Lysosomes and Autophagosome) are all interconnected, in our opinion, it might be quite challenging to make such a definitive statement.
Such assertion would require extensive further investigation to relate each organelle to the LDs and/or sEVs. However, with our research, we hope to open the door to a new era of investigations regarding the sEV – LDs connection.
OTHER COMMENTS
1) Which cell line is used for sEV analysis (markers vs contaminants (Fig S1B)? In any case, the data should be shown for both cell types.
Our method to isolate sEVs is a standardized method that was already published by our group and collaborators in 2020 (M. Bordas, et al., Optimized Protocol for Isolation of Small Extracellular Vesicles from Human and Murine Lymphoid Tissues. Int J Mol Sci (2020) https:/doi.org/10.3390/ijms21155586.). This protocol was validated on human and mouse tissues, much more complex samples than cell culture supernatant.
Figure S1C was modified, as requested by the Reviewer, including new data for HT29, Panc01 and MCF7 cell lines to broaden the panel. Those results confirmed the good purity of sEV samples isolated from cell culture supernatant.
2) The Tsg101 blot is not impressive (Fig S1B): the difference between cells and sEVs is not easy to see. It would be nice if blots were quantified.
Indeed, the signal obtained for TSG101 for sEVs derived from Panc01 cell line is quite weak. It is important to remember that not all sEV markers are highly expressed in all cell lines and their derived sEVs. Some cell line-derived sEVs show a low or high expression of the diverse sEV markers. To answer the Reviewer #1’s comment, we quantified the expression of TSG101 in Panc01-derived sEVs. The quantification showed that TSG101 is 6.8 times more expressed on Panc01-dervied sEVs as compared to the cell line. However, since the expression is quite low, this quantification should be taken with some caution.
In light of the Reviewer ‘comment, we have performed the Western Blot analysis on other cell lines (HT29 and MCF7), and we have replaced TSG101 marker with CD9 marker (Figure S1C).
3) From Fig 1B it cannot be concluded that the size of sEVs ranges from 30 to 200nm: the micrograph only shows a few structures.
We appreciate the Reviewer's comment and have attempted to provide more clarity. Firstly, we want to highlight that TEM micrographs of sEVs typically show the donut shape, a unique feature of sEVs imaged with TEM, as well as a size range. In Figure 1B micrograph, the sEV size is approximately 100 nm. The size distribution of LoVo and HT29-derived sEVs can be observed from the NTA size measurements in Figure S1B. Indeed, the peak size is 148 nm for LoVo-derived sEVs and 135 nm for HT29, which aligns with the sEV sizes presented in Figure 1B. We have also included multiple micrographs here under. As the number of Supplementary Figures is already large, we have decided to not include those micrographs in the manuscript. The average size of LoVo-derived sEVs, based on TEM micrograph analysis, was 94 ± 41.10 nm, while the average size of HT29-derived sEVs was 76.41 ± 44.22 nm. The size discrepancy between the two methods (NTA versus TEM) can be ascribed to the dehydration step required for TEM, which results in a reduction of the actual sEV size.
4) HT29 cells contain far more LDs than LoVo cells (Fig 1A). Similarly, sEV proteins (CD63, CD81, CD9, Hsc-70) are more abundant in HT29 sEV____s____ than in LoVo sEVs (Fig 1D). However, the sEV preparation from HT29 cells contains only approx. 50% more total protein than LoVo sEVs (Fig S1D-E). Are sEVs prepared from LoVo cells far more contaminated with cell debris etc.. than sEV fractions from HT29 cells?
We are confident that our EV isolation method allows us to achieve high yield and excellent purity. It is possible that a lower number of sEVs in samples may lead to increased protein contamination during ultracentrifugation. However, size exclusion chromatography should minimize this protein contamination. It is important to note that the NTA method is significantly more sensitive and accurate than Qubit protein quantification. Consequently, protein concentration and particle concentration should not be directly compared.
5) LD staining should be shown for the corresponding populations of cells with high/low CD63 (Fig 1E). Cells in culture can be somewhat heterogeneous, but the difference between low and high CD63 is quite extreme (Fig 1E). Is such high heterogeneity also observed with other proteins of the endocytic and biosynthetic pathways? Authors conclude that cells containing high CD63 levels also contain more MVBs (Fig 1E): are all late endosomal proteins (e.g. LAMP1, RAB7) upregulated in cells with high CD63?
We thank the Reviewer for this comment, and we totally agree with the Reviewer that it would be better to have the LD and CD63 staining on the same images. Unfortunately, the staining for CD63 on LD540-sorted HT29 cells requires a permeabilization step that interferes with the cellular lipid part and could therefore negatively affect the LD imaging by confocal microscopy. To prove that the HT29 LDHigh and HT29 LDLowcontain high and low LD amount respectively, we sorted HT29 cells based on the LD content and, soon after, we observed them at the confocal microscopy. We thus added new images in Figure S1F, corresponding to the LD fluorescence detection. The readers will also appreciate the explanation regarding the inability of observing both LDs and CD63 staining on the same confocal images under the line 165 – 166:
“As the staining for CD63 required a permeabilization step, and therefore lipid digestion, it was not possible to assess both LDs and CD+MVBs on the same micrographs “.
In addition, we have added confocal images representing HT29 cells sorted based on their LD content and stained with Hoechst and Lysotracker. A quantification of the Lysotracker fluorescence per cell and the correlation with the number of LDs can also be appreciated in Figure S8A-B.
Finally, we performed Western Blot analysis to examine Rab7a expression under various conditions described in our manuscript (Figure S8C). In general, Rab7 expression corresponded with LD content, indicating that cells with high LD content exhibited higher Rab7 expression, while cells with low LD amount showed lower Rab7 expression, except for Triacsin-C. The Reviewer can now appreciate the quantification in the graphs provided below (not included in the manuscript).
Regarding the heterogeneity of LDs, CD63+MVBs, or lysotracker among the cell population, we have indeed noticed heterogeneity observable in these three types of staining in HT29, particularly in the HT29 LDHighpopulation.
6) Inhibitors of lipid synthesis reduce LD formation (Fig 2B), sEV production and CD63 / CD81/ CD9 secretion (Fig2C-D, Fig S2B). Are the cellular levels of these (and other endosomal) proteins also reduced after inhibitor treatment? Does the stimulation of LD formation with oleic acid also stimulate CD63 synthesis and sEV production?
We thank the Reviewer for this very interesting comment. To answer this question, we have added a supplementary figure (Figure S2A, S2B) showing the cellular expression of CD63 upon LD inhibition or stimulation.
During the planning of our experiments, we discussed about the possibility of using oleic acid to induce the formation of Lipid Droplets, which was ultimately not done. This is because the use of oleic acid would have more strongly stimulated the triglyceride pathway, as extensively discussed elsewhere (Mejhert N. et al., The lipid droplet knowledge portal: a resource for systematic analyses of lipid droplet biology, Developmental Cell, 2022). Since Lipid Droplets are made by cholesterol esters and triglycerides, we preferred to use other stimuli (hypoxia, radiation), all of them already discussed in literature, to induce both pathways simultaneously, resulting in the Lipid Droplet formation/induction.
7) It seems that pH and irradiation increase sEV markers far more significantly (Fig 4 B-C and Fig S4A-E) than FTH1 depletion decreases sEV markers (Fig 3 D-E). In fact, authors mention that they cannot exclude a contamination of sEVs with small apoptotic / autophagic vesicles after irradiation (Fig 4). To facilitate comparison, it would be nice to also show the number of secreted particles per cell (like after FTH1 depletion Fig 3D), as well as the distribution of possible contaminants (e.g. Fig S1). Also, authors state that the increase in the number CD63+ MVBs after irradiation is shown, but this is not the case.
We apologize to the Reviewer because, in fact, one figure was missing (Figure 4). We have rectified this by increasing the quality of Figure 4 and have added representative images for each acquisition of the number of MVBs, either positive for CD63 or Alix, in transfected Panc01 cells X-ray irradiated (8 Gy) or not (0Gy). In addition, a similar experiment was performed in MCF7 cells transduced with shRNA or shFTH1. CD63+ MVBs were assessed in both cell line and the number of CD63+ puncta (MVBs) were quantified by ImageJ. The results, although not significative, illustrated a trend for MCF7 shFTH1 to contain less CD63+ MVBs than MCF7 shRNA. Furthermore, the quantification of sEVs released in the conditioned media was performed in three independent experiments and demonstrated that significantly less particles (sEVs) were released by MCF7 shFTH1 than MCF7 shRNA.
8) Are the proteomic data (Fig 6) with LDlow and LDhigh cells obtained after cell sorting, as in Fig 1E? Did authors compare the proteome of LoVo and HT29 sEVs? How do the protein profiles (in particular proteins involved in lipid metabolism) obtained under different conditions compare with each other, in particular after irradiation (Fig4N) and knockdown of ferritin heavy chain (Fig 3, Fig S3)? It would also be interesting to compare these data with the data obtained in CR-CSCs culture under hypoxia (Fig S5). Are common proteins involved in sEV production and LD biosynthesis identified in the analysis of these biological processes? Is there a common set of proteins/genes revealed by this analysis, which may potentially control sEV production and LD biosynthesis?
We thank the reviewer for this interesting comment.
Proteomic analyses have been performed on the following conditions:
- Panc01 (0 Gy – 6 Gy – 8 Gy) for sEV samples
- MCF7 (shFTH1 and MCF7 shRNA)
- MCF7 (0 Gy and 6 Gy)
- MCF7 (Normoxia and Hypoxia)
- H460 (0 Gy and 6 Gy)
-
H460 (Normoxia and Hypoxia) RNA sequencing was performed on the following conditions:
-
CR-CSCs (#4, #8, #21) Based on all those data, we have analyzed the sEV pathway and how this pathway was modulated in the conditions with high LD content and low LD content. We therefore came up with several proteins, presented in Figure S7. Based on this analysis, we have decided to further investigate the role of RAB18, RAB5c and RAB7a in the connection between LDs and sEVs. Those additional results can be found in Figure 6 and Figure S7A (originally Figure 6). We have found that RAB5c, but not RAB7a or RAB18, seems to be a good candidate to intervene in the LD – sEV connection.
Minor comments
1) Some parts of the text are still a bit rough, and should be read and corrected carefully. For example: i) isn't it obvious that a common source of lipids builds up the membrane of sEVS, much like any other membrane (line 90, p.2); ii) what does this sentence mean: "LD have been considered as mere fat storage organelles for a long time, although important evidence could be traced back to the early 1960's". Important evidence for what? iii) why is the acronym AdExo used? iv) (line 138) the text should probably be "sEVs released during 72h were studied" and not "released sEVs were studied ... 72 h after seeding".
We apologize to the Reviewer if some parts of the paper were a bit rough. We have re-read the entire manuscript and corrected all the parts that needed revision work.
2) The captions are far too small in most figures and diagrams (for example ____X____ and Y axis in Fig 1C-D, text in Fig 1E; Fig S1; Fig 3C proteins in the heatmap).
We agree with the Reviewer. All images and their captions were properly revised.
3) The color code for LoVO and HT29 cells is reversed in Fig S1D-E
The mistake was corrected.
4) In Fig 1D, I cannot see CD81 in the LoVo blot.
In the image below, it is possible to see the LoVo blot.
5) Wording is not adequate in following sentences: "62.7% of proteins related to the exosomal pathway are downregulated in MCF7 shFTH1 cells" (line 233) and a few lines below: ".. the expression of almost all exosomal markers was downregulated in MCF7 shFTH1 cells" (line 239). Does 62.7% represent all proteins?
We apologize to the reviewer for the mistake. We rephrased this sentence.
6) In Fig 3E authors compare sEV markers secreted by cells treated with shFTH1 or control shRNA. The Anx5 and CD63 blots are not very convincing (quantification would be helpful).
We apologize to the Reviewer for this issue. These Western Blot analyses were performed only once, therefore a quantification in the manuscript would not be relevant. However, we report here the results of the quantification. The expression of Annexin V was 1.58 times higher in MCF7 shRNA than MCF7 shFTH1, while the expression of CD63 was 1.34 time higher in MCF shRNA as compared to MCF7 shFTH1.
7) The micrographs in Fig 4L are too small: gold particles cannot be seen, even in the high magnification views.
We thank the Reviewer for her/his comment. We have moved the micrograph and the quantification histogram to the Figure S6. Now, it is possible to discriminate easily gold nanoparticles.
8) The micrographs showing ALIX and CD63 (Fig 4J) in irradiated and unirradiated Panc01 cells should be shown for comparison.
We followed the Reviewer’ suggestion as it is possible to note in the Figure below.
Reviewer #2
This manuscript describes a relationship between lipid droplet presence in cells and small EV secretion. First, correlations are done between number of lipid droplets and numbers of EVs secreted. Then chemical inhibitors of lipid droplet biosynthesis pathways were shown to reduce small EV secretion. Then various processes known to target lipid droplets, including iron metabolism, irradiation, hypoxia, low pH are used to show concordant effects on lipid droplets and small EV secretion. Proteomic analysis of EVs and cells subjected to some of the treatments are also performed. Overall, it is an interesting line of investigation and the data overall seem solid. Several flaws exist, which can probably be fixed. These include the use of different cell lines for different experiments. It makes it a bit difficult to connect everything together. It could be fixed by adding some extra cell lines to some experiments - for example taking the MCF7 and Panc-01 cells for which proteomics was performed and redoing some of the correlative and causative experiments from Figs 1 and 2.
We appreciate the Reviewer's insightful observation. Following her/his suggestion, we have conducted additional experiments on MCF7, H460 and PANC-01 cell lines to enhance data consistency and facilitate a smoother transition between different sections of the paper.
It also would be good to have some more direct evidence of the connection between lipid droplets and EV secretion - one could argue that this was already done in Fig 2 with the chemical inhibitors, I wonder if there is a genetic way to do it too?
We totally agree with the Reviewer. Indeed, starting from our proteomic data we highlighted some genes belonging to the RAB family as potential candidates to interfere with the LD – sEV connection. The Reviewer can now appreciate in Figure 6 and Figure S7, the results from the additional experiments we carried out on RAB5c, RAB7a and RAB18 silencing in HT29 cells. The former Figure 6 has been moved in the Supplementary part (Figure S7).
Some tightening up of the writing (especially the Discussion) and the resolution of the figures would also improve the manuscript.
We apologize to the Reviewer for this issue. We have now re-prepared all Figures by increasing their resolution, as well as reviewing the entire manuscript with the aim of making the reading smoother and simpler.
__Overall, it is a nice piece of work but there are many minor things to be fixed. __
__Specific Comments: __
The sentence in the Introduction: "The non-endosomal pathway generates sEVs devoid of CD63, CD81 and CD9 or sEVs enriched in ECM and serum-derived factors (7)." is not well-supported and should be removed. The idea that you can classify membrane of origin based on markers has not been proven, but rather assumed.
We agree with the Reviewer. We have rephrased the sentence.
We thank the Reviewer for this comment. In response to this, we have generated correlation graphs for several of our experiments:
- HT29 (CTL – Triacsin-C - PF-06424439) in Figure 2E
- PANC-01 (CTL – 2 – 4 – 6 – 8 Gy) in Figure 4K
- CR-CSCs (#4, #8, #21) in Figure 5E
__The Method used for EV purification should be stated in the Results rather than referring to a reference and a Supplemental Figure (S1A) that is too low of a resolution to see. __
Our method to isolate sEVs is a standardized methods that was already published by our group and collaborators in 2020 (M. Bordas, et al., Optimized Protocol for Isolation of Small Extracellular Vesicles from Human and Murine Lymphoid Tissues. Int J Mol Sci (2020) https:/doi.org/10.3390/ijms21155586.). This protocol was validated on human and mouse tissues, much more complex samples than cell culture supernatant.
In regard to the Reviewer’s comment, we have added a better description of the protocol in the Results part, referring to the Material and Method. For this reason, we decided to keep the sEV protocol in the SI section. We apologize for the low quality of the Figure S1. In agreement with the Reviewer suggestion, we have modified the image by increasing its quality.
__Fig 1B would be better to have an image in which the EVs are not aggregated. __
We thank the Reviewer for this comment and have modified the Figure accordingly.
__Fig 3 is interesting but jumps cell lines. For better continuity, some of the experiments from Figs 1 and 2 should be repeated in the MCF7 cells to connect with the proteomics. __
In agreement with the Reviewer’ comment, we decided to perform additional experiment on MCF7, using Triacsin-C. The Reviewer can now appreciate the results in Figure 2F, Figure 2G and Figure S2E.
__Fig 3C is too low resolution to read, please export at higher resolution. __
We are sorry for the low-quality Figure. We have modified the image accordingly.
__Please provide all the raw proteomics data as a supplementary spreadsheet____. __
We have provided all the raw data regarding our proteomic analyses.
__Fig 4 panels are low resolution __
We apologize for the low-resolution Figure. We have modified the figure by increasing the quality.
Fig 4 again adds new cell lines with H460 and Panc-01
We thank the reviewer for this comment. In this regard, we have performed additional experiment:
- Western Blot: comparison cellular and exosomal markers (Figure S1C)
- MCF7 (CTL - Triacsin) (Figure 2F, Figure 2G and Figure S2E)
- Western Blot: analysis of RAB7a, GM130
__The images corresponding to 4J should be shown in a Supp Figure somewhere __
We thank the reviewer for pointing out this oversight. We have added the confocal images corresponding to the Figure 4J below the quantification.
The statements: "In addition, the exosomal nature of Panc01-derived vesicles was demonstrated by an analysis of CD63+ or Alix+ multivesicular bodies (MVBs) in unirradiated (0 Gy) or irradiated (8 Gy) pancreatic cancer cells (Fig 4J). Moreover, we confirmed a clear correlation between cellular LD content and sEV biogenesis, as represented in Fig 4K." are overly conclusive. For 4J, one can make a statement about the MVBs but not the EVs as that's not what was measured there. Likewise for 4K, what was measured was how many EVs were released not how many were formed. While the data are suggestive of alteration of exosome biogenesis, they are not conclusive.
We agree with the reviewer and have performed the necessary changes in the manuscript. The reviewer can see the changes under the lines 282 – 284:
“In addition, the analysis of CD63+ or Alix+ multivesicular bodies (MVBs) in unirradiated (0 Gy) or irradiated (8 Gy) pancreatic cancer cells revealed an increased number of MVBs after irradiation (Fig____ure 4J).”
__Western blot is always capitalized by convention - Western not western. __
We have corrected it accordingly.
__Fig 5A is too small and low resolution - suggest eliminating and just put info in methods. __
We are sorry for the low-resolution image. We have followed the Reviewer suggestion. The graphical method has been now moved to the Supplementary Figure S6.
Fig 5G, many of the genes shown are frequently EV cargoes but most not involved in exosome biogenesis - not sure where the label of Exosome pathway came from but it is not very compelling. Only ANXA2, Arf6, and Rab5C seem related and they are barely elevated.
We completely agree with the Reviewer's comment. As a result, we have revised the heatmap title to "Exosomal Cargoes and Pathways" instead of "Exosomal Pathway".
__Most main figures and all supplementary figures are extremely low res - please fix. __
We are very sorry for the low-quality figures. We have revised all Figures (main text and SI) by increasing their quality.
__Fig 6 is first mentioned in the Discussion - it should be described in the Results before that (or alternatively removed). __
We agree with the Reviewer. Our initial idea was to mention perspectives of analyses that could be carried ulteriorly. Nevertheless, we have performed additional experiments in order to get insight on the mechanism involved in the LD – sEV connection. Indeed, based on our proteomic data, we have analyzed the sEV pathway and how this pathway was modulated in the conditions with high LD content and low LD content. We therefore came up with several proteins, presented in Figure S7A (originally Figure 6). Based on this analysis, we have decided to further investigate the role of RAB18, RAB5c and RAB7a in the connection between LDs and sEVs. Those additional results can be found in Figure 6 and Figure S7 in the Results section. We have found that RAB5c, but not RAB7a or RAB18, seems to be a good candidate to intervene in the LD – sEV connection.
Table S1, also first mentioned in the Discussion, is missing. Either describe in the Results section or remove the callout to it.
Our apologies for that. The Table S1 has been now mentioned in the Results section and has been properly uploaded.
__The discussion is too dense with too many trains of thought, often many different directions in the same paragraph. It needs to be streamlined, with a central thought for each paragraph and good transitions between the paragraphs. __
We apologize to the Reviewer if the Discussion part was a bit confusing. We rewrote the paragraph, streamlining it and making the transitions between its paragraphs smoother.
Reviewer #2 (Significance (Required)):
__ Strengths of this manuscript are the interesting connection between lipid droplets and exosomes and the number of experiments to address it. __
__ Limitations: use of different cell lines for different figures, overall descriptive nature with regard to direct demonstration of connection to lipid droplets -- it's kind of done in Fig 2, but could be possibly bolstered. __
-
-
www.biorxiv.org www.biorxiv.org
-
Note: This response was posted by the corresponding author to Review Commons. The content has not been altered except for formatting.
Learn more at Review Commons
Reply to the reviewers
Reviewer #1
Evidence, reproducibility and clarity
In this manuscript, Hoskins et al describe analyses of the effects of sequence variation on RNA levels, protein levels, and ribosome loading for the COMT gene. They use multiple experimental approaches to assay these levels and report on how sequence differences affect expression. Overall, the paper is interesting in that it presents a very deep dive into the effects of sequence variation on gene expression, including in coding sequences. However, there are some issues with the polysome loading assay technique and there are substantial issues with the figure presentation, which is often confusing.
__Response: __Thanks for the positive assessment of our manuscript and the constructive feedback regarding the issues with the figure presentation. We have addressed all of these below and they have significantly improved the clarity.
-
Major comments:*
-
1) Figures:*
-
--Fig 1C needs a cartoon description to show where the UTRs are. Y-axis should say "Ribo-seq CPM"*
__Response: __Fig 1C now includes a schematic and the y-axis is updated. Locations of the uORFs are also now included in Fig 1A.
- --Sup Fig 1A confusing, what is "start" what is the point of this panel?*
__Response: __We apologize for the confusing labeling of the panels in Sup Fig 1. “Start” refers to the MB-COMT start codon. We removed this annotation as it is irrelevant to the figure. We included Supplementary Figure 1A to show RNA probing data for the entire transcript. Figure 1A and B only show the regions that encompass the variants assayed in our study.
- --Sup Fig 1B what is PCBP del?*
Response: “PCBP del” refers to deletion of PCBP1/PCBP2 RNA binding protein motifs. The legend now specifies this.
- --Sup Fig 1C what is "uORF B restore"? The description in the figure legend is not interpretable. Draw diagrams of the mutations that tell the reader what was assayed and why it was assayed. Why are there multiplication factors listed (e.g. 1.33X)? The data are depicted on a log scale, which makes it difficult to appreciate the fold-effects of the mutations (e.g. does uORFA mutation increase expression 1.5-fold?). Please calculate median expression values and report them on a bar graph or something like that so readers can interpret the results.*
Response: “uORF B restore” refers to restoration of the endogenous uORF B frame with a silent variant in the Flag tag of the transgene. The multiplication factors listed were the fold change in median fluorescence between each mutant and the template (wild-type) transgene. We retained the figures as they show the raw distribution of fluorescence in each cell line, but in response to the reviewer’s suggestion we included a new figure displaying the effects as a bar graph (Supplementary Figure 1E).
- --Fig 2A. It's hard to understand the cartoon diagram of the expression reporter construct. Why is +Dox shown here? Does that induce transcription?*
__Response: __The reviewer is correct. “+Dox” indicated addition of Doxycycline to induce transcription before the data collection step. We agree that there may have been too much detail in this diagram and have now removed this for simplicity and indicated this in the Methods section.
- --Fig 2B. What's on the x-axis? is it Log2(RNA/gDNA) from sequencing? is it Log2 or Log10 or Ln?*
__Response: __Variant effects in each figure were derived from ALDEx2 analysis, which reports effect size as the median standardized difference between groups. The effect size is not directly interpretable as a log fold change; it takes into account the difference between groups as well as the dispersion. This analysis strategy has been previously demonstrated for analysis of SELEX experiments (Fernandes et al. 2014), which are used to select small populations of cells with specific phenotypes.
ALDEx2 is a robust and principled choice for the analysis of count-compositional datasets, particularly after selection (e.g. sorted cell populations or low-input RNA fractions arising from polysome profiling). While we understand that this choice leads to less easily interpretable effect sizes, the mathematical advantages make ALDEx2 a more appropriate choice for this type of data. In the past, we had used other methods to analyze log frequencies (limma, a frequency based normalization-dependent analysis, as previously employed in Hoskins et al. 2023. Genome Biology) that directly reported fold changes. In our experience, the ALDEx2-derived effect sizes are well-correlated with those estimates (Pearson correlation 0.93 for variants significant at a FDR
- --Fig 2C. What's on the y-axis (same question). I think it's LogX(mutant/wt)RNA level?*
__Response: __For consistency with other figures, we replaced Figure 2C to report the effect size statistic as described above.
- --Fig 2D. What's on the y-axis now? Fold-difference (not log transformed)?*
__Response: __Please see our response above.
- --Fig 2E. The scale bar is flipped vs. normal convention. This is also log transformed, but it's not labeled. Please label as log(whatever) and put the negative values on the left side of the bar (red on the left, blue on the right).*
__Response: __Thanks for the suggestion, we have now updated the scale bar.
--Fig 2F y-axis should say Ribo-seq CPM.
__Response: __Done
- --Fig 3A - please separate the graphs more. Did you sort cells from ROI2 into populations, or just cells from ROI1?*
__Response: __Thanks for the suggestion, we now separate the graphs further. Cells were sorted for both ROI 1 and ROI 2 libraries.
- --Fig3C-F What's the "effect size" mean on these graphs?*
__Response: __Please see the response above regarding the effect size estimate from ALDEx2.
-
--Fig3D It looks like the colors have switched for positive / negative "effects" on the heat map*
-
compared to Figure 2E. Please define what "median effect" means and be consistent with*
-
comparison to figure 2E.*
__Response: __We intentionally inverted colors for Figure 3. The rationale is that a variant causing low protein abundance corresponds to enrichment in P3 compared to gDNA, as opposed to depletion in P3. On the other hand, for effects on RNA abundance and ribosome load, a variant leading to low abundance for these measures is depleted.
- --Figure 4 what does effect size mean, what's the log-transformed scale (log2, 10, etc) same issues from earlier figures.*
__Response: __Please see response above.
- --Figure 5 "effect size"*
__Response: __The same definition of effect size was used with the exception that effect sizes are multiplied by -1 so that color schemes are consistent for deleterious effects.
- 2) "Codon stability" should always be "Codon Stability Coefficient", maybe use "CSC". Otherwise it's confusing.*
__Response: __Thanks for the suggestion. This has been updated throughout the manuscript.
3) Flow cytometry section talks about "RNA fluorescence", which is confusing. You need to explain that it's IRES-driven mCherry as a proxy for the level of RNA first. It would also help to state explicitly that you sorted the cells into four populations, and define them all first before describing the results.
__Response: __We apologize for the use of imprecise language with respect to this reporter. We revised the text to emphasize that mCherry is a proxy for RNA abundance and described the populations first as suggested.
4) What are DeMask scores? How are they related to conservation or amino acid properties? If you define these, you can help the reader interpret the result.
__Response: __Thanks for the suggestion. We now include a conceptual interpretation of the DeMask score in the relevant section. We also include a comparison to a recent large language model for variant effect prediction (ESM1b, Brandes et al. 2023) which is now reported in Supplementary Figure 5C.
5) There are several issues with the Polysome gradient fractionation. The gradients did not separate 40S, 60S, and monosomal fractions, so it's hard to tell how many ribosomes correspond to each peak on the gradient graph in Figure S5. This is probably because the authors used a 20-50% gradient instead of a lower percentage on top. More significantly, variations in the coding region of COMT are likely affecting the polysome association in ways the authors didn't consider. Nonsense codons will simply make the orf a lot shorter, hence fewer ribosomes. This may have nothing to do with NMD. Silent and missense variants may have unpredictable effects because they may make translation faster (fewer ribosomes) or slower (more ribosomes) on the reporter. This could lead to more ribosomes with less protein or fewer ribosomes with more protein. The reporter RNA also has an IRES loading mCherry on it, which probably helps blunt or dampen the effects of the COMT sequence variants on polysome location distribution. Overall, the design of the polysome assay is probably very limited in power to detect changes in ribosome loading (four fractions, limited separation by 20-50 gradient, IRES loading, etc). This is partially addressed in the limitations section, but these issues could be discussed in more detail.
__Response: __Given high polysomal association of endogenous COMT and our COMT transgene (Supplementary Figure 2B, Supplementary Figure 5B-C), we chose a 20-50% sucrose gradient to better resolve changes in ribosome load among heavy polysomes.
We thank the reviewer for offering another valid explanation regarding the depletion of nonsensense variants. We have now included a sentence in the discussion to indicate lower ribosome load for nonsense variants may be due to a shorter ORF as opposed to NMD. We further include the potential limitation of the assay due to the presence of the IRES-mCherry.
We agree that variants may have unpredictable effects due to effects on the dynamics of translation elongation. To address this potential limitation, we attempted to devise a selective ribosome profiling strategy by immunoprecipitating N-terminal Flag tagged peptides to enrich ribosomes translating COMT. However, we were unable to achieve significant enrichment, limiting our ability to measure variant effects on elongation in a high-throughput manner.
Significance
The study is novel in that it assays both 5' UTR and a wide range of protein coding sequence variants for effects on RNA and protein levels from a clinically important gene, COMT. The manuscript reports that most protein coding variants have modest effects on RNA levels, and that the minority of variants that do affect RNA levels are not predictable due to their affect on codon usage. The work also determines the distribution of effects of variants on protein levels, finding a variety of effects on expression. Interestingly, the authors found SNPs that affect ribosome loading generally affect RNA structure of the COMT coding region, rather than affecting codon usage.
This should appeal to many different communities of biologists - gene expression experts, geneticists, and clinical neurobiologists who focus on COMT. So there is a potential for fairly broad interest. The main limitations to the work are in a lack of clarity in the figures and perhaps in the underdeveloped nature of the discussion section. The discussion section reports new results (SNP associations that affect expression). These would make more sense in the results section, such that the discussion could do a better job relating the impact of sequence variants on expression levels to prior work to highlight the novelty.
__Response: __We thank reviewer #1 for their positive assessment of the broad significance of our study. We also thank them for constructive suggestions that led to increased clarity in the presentation. We have moved the analysis of gnomAD variants to the Results section and expanded the discussion.
Reviewer #2
Evidence, reproducibility and clarity
Summary:
Hoskins and colleagues expressed a reporter containing all silent, missense, and nonsense codons at 58 amino acid positions in the human COMT gene in HEK293T cells and measured levels of DNA, bulk RNA, and pooled polysomal mRNA. They included a C-terminal translational GFP fusion and a downstream transcriptional mCherry fusion in the reporter in order to also bin variants by their relative protein and mRNA levels by flow cytometry. They hypothesized that RNA structure, in-part by mediating uORF translation, influences COMT gene expression. The authors conclude by identifying previously-uncharacterized COMT variants that, in this reporter system, affect RNA abundance and ribosome load. We generally found the results of this paper convincing and clear. We do not have major comments, but have many minor comments that we hope the authors can address. These comments mostly deal with clarification on analysis metrics and giving recommendations on data presentation.
__Response: __Thanks for highlighting the strengths of our study and the constructive suggestions to improve the presentation.
Minor comments:
In Figure 2C, the vertical axis reads "Median between-group difference". How was this metric calculated and normalized? We also agree that nonsense mutations having consistently-detrimental effects on RNA abundance is reassuring, but recommend more explanation as to why the difference in the effects of silence and missense mutations between regions may be biologically relevant.
__Response: __Variant effects in each figure derive from ALDEx2 analysis, which reports effect size as the median standardized difference between groups. In particular, to avoid any distributional assumptions for standardization, ALDEx2 uses a permutation based non-parametric estimate of dispersion. The effect size is not directly interpretable as a log fold change; it takes into account the difference between groups as well as the max dispersion of the groups. We have now provided explicit references to the specific R functions that were used to calculate the effect size.
ALDEx2 is robust for analysis of count-compositional datasets, particularly after selection and bottlenecking (e.g. sorted cell populations or low-input RNA fractions arising from polysome profiling). While we have used other methods to analyze log frequencies (limma, a frequency based normalization-dependent analysis, as previously employed in Hoskins et al. 2023. Genome Biology), we opted for the less-interpretable but more robust ALDEx2 analysis to report variant effects between varying nucleic acid inputs.
We currently lack a mechanistic interpretation for the difference in RNA abundance effects between ROI 1 and 2. However, we observed consistent results using a different analysis framework, which makes use of variant frequencies (as in Hoskins et al. 2023 Genome Biology) instead of the centered log ratios used in ALDEx2 analysis, further supporting a biological difference between the two.
In Figure 3, we believe that the authors are claiming that lower RNA abundance causes lower protein abundance in some variants. However, this data only reports on protein abundance relative to transcript abundance, not absolute protein abundance. We think the claim should be revised to (1) clarify that the authors are measuring protein per mRNA, and (2) express that lower mRNA amounts are more likely to co-occur with lower protein amounts, but that this data does not support any causative model.
__Response: __Thanks for the suggestion. We have now included an explicit description of the experimental design in the results section and noted that we are unable to assign protein abundance effects to underlying RNA abundance effects. In the current setup, we did not sort cells based on the ratio of moxGFP/mCherry fluorescence (protein per mRNA), but rather we defined gates based on the 2D plot of moxGFP versus mCherry. This is explicitly marked in Figure 3A.
On page 9, the authors claim that their data supports a model that rs4633 increases RNA
abundance, leading to higher COMT expression. Can the authors rule out a model whereby rs4633 facilitates translation initiation, as suggested by Tsao et al. 2011, leading to both an increase in mRNA and protein abundance?
__Response: __Thanks for this question and opportunity to clarify. We have now added a sentence to the Discussion and included the following paragraph in the Supplementary Note:
“Importantly, our study does not rule out a model where rs4633 facilitates translation initiation. Nevertheless, our data suggest a potential concurrent mechanism where rs4633 leads to higher protein abundance in human cell lines and in an in vitro translation assay (Tsao et al. 2011) by increasing RNA abundance. We note that Tsao et al did not directly measure RNA abundance in their study. In Supplementary Figure 3A of Nackley et al 2006, the APS haplotype containing rs4633 C>T showed slightly higher total RNA abundance compared to the LPS haplotype (in our study, the wild-type template). However, this was not statistically significant and was only observed for the S-COMT isoform. It is possible that our observations are compatible with the conclusions in Tsao et al. 2011. For example, increased translation of rs4633 C>T may lead to stabilization of the RNA.”
The paper references "effect size" at multiple points (e.g. "polysome effect size") but we could not find this term explicitly defined (for example: for the polysome effect size, were RNA counts for each polysome fraction divided by the relative abundance of that RNA in total RNA?)
__Response: __We apologize for this confusion. Please see our response above. We have also stated the definition of effect size explicitly in the revised manuscript.
Could you elaborate on how you define "protein abundance and "effect size: in Figure 5G? How is enrichment in P3 or P1 calculated?
__Response: __Effect size is defined as described above. Enrichment in P3 or P1 is calculated with respect to the abundance in gDNA (unsorted cells).
Were 3396 variants considered for all readouts in this paper? How many of these variants were present in each ROI? It may be worth clarifying sample sizes.
__Response: __Thanks for the suggestion. The reviewer is correct: 3396 variants were present in all biological replicates and all readouts (after excluding polysome metafractions 1 and 2 and flow cytometry population 4). The Methods were updated to include all readouts that were dropped. The number of variants in each ROI are now included in this section of the main text.
How did Twist generate these mutagenized sequences? We assumed that they used error-prone PCR due to the mention of multiple nucleotide polymorphisms, but couldn't find an explicit answer.
__Response: __Twist generates these mutagenized inserts using degenerate primers. This allows all alternate codons to be assayed (all silent, missense changes). This is now noted in the Methods.
In the methods, it may be worth elaborating on the composition of the HsCD00617865 plasmid. For example: this COMT reporter is under the control of a constitutively-expressed T7 promoter, correct?
__Response: __The HsCD00617865 plasmid was only used as a template for PCR amplification and generation of the transgene. The transgene is cloned into a vector containing attB sites for recombination into the landing pad cell line (Matreyek et al 2020). Transcription is induced by Doxycycline from the landing pad locus. Plasmid maps used for transfection into the landing pad line are now included in the GitHub repository.
In Supplementary Figures 4 and 5, it would be helpful to explicitly say that you are reporting Pearson correlations between biological replicates.
__Response: __Thanks for the suggestion. The legends have been updated accordingly.
"After summarizing biological replicates (N=4) for each readout...": how did the authors summarize biological replicates? Were counts averaged?
__Response: __Biological replicates were summarized using the median. This is now clarified in the Methods.
The authors used pairwise correlations between flow cytometry fractions, polysome fractions, and total RNA/gDNA as indications of data quality. Do the authors expect for these counts to be strongly correlated? We would not necessarily expect to see a strong correlation between ribosome load and RNA/gDNA.
__Response: __We used replicate correlation as an indicator of data quality. Our readouts of ribosome load reflect the abundance of a variant in a particular polysome fraction. Given that variants that are highly abundant in the RNA pool will on average be more highly represented in polysome fractions, we would expect a correlation between the abundance of a variant in total RNA and in polysome fractions.
The authors may need to check that their standard deviations on fold changes are properly reported.
__Response: __iIn the Figures and the main text, we specified the confidence intervals as calculated by ALDEx2 method instead of reporting standard deviations on fold changes,. Specifically, the confidence intervals were determined by Monte Carlo methods that produce a posterior probability distribution of the observed data given repeated sampling. Variants in which the confidence intervals do not cross 0 are considered true discoveries (section 5.4.1 of the ALDEx2 vignette on Bioconductor).
We would expect standard deviation bounds to be symmetric for log fold changes, but not on unlogged fold changes - for example see page 8, for the sentence "our point estimate for nonsense variant effects on COMT RNA abundance was approximately a two-fold decrease relative to the gDNA frequency (fold change of 0.43 +/- 0.13; mean +/- standard deviation; Methods)."
__Response: __Thanks for the suggestion. To avoid any confusion about the symmetry, we replaced the +/- notation, and explicitly noted the mean and standard deviation. To help the reader gain an intuition of the magnitude of variant effects, we conducted a frequency based normalization-dependent analysis using limma (as previously employed in Hoskins et al. 2023. Genome Biology). We now report a fold change (unlogged) for RNA abundance compared to gDNA abundance. The point estimate is the mean and s.d. across all nonsense variants.
On page 10, the authors say that their data suggests that hydrophobicity in the early coding region of COMT may be important for COMT folding. If this is the case, would we expect to see this effect in flow cytometry data (which is affected by protein degradation) and not polysome profiling (which is unaffected by post-translational protein degradation)?
__Response: __We apologize as we are uncertain about the reviewer’s intended question. The section that refers to the importance of hydrophobicity indeed refers to the flow cytometry data. While there are specific instances in which the amino acid properties encoded by the mRNA influences translation dynamics, these are not universally true. Consequently, we did not expect these impacts to be observed at the level of polysome profiling.
We believe that we would have some trouble replicating the analysis from this paper from the raw data, given that the bulk of the analysis on GitHub is presented as a single R Markdown file, with references to local files to which we do not have access. We recommend that the authors add additional documentation to their repository to facilitate re-analysis.
__Response: __Thanks for the opportunity to address this issue of critical importance. To facilitate replication, we have now deposited all analysis files to Zenodo and refactored the code to enable replication by simply running a markdown file.
In Figure 1B, indicating that more signal indicates less structure (in the legend or the figure itself) may assist readers who are unfamiliar with DMS-seq.
__Response: __Thanks for the suggestion. This is now updated.
Figure 1C does a great job presenting evidence for the translation of uORFs, but does not seem to flow with the overall argument of the paper, so may fit better in the supplement.
__Response: __We considered this suggestion, and opted for keeping its placement as it gives evidence that our transgene is translated primarily as the MB-COMT isoform. This ensures that, for variants upstream of the S-COMT isoform, we can assay effects on ribosome load that are tied to mechanisms of translation elongation and codon stability.
We believe there is a typo in the Figure 1 legend that should read "K562" instead of "H562".
__Response: __Thank you, this was indeed a typo.
You also gated to separate into P1-P4, correct? Can you also show the bounds of that gating
strategy in Figure 3A?
__Response: __This has been updated. We also added the gating strategy in response to comments from reviewer #1.
We find Figure 3F very compelling. Do you have any theories as to why mutating I59-H66 to
nonpolar, uncharged residues leads to increased COMT expression?
__Response: __We do not have any theories for why this may be. However, we noted that with the exception of V63, residues I59-H66 are not evolutionarily constrained (based on DeMask entropy values). This suggests mutational tolerance for nonpolar, uncharged residues in this region (with the exception of V63 and H66; see Figure 3D).
There appears to be a non-negligible proportion of di- and tri- nucleotide polymorphisms in Supplementary Figure 4. Were these excluded in downstream analyses?
__Response: __These variants are expected from the Twist mutagenesis strategy and included in analysis. We believe they are at lower frequency compared to SNPs due to less favorable annealing of the degenerate primers.
A minor typo in the discussion reads "fluoresce".
__Response: __Done
Significance
Describe the nature and significance of the advance (e.g. conceptual, technical, clinical) for the field.
This work investigated the regulatory effects of thousands of coding variants in the COMT gene, focusing on two regions with clinical significance, by using high-throughput reporter assays. The results from this will be useful for clinical scientists interested in understanding the impacts of COMT mutations and be a useful framework for other systems/computational biologists to understand the impacts of coding mutations across different levels of regulatory function. Mutations in protein regions, if having a function, are classically known to interfere with protein function. There are fewer large-scale efforts to understand the impacts of coding mutations affecting expression through potentially changing of RNA structure or codon optimization - this work has contributed towards that frontier.
Place the work in the context of the existing literature (provide references, where appropriate). This is (as far as I am aware) the first paper that has integrated high-throughput screens massively parallel reporter assays from RNA degradation, ribosomal load, and flow cytometry. Previous papers have tended to measure on expression regulation on only one dimension (i.e. Greisemer et al. 2023 on RNA degradation, Sample et al. 2019 on ribosomal load, and de Boer at al. 2020 on protein expression).
__Response: __Thanks for highlighting the novelty of our approach compared to existing strategies in the literature.
State what audience might be interested in and influenced by the reported findings.
Clinicians/researchers interested in COMT, computational biologists, geneticists and potentially structural biologists interested in understanding the consequences of amino acid mutations on RNA/protein expression
__Response: __Thanks for noting the broad significance of our study.
Define your field of expertise with a few keywords to help the authors contextualize your point of view. Indicate if there are any parts of the paper that you do not have sufficient expertise to evaluate.
Genomics, Massively parallel reporter assays, High-throughput regulatory screens.
Reviewer #3 (Evidence, reproducibility and clarity (Required)):
*This manuscript reports on transcript sequence variants that affect expression of the gene COMT. Targeted analysis of SNPs identifies 5' UTR variants that affect COMT, leading to the identification of translated uORFs. Common coding sequence SNPs do not affect COMT expression, however. Massively parallel analyses of mRNA abundance, protein abundance, and translation are combined to look more broadly at coding sequence variants. These analyses focus on regions of predicted structure in the COMT transcript. Both silent and missense mutations that increase mRNA abundance are identified. Protein abundance is then measured and many missense mutations are found to change protein levels. To address translation directly, analysis of polysome loading is performed and significant differences are identified, although technical challenges limit data quality in these experiments. These different experiments are then analyzed jointly to classify mutation effects and identify a class of silent mutations with expression effects, leading to a proposal that these act through structure. *
*The joint, integrative analysis of COMT variants through a range of methods allows clearer insights into interconnected post-transcriptional effects. The massively parallel experiments generate high-quality data, although targeted validation of key results would strengthen the work. The findings advance our understanding of silent variant effects, which remains an open question, and technical innovations could find broader applications. *
__Response: __Thanks for the positive assessment of the quality of the data generated and the potential for the broader application of the technical innovations.
*I do have concerns with the present version of this work. *
-
- There is no validation presented for high-throughput experimental data. I would say that validating the effects of M152T and V63V variants from Figure 2B would substantially strengthen the work and support key conclusions. * __Response: __Our experiments collectively enabled nearly 10,000 measurements of variant effect (summed over three layers of gene expression). The goal of our study was to identify broad mechanisms of variant effect. While we are excited about the specific variants uncovered, targeted experimental methods for validating changes to RNA abundance, such as RT-qPCR, are unlikely to be sufficiently sensitive. For example, RNA abundance effects in our study had a median effect size of 1.47 for variants up in RNA, and 0.4 for variants down in RNA. This likely corresponds to less than one Ct difference between the variant and the reference allele. Indeed, previous studies such as Findlay et al., 2018 Nature that reported similar effect sizes (FGF7 and FOS, respectively (Figure 4B).
Thus, for time and cost concerns, we respectfully suggest that targeted experiments involving V63V and M152T are beyond the scope of our study. Nevertheless, to further strengthen our conclusions, we have computationally confirmed our findings using a different analysis framework. We found 75/76 of the variants significant by ALDEx2 analysis were also significant by limma analysis (a frequency based normalization-dependent analysis, as previously employed in Hoskins et al. 2023. Genome Biology) using the same FDR (0.1).
- In the fluorescent reporter scheme, it seems that variants reducing mRNA abundance should be enriched in the "P2" gate region relative to "P1", as they would have lower mRNA abundance and correspondingly lower protein abundance. However, this analysis is not performed, and instead P1 and P3 are compared (Figure 3G), which would seem to focus on protein-level effects. *
__Response: __Our initial hesitation in comparing P2 to P1 is that the P2 population may be enriched for cells that underwent inefficient induction of transcription with Doxycycline. Hence technical factors as opposed to the effect of the variants may dominate this comparison. In response to the reviewer’s comments, we carried out the suggested analysis (new Supplementary Figure 5B). We found that variants that are down in RNA are enriched in P2 relative to P1 as expected. This is now noted in the Results section.
- In general the work classifies variants in several different ways and it would help to be a little clearer in naming these classes. For instance, in describing the FACS-based analysis of variant expression it is written, "protein fluorescence conditioned on RNA fluorescence" which is confusing at best-it's a fluorescence-based measurement that is used indirectly to measure COMT reporter abundance. *
__Response: __Thanks for the suggestion. We agree that our initial word-choice was imprecise. We rewrote this section to indicate mCherry fluorescence is an indirect proxy for RNA abundance.
- Likewise, the populations with shifted GFP/mCherry ratio in this assay are described as "uncorrelated" populations, which is opaque and somewhat inaccurate-there seems to be a correlation in this group but at a different ratio. *
__Response: __We have revised the language in the manuscript. We opted for “low or high RNA/protein abundance” to indicate the relationship between GFP and mCherry fluorescence in populations P3 and P4.
- In the same way, "deleterious variants" is used to describe protein abundance changes, but this term implies a fitness effect and is not very specific. *
__Response: __We apologize for the confusing word choice. We did away with this term in favor of “variants with low protein abundance”.
- In discussing the effects of missense COMT variants on protein levels, there is an implicit assumption that degradation of mis-folded protein (or perhaps properly-folded protein with excess hydrophobic exposure?) explains these effects. This is plausible, but it would help to lay out this reasoning more clearly. *
__Response: __Thanks for the suggestion. We have added a sentence at the end of the section that specifies this assumption and cites a recent study reporting that rare missense variants in COMT may be misfolded and degraded by the proteasome (Larsen et al. 2023).
- It is written that,"In line with codon stability as a predictor of translational efficiency (Presnyak et al., 2015), variants with low codon optimality were depleted from polysomes compared to variants with optimal codons". However, this mis-states the conclusions of the cited study, which notes, "Importantly, under normal conditions the ribosome occupancy of the HIS3 opt and non-opt constructs was determined to be similar (Fig. 6B)". *
__Response: __We apologize for mis-stating the conclusions of Presnyak et al. 2015. We have now revisited the relevant literature to more accurately place our conclusions in the context of literature. While Presnyak et al. and several other studies (Bazzini et al., 2016; Mauger et al., 2019) have clearly linked the association between codon choice and mRNA stability. We now reference Mauger et al. 2019 who used elegant experiments to demonstrate that mRNA secondary structure is a driver of increased protein production and synergizes with codon optimality (Figure 5B). Their results further support the role of codon optimality on RNA stability while providing evidence of additive impact on translation efficiency.
- It is written that, "One intriguing possibility is to develop multiplexed assays of variant effect on RNA folding, using mutational profiling RNA probing methods (Weng et al., 2020; Zubradt et al., 2017)." How would this differ from the "Mutate and Map" approach in doi://10.1038/nchem.1176 and subsequent work from the same group? *
__Response: __Thanks for pointing out the more recent work following the initial papers in 2010-2011. We have missed the work from the Das lab that extended the Mutate and Map approach to utilize mutational profiling (Cheng and Kladwang et al., 2017). We updated our Discussion to indicate that the proposed assay has been pioneered and is a viable approach for high-throughput determination of variant effects on RNA folding.
Because mutational profiling methods leverage reverse transcriptase readthrough and mismatch incorporation, they enable deeper and more uniform coverage of sequencing reads, particularly for longer transcripts. A key design principle of the proposed assay is to mutagenize only certain types of variants in the library such that they do not overlap RT mismatch signatures arising from the RNA probing reagent/RT enzyme. For example, readthrough of DMS base adducts largely generates A>N or C>N mismatches, so a variant library would be designed to only contain variants at G or T bases. This ensures variants in the library can be differentiated from signals of the RNA probing method.
***Referees cross-commenting** *
*I generally agree with the other reviewers and found that many small points on the figures were confusing, and in some cases the values being computed and displayed were under-specified. *
*I agree with Reviewer 1 that the polysome fractionation probably has limited power due to experimental design, and that the interpretation of changed ribosome loading is subtle. *
__Response: __In response to these helpful comments, we have clarified the points highlighted by the reviewers and expanded the limitations section related to the ribosome loading assay. Thanks for these constructive suggestions to strengthen our study.
*Reviewer #3 (Significance (Required)): *
*The joint, integrative analysis of COMT variants through a range of methods allows clearer insights into interconnected post-transcriptional effects. The massively parallel experiments generate high-quality data, although targeted validation of key results would strengthen the work. The findings advance our understanding of silent variant effects, which remains an open question, and technical innovations could find broader applications. *
__Response: __Thanks for pointing out the high-quality of the generated data and the broad significance of our study. The goal of our study was to identify broad mechanisms of variant effect instead of focusing on differential expression for any specific variants.
-
-
-
www.biorxiv.org www.biorxiv.org
-
Note: This preprint has been reviewed by subject experts for Review Commons. Content has not been altered except for formatting.
Learn more at Review Commons
Referee #3
Evidence, reproducibility and clarity
Summary:
Provide a short summary of the findings and key conclusions (including methodology and model system(s) where appropriate). Please place your comments about significance in section 2.
In the manuscript "Regulation of adaptive growth decisions via phosphorylation of the TRAPPII complex in Arabidopsis" the authors investigate the TRAPPII interactome carried out by an already published IP-MS screen. They study previously identified shaggy-like kinases SK as TRAPII interactors and the phosphorylation sites by Y2H (interactions of wild type, deletion mutants and phosphomutants) and kinase assays (in vitro) and pharmacological inhibition in the subunit AtTRS120. The authors provide a deeper phenotypical analysis of trapii null mutant lines and classification as "decision mutants", based on "limited budget" and "conflict of interest" experiments (previously described) as a starting point of investigations of TGN function in comparison with hormone mutants. Cell elongation is used as a response phenotype. Authors focus on mainly TRS120 and phosphorylation by SK and partly on another TRAPP component, CLUB. Authors study the assays with differing kinases, e.g. Y2H with BIN2, phosphorylation with SK11.
Major comments:
- Are the claims and the conclusions supported by the data or do they require additional experiments or analyses to support them?
A major issue is that new claims and conclusions are not supported by the new data provided here. The title says "in Arabidopsis", but only components are from Arabidopsis. Interactions, instead, are studied in this manuscript by Y2H in yeast, and phosphorylation in vitro. The abstract is very misleading and does not distinguish which aspects are studied in vitro and which in vivo. The Abstract does not mention that this study is based on previously identified interactome data.
o The figure legends are often not sufficiently detailed to understand what exactly is represented.
Therefore, it is not possible to judge in every case whether experiments are supported by data. E.g.
Fig. 1: A, describe which data were used and which control for IP-MS had been taken into account. B, this is a plot, please describe what is represented. Explain better why Shaggy kinases were chosen. C, explain the principle and what is represented. How is this experiment controlled and how is it ensured that negative results are not caused by absent proteins.
Fig. 2: Indicate the phosphorylation sites in the other subfigures. Fig. 2E: How was it generated, explain what is seen. Since this is the only figure illustrating the protein complex of TRAPP, this figure should be more thoroughly prepared and labeled. I recommend a better visualized protein complex. As before, Fig. 2F remains unclear.
Fig. 3: Please add a figure illustrating the mutations. 3C: what has been diluted? Other examples are found in other figures.
Fig. 4: Shouldn't the wild type be compared with all the mutants? Then statistics have to be conducted accordingly. Better explain G and H. If there are quotients, explain of what exactly.
Fig. 5. Same as before. How do I see that there is a phenotype? There is no comparison with wild type. It is also unclear to which values the statistics refer to.
Fig. 6: Please guide the reader through the figure and experiment.
Fig. 8: I miss the connection with other shaggy-like kinases. This summary could be more complete. What about phosphorylation sites?
o Line 133-134: "we focus on the TRAPPII complex as a starting point as it is required for all aspects of TGN function, including the sorting of proteins such as PINs to distinct membrane domains" I did not find an obvious connection to the PIN transporters as well as clear data to TGN functions. This sentence was for me misleading about the context of this manuscript.
o Figure 1C: A supporting Western Blot control is needed, to fully validate the missing interaction of BIN2 with the truncated variants of TRS120 and CLUB. Additionally, swapping the constructs from DB to AD and vice versa will provide a better set-up of the interaction screen. This should be easily done in a few weeks.
o Line 431-432: "This presents intriguing implications regarding the potential role of the AtSK-TRAPPII module in meeting the unique demands of endomembrane traffic in plants." Why do the authors come to this assumption? Further discussion is needed here.
o Figure 2F: What serves as positive controls? What is the purpose of showing every panel between each TRS120-T2 variant with CLUB-C2, CLUB-C3, TRS120-T1 and TRS120-T3 and not only interactions between BIN2 and the TRS120-T2 variants? Why are there six negative controls as it is every time the same control? - Please request additional experiments only if they are essential for the conclusions. Alternatively, ask the authors to qualify their claims as preliminary or speculative, or to remove them altogether.
Clearly, title, abstract and statements have to be formulated differently. The discussion should contain a limitations paragraph in which the authors detail that conclusions are based on in vitro, yeast and plant IP-MS screening data, and they should describe approaches how the study can be continued in the future. Which alternative explanations are possible. Are SKs and TRAPP expressed and present in the same locations? - If you have constructive further reaching suggestions that could significantly improve the study but would open new lines of investigations, please label them as "OPTIONAL". - Demonstrating interactions and phosphorylation by other approaches in vivo - demonstrating effects of TRAPP phosphomutants and lack of kinases in vivo - Are the suggested experiments realistic in terms of time and resources? It would help if you could add an estimated time investment for substantial experiments. - Are the data and the methods presented in such a way that they can be reproduced?
o Figure S9: Is it not a loss of chlorophyll instead of GFP? Does not look like a fluorescent image.
o Lacking information of pH of in vitro kinase assay solution with Mass-spectrometry.
o What is the purpose of transferring 10 days old seedlings to fresh plates for scanning? Needs additional information for understanding, at the moment it sounds more like unnecessary extra stress for the seedlings.
o Why are seedlings grown under constant light? - Are the experiments adequately replicated and statistical analysis adequate?
o Figure 5: It will be good to use ANOVA for statistics here. I personally doubt the high significance of some parameters, e.g. for club-2 cell width and cell surface area between dark and darkW due to the high standard errors. Rechecking with the original values is necessary. Why is there no comparison between wild-type and the two mutants?
o Figure 7A - C, statistic is probably not correct. For example: in A statistical differences with P<0.001 between wild-type (~100 %) and TRS120SαβγD (~80 %), in C statistical difference of only P<0.05 between wild-type type (90 %) and TRS120SαβγD (60 %)
o No information on IP-MS replicate numbers mentioned.
o Also see comments above to figures
Minor comments:
- Specific experimental issues that are easily addressable.
- Specific experimental issues that are easily addressable.
o Figure 4, S7, S8, S11 and S12: It will be helpful to support the data with images of the seedlings. - Are the text and figures clear and accurate? Do you have suggestions that would help the authors improve the presentation of their data and conclusions?
o The introduction is quite lengthy with unnecessary information, e.g. about PIN transporters, but useful information about shaggy-like kinases and connection to brassinosteroid signaling is lacking.
o Figure 1C: In the figure legend is no explanation of abbreviation "Co"; no explanation of BET3, TRS31, Tca17 and TRIPP; no indication that spots come from different plates (just visible by different brightness of the squares). Why are there eleven negative controls as it is every time the same control?
o Figure 1C is specifically for BIN2, but BIN2 was not identified in the IP-MS screen represented in Figure 1B. Why does 1C not focus on SK11/12/32, identified in 1B?
o Figure 1C shows several truncated variants of TRS120 and CLUB, a schematic overview as represented in Figure 2A will be helpful for the understanding of 1C. Order of variants should be the same (now: in 1C first TRS than CLUB in 2A first CLUB than TRS).
o Figure 1C: Interaction of TRS120 full-length with BIN2 is missing in this figure but is presented in Figure 2F.
o Result of Figure 2F is described after Figure 3. Better arrangement of Figures or text is needed here.
o Figure 3A: Why was AtSK11 and not BIN2 used for the main figure? Better change Figure 3A with Figure S4 to keep the focus on BIN2. No explanation of the result in the text.
o Figure 3A: In the figure legend is no explanation of abbreviation CBB. What are the non-phosporylated variants? Where are they shown? Description sounds that only TRS120-T2-SαβγA versus TRS120-T2 WT was tested by t-test is this correct? And if yes, why?
o No need for Figure 3B, information was already given in Figure 2A + B.
o Figure 3C: Why BIL2 for Clade II and not BIN2?
o Figure 4: Why are A-E not directly compared to wild-type but trs120-4 as seen in 4F? What is the purpose of using different types of diagram?
o Figure 4H: Why are phyAphyBcry1cry2 and pyrpyl1pyl2pyl4 depicted? No description in the text.
o Figure 6: Confusing order of given information in the figure legend. Sentence one belongs to D and H only, second sentence describes whole figure. o Figure 6D + H, color difference between black and blue is hard to see, better change one into e.g. red.
o Figure 7D - F wrong indication of D to F, named in the description as A) - C). Why is E different to D in F (D and F: 0-1 is attenuated, >1 enhanced; E the other way around).
o Figure S9A: Indication of protein size on the Coomassie gel is missing and the respective position of 160 kDa is not visible on the gel.
o Figure S12D: No explanation of the color code in the figure legend.
o Consistent labelling and layout of all Figures and Supplemental Figures will be helpful. E.g., Figure 3A and S4; in S8A-E + S11A-C bars of different conditions have the same color. Most of the figure legends are quite shortly described and lack information about what kind of data is presented.
o YFP parameters are described in material and methods, but no YFP construct appeared in the manuscript to my knowledge. - Are prior studies referenced appropriately? - Lines 243-245: Text is nearly identical to Kalbfuß et al., 2022. - Lines 246-254: Text is identical to Kalbfuß et al., 2022. - Are the text and figures clear and accurate?
Please see the above and below comments to figures and figure legends. - Do you have suggestions that would help the authors improve the presentation of their data and conclusions?
Overall, the manuscript may have very interesting data and new findings. It is very interesting that the authors study the regulation of a protein complex that may mediate environment responses and intracellular Golgi functions. However, it is very difficult to follow and understand the ideas and concept of the manuscript. This manuscript is based on a previously published interactome study by Rybek et al. 2014, Steiner et al. 2016, Kalde et al. 209. Moreover, a physiological approach is published in Kalbfuß et al. 2022. The outcomes and conclusions from these previously published manuscripts and the emanating open questions addressed here should be clearly described in the introduction. This is currently not the case. Moreover, many experimental approaches and results (e.g. figures, figure legends) are not properly described. Overall, it is therefore not possible to understand the manuscript without studying in depth all other manuscripts. Before the manuscript can be more thoroughly judged, it is necessary that the authors rewrite the manuscript, reorganize it and explain better their ideas and approaches. It is also necessary to explain and define unusual terms such as "decision mutants", "limited budget" and "conflict of interest" experiments, which are crucial for the understanding. The importance of the TRAPPII complex should be illustrated using specific physiological examples and the context in which this complex is studied here has to be explained. Before this is not corrected, the following assessment will remain rather incomplete. Another complication is that two subunits of TRAPP were studied and different types of SKs, however, authors did not systematically analyze all interactions. At least it should be thoroughly described, and a flow chart would be helpful as supplemental figure clearly describe which types of proteins were tested in the different assays. The introduction is not well written. It is very lengthy, however the important messages from previous publications are left out. Thus the open question is not understandable (see above). Instead, the results parts start with introduction again. Explanations are also lacking in every result paragraph on the approach and expected data. The Discussion is also not very well written. It is much focused on physiological and molecular actions and consequences in plants. However, there should be at first a technical discussion on the relevance since in the study is based on in vitro and heterologous expression data, and the physiological analysis was only conducted with knockouts but not phosphomutants. Therefore, the link between the protein interaction and physiological functions needs to be worked out.
Referees cross-commenting
My colleague and I have read thoroughly the manuscript and found a number of issues which we indicated in our review. These points can be fixed by the authors, if they formulate more carefully and remove the overstatements. They should also work on reorganizing and including more explanations.
Significance
Provide contextual information to readers (editors and researchers) about the novelty of the study, its value for the field and the communities that might be interested.
The following aspects are important:
One new aspect of this story is the validation of interaction of TRAPII subunits as substrate for AtSKs and their action as phosphorylation agents shown in vitro. The other new aspect is the phenotypical characterization of trapii mutants under stress-conditions (grown in darkness) and additive stress (with additional drought stress). The potential interaction with brassinosteroid signaling via BIN2 is intriguing.
- General assessment: provide a summary of the strengths and limitations of the study. What are the strongest and most important aspects? What aspects of the study should be improved or could be developed? A strength is that a new interaction is further studied. A weakness is that the studies are primarily conducted in yeast and in vitro, leaving open how relevant this process is in plants. A strength is further studies and phenotypic analysis of trapii mutant effects. A weakness is that this mutant analysis is disconnected from the action of SKs.
Further, the writing should be improved and more clear (see comments above). - Advance: compare the study to the closest related results in the literature or highlight results reported for the first time to your knowledge; does the study extend the knowledge in the field and in which way? Describe the nature of the advance and the resulting insights (for example: conceptual, technical, clinical, mechanistic, functional,...). The introduction gives the impression of a stronger investigation of TGN function, which is from my point of view not the case and should be reformulated and/or put into a deeper context with known literature. The authors switch several times between the different TRAPPII subunits and shaggy-like kinases in the main figures which made it for me very confusing. I believe that rearranging some data/figures will improve the understanding of the story. The text is also lacking explanations of many abbreviations and gene names which caused more difficulties in understanding the story and slowed down the reviewing process. From my point of view it seems to be necessary to read the often cited Kalbfuß et al., 2022 publication before, as many important technical aspects and scientific background, e.g. the reason to use specific control mutants, are well explained there, but are lacking in this manuscript and needs improvement. - Audience: describe the type of audience ("specialized", "broad", "basic research", "translational/clinical", etc...) that will be interested or influenced by this research; how will this research be used by others; will it be of interest beyond the specific field? Based on the cited literature in this manuscript the direction of the story with "limited budget" and "conflict of interest" situations to classify mutants methodically seems to be a recently emerged approach. Apart from that this manuscript provides only new impact on TRAPII and AtSKs specific knowledge based on well-established and frequently used techniques that address the problem in vitro and in a heterologous system. Therefore, this story will be interesting for researchers specialized in stress responses, TGN and growth defects as well as important for basic research. Limitations in interpretation are present. - Please define your field of expertise with a few keywords to help the authors contextualize your point of view. Indicate if there are any parts of the paper that you do not have sufficient expertise to evaluate.
Our field of research is related to nutrition-regulated processes especially in Arabidopsis with a strong methodological background in interactomics, physiological, morphological and molecular responses and biochemical approaches and microscopy.
-
-
www.biorxiv.org www.biorxiv.org
-
Note: This response was posted by the corresponding author to Review Commons. The content has not been altered except for formatting.
Learn more at Review Commons
Reply to the reviewers
Replies to Reviewers
Thank you for inviting us to submit our revised manuscript titled, “Diffusive mediator feedbacks control the health-to-disease transition of skin inflammation.” We appreciate the time and effort the editor and each of the reviewers have dedicated to providing insightful feedback on ways to strengthen our manuscript. The revisions in the main text in response to the detailed comments are highlighted in red and were proofread by professional English editors. We hope that our revision and responses address all the concerns raised by the reviewer, and we look forward to hearing from you regarding this submission.
Reviewer #1 (Evidence, reproducibility and clarity (Required)):
The manuscript provides a model of interacting populations of pro- and anti-inflammatory mediators to explain spatial patterns associated with various inflammatory conditions. The work is robust and articulated well, and is certainly scientifically relevant.
Authors: Thank you for your positive evaluation and many insightful comments on our manuscript. We have incorporated your feedback, and hope that our revisions satisfy all the comments.
Minor amendments:
Personally, I feel that the model should be reported prior to the results, as the choice of model is likely to have great significance on the observations. It would be preferable for the reader to have a clear picture of the governing equations in their mind as they digest the results.
Au: Following this reviewer's suggestion, we have relocated the Method section including the model description to be written prior to the Result section (p.9-14 lines 152-232; revised manuscript).
The literature review is largely relatively thorough; however, I think it is important that the previous works of Joanne Dunster (University of Reading) and collaborators are included, as these are very closely related to this work. In particular, the authors should note the following two papers, which take a spatial approach:
-
Bayani, A., Dunster, J.L., Crofts, J.J. et al. Mechanisms and Points of Control in the Spread of Inflammation: A Mathematical Investigation. Bull Math Biol 82, 45 (2020). https://doi.org/10.1007/s11538-020-00709-y
-
Bayani A, Dunster JL, Crofts JJ, Nelson MR (2020) Spatial considerations in the resolution of inflammation: Elucidating leukocyte interactions via an experimentally-calibrated agent-based model. PLoS Comput Biol 16(11): e1008413. https://doi.org/10.1371/journal.pcbi.1008413
Au: We have incorporated this comment by adding the two suggested papers to the relevant sentences in the literature review (p.6 line 118-119; revised manuscript) as follows: “Previous reaction-diffusion models, including chemotactic cells, have reproduced the resolution of inflammation in the lung [Bayani et al. 2020a, Bayani et al. 2020b]”
One key point that should be mentioned in the discussion is that the model neglects any immune cells (e.g. neutrophils, macrophages) which contribute greatly to the inflammatory condition. Since these cells are motile, and also can contribute both pro- and anti-inflammatory effects, they are likely to influence spatial patterns significantly. It is not necessarily a problem that these aren't included in the model, but I feel that it is important that their omission be discussed in the manuscript.
Au: We have now discussed the immune cells in the “Future implications” as the reviewer suggested (p.29 line 477-483; revised manuscript) as follows: “This is probably because the present model focuses on the non-chemotactic cells (e.g., including keratinocytes), whereas chemotactic cells (e.g., macrophages and neutrophils) also contribute to skin inflammation [Zhang and An 2007, Coondoo 2011]. Moreover, the present model focuses on the innate immune response, whereas the skin initiates an acquired immune response in the persistence of the innate immune response. Therefore, incorporating the chemotactic cells and acquired immune response into the model will reproduce the end of the expansion.”
Reviewer #1 (Significance (Required)):
The manuscript advances our current understanding of spatially spreading inflammation and corresponding patterns, but needs to be contextualized against existing literature as described above.
This manuscript will appeal to theoreticians (Mathematicians) and clinicians/experimentalists alike.
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
The authors propose a minimal mechanistic mathematical model able to reproduce qualitatively different spatial patterns observed in healthy and disease epidermis. The starting point is a systematic review of medical images of different dermatological conditions, which they classify and successfully capture according to the spatial patterns. It is an interesting piece of work, but I consider that it will gain significance if the theoretical results are compared again with the clinical data. Specifically, the authors show a very interesting map between parameter regions and different spatial patterns; this result should be compared back to clinical data, to confirm that specific changes in spatial patterns indeed result from predicted changes in a specific parameter (e.g., due to a genetic condition that affects a feedback strength).
Authors: We thank you for providing your valuable comments on our manuscript.
Following your suggestion about the comparison of theoretical results with the clinical data, we have predicted which specific parameters including the feedback strength cause specific transitions of spatial patterns in the respective diseases. The discussion was added on p.26 lines 415-438 in the revised manuscript as follows: “The parameter-to-patterning correspondence (Fig. 4A, B, S2 Fig., and S3 Fig.) allows us to infer the pathogenesis mechanism in various diseases exhibiting each of diverse expanding patterns (seen in Table 2). For instance, psoriasis exhibits all five expanding patterns (Table 2) and increased levels of pro-inflammatory mediator (TNF-α) [Ringham et al. 2019], which is consistent with our theoretical results. The elevated pro-inflammatory mediator in psoriatic skin has been suggested to be caused by genetic mutations affecting regulatory feedback [Valeyev et al. 2010]. Considering these previous studies, our model predicts a psoriasis progression where fading pattern transits to arcuate, polycyclic, gyrate, annular, and circular pattern where increase in the TNF-α level is possibly due to mutation-induced alteration in the feedback parameters, e.g., increase of the production of pro-inflammatory mediator qa (Fig. 4A). Alternatively, Lyme disease exhibits circular, annular, and polycyclic patterns (Table 2). A clinical report showed that patients in Missouri predominantly exhibit an annular pattern without prognostic symptoms, while those in New York tend to exhibit a circular pattern with prognostic symptoms following the same treatment [Wormser et al. 2005]. Considering our theoretical result that the overproduction of pro-inflammatory mediators and the depletion of anti-inflammatory mediators leads to the annular and circular pattern, respectively (Fig 4, 5A, and B), altered levels of pro-inflammatory and anti-inflammatory mediators may significantly impact the development and prognosis of Lyme disease in Missouri and New York patients, respectively.
These qualitative parameter estimations will be verified in the future through parameter quantification in each diseased skin exhibiting any expanding patterns. By incorporating this quantitative correspondence between patterns and parameters measured in each disease into the present model, we would develop each disease-specific model with a quantitative predictability of how much change of the skin parameters transit from healthy to diseased pattern or vice versa. Therefore, this study provides the first step to controlling the healthy-to-diseased transition of skin inflammation via diffusive mediator feedback.”
Another shortcoming of this work is that some of the conclusions are rushed: the parameter-to-spatial patterns analysis would strongly benefit from adding a quantitative to the qualitative description, e.g., mapping how changes in a given parameter value results in gradual changes in fading speed. Along the same line, the stability analysis for the different fading pattens was performed only for selected parameter values, it is not clear how variations in parameter values affect the sizes of the basins of attraction of the different steady states; we want to make sure that the parameter values were not cherry-picked. Further, given that the authors show bistability for some parameter values, then the dependency on initial conditions on the final spatial pattern should be more extensively investigated.
Au: We have incorporated these comments by adding a quantitative description including new results and future research strategies following each of the three constructive suggestions raised by the reviewer.
First, regarding “the fading speed” the reviewer suggested, fading speed is affected by changes in parameters involved in mediator production. In particular, the speed is reduced by an increase in the production parameters of pro-inflammatory mediators (pa, qa) and a decrease in those of anti-inflammatory mediators (pi, qi) (Fig.2. C and D). Moreover, “the size of the basins” the reviewer pointed out corresponds to the distance between ST (Threshold) and SH (Healthy state) in the cases with excitability. The distance between ST and SH becomes closer indicating the health state being less stable when pro-inflammatory mediators (pa, qa) increase or anti-inflammatory mediators (pi, qi) decrease from the healthy fading pattern. The imbalance of the mediator production transits the fast fading pattern with a small trajectory into a slow fading pattern with a larger trajectory. As imbalance goes on, the expanding pattern appears in the order of arcuate, polycyclic, and gyrate (Fig. 5). In cases with bistability, the size of basins corresponds to the relative distance ST to SH and ST to SI (Inflamed state). The circular and annular patterns appear when the distance between ST and SH is closer. On the other hand, when the distance between ST and SI was closer, the inflamed area shrank rather than expanded. The shrinking pattern appeared by reducing the production of pro-inflammatory mediators (pa, qa) or increasing the production of anti-inflammatory mediators (pi, qi) under conditions of stability. We have added a new figure and described this finding in Results (p.24 lines 384-388; revised manuscript) as follows: “As a result, we found that the distance between the healthy state (SH) and the threshold state (ST, a closer unstable steady state to SH) was the smallest in the gyrate pattern and increased in the order of polycyclic, arcuate, slow fading pattern, and fast fading pattern (Fig. 5C–F, S4 Fig. B and C). The fast fading pattern showed a smaller trajectory (green curve in S4 Fig. B and C) of change in the mediator concentration than the slow fading pattern.”
Second, regarding “the dependency on initial conditions”, we have further added a new result (p.24 line 374-382; revised manuscript) as follows: “The number of stable states determines the pattern regardless of the initial condition in the spatial distribution of mediator concentration. Similar to the fading pattern (Fig. 2), the arcuate, polycyclic, and gyrate patterns with the excitability appeared reproducibly, independently of the initial conditions due to a single stable state SH (Fig. 5C-F). Even in circular and annular patterns with bistability where the threshold ST was closer to the inflamed state SI than the healthy state SH (Fig. 5A-B), the final spatial pattern was dominated by the SI independently of the initial condition. On the contrary, when ST was closer to the SH than the SI, the inflamed area shrank rather than fading (S4 Fig. A). These results are general outcomes of the traveling wave of bistable systems [Murray 2002], and consistent with the previous theoretical studies on inflammations [Sudo and Fujimoto 2022, Volpert 2009]. ”
Finally, we have added “a quantitative to the qualitative description as a future research strategy (p.27 line 432-438; revised manuscript) as follows: “These qualitative parameter estimations will be verified in the future through parameter quantification in each diseased skin exhibiting any expanding patterns. By incorporating this quantitative correspondence between patterns and parameters measured in each disease into the present model, we would develop each disease-specific model with a quantitative predictability of how much change of the skin parameters transit from healthy to diseased pattern or vice versa. Therefore, this study provides the first step to controlling the healthy-to-diseased transition of skin inflammation via diffusive mediator feedback.”
For reproducibility it is essential that the authors add a much more detailed description of the methods, including the software tools / numerical analysis tools used. Making the code publicly available would also be very beneficial to ensure the reproducibility of the results.
Au: Following your suggestion, we have added a description of the methods, including the simulation code, to the “Methods” (p.13 lines 231-232; revised manuscript) as follows: “A simulation code written in C language is available from GitHub: https://github.com/MakiSudo/Erythema-Patterns/blob/main/AInondim.c.”
In conclusion, the work is very interesting and worth publishing, but requires (a) to come back to the clinical data for validation of model predictions, (b) a more thorough and quantitative investigation of the effects of parameter variations on model behaviors, (c) a more rigorous and systematic presentation of the methods, (d) carefully explaining how the proposed model is similar / differs to the classical activator -inhibitor model proposed by Turing, and (e) discussing / showing if the fading patterns result from a turning instability.
Au: For (a) “validation of model predictions,” (b) “model behaviors,” and (c) “a more rigorous and systematic presentation of the methods,” we have reflected your suggestions in the revised manuscript as described above.
Regarding (d) and (e), we have added an explanation of “how the proposed model is similar/differs to the classical activator–inhibitor model” and “if the fading patterns result from Turing instability” after the model construction in Methods (p.11-12 line 210-216; revised manuscript) as follows: “Reaction terms of this model are similar to the classical activator-inhibitor model proposed by Turing [Turing 1952], which includes the negative feedback of the activator through the inhibitor and the positive feedback of the activator. These reaction terms potentially result in Turing instability. However, the present model setting does not show Turing instability. The reason is that Turing instability requires a large difference between the diffusion coefficients of the activator and inhibitor [Murray 2002], whereas these coefficients in the present model were set to be equal based on molecular findings that these molecular weights are close in proximity [Coondoo 2011]. ”
**Referees cross-commenting**
I agree with the comments from Reviewer #1.
Reviewer #2 (Significance (Required)):
The work aims to bridge mathematical modelling to dermatological practice, which is much needed to enable the use of theoretical and computational tools to clinical decision-making. While some mathematical models of skin inflammation have been proposed in the past (refer to papers from the RJ Tanaka group in systems dermatology), most of these do not consider explicitly the spatial component, which is crucial for modelling the clinically visible spatial patterns. Potentially interested audience includes biomathematicians, systems biologists, systems dermatologists, and, if the validation of the model predictions is achieved (as suggested above), also dermatologists.
I am a systems biologists working on multi-scale mechanistic mathematical modelling of epithelial tissue diseases. The work I just reviewed falls exactly within my area of expertise.
-
-
Note: This preprint has been reviewed by subject experts for Review Commons. Content has not been altered except for formatting.
Learn more at Review Commons
Referee #2
Evidence, reproducibility and clarity
The authors propose a minimal mechanistic mathematical model able to reproduce qualitatively different spatial patterns observed in healthy and disease epidermis. The starting point is a systematic review of medical images of different dermatological conditions, which they classify and successfully capture according to the spatial patterns. It is an interesting piece of work, but I consider that it will gain significance if the theoretical results are compared again with the clinical data. Specifically, the authors show a very interesting map between parameter regions and different spatial patterns; this result should be compared back to clinical data, to confirm that specific changes in spatial patterns indeed result from predicted changes in a specific parameter (e.g., due to a genetic condition that affects a feedback strength). Another shortcoming of this work is that some of the conclusions are rushed: the parameter-to-spatial patterns analysis would strongly benefit from adding a quantitative to the qualitative description, e.g., mapping how changes in a given parameter value results in gradual changes in fading speed. Along the same line, the stability analysis for the different fading pattens was performed only for selected parameter values, it is not clear how variations in parameter values affect the sizes of the basins of attraction of the different steady states; we want to make sure that the parameter values were not cherry-picked. Further, given that the authors show bistability for some parameter values, then the dependency on initial conditions on the final spatial pattern should be more extensively investigated.
For reproducibility it is essential that the authors add a much more detailed description of the methods, including the software tools / numerical analysis tools used. Making the code publicly available would also be very beneficial to ensure the reproducibility of the results.
In conclusion, the work is very interesting and worth publishing, but requires (a) to come back to the clinical data for validation of model predictions, (b) a more thorough and quantitative investigation of the effects of parameter variations on model behaviors, (c) a more rigorous and systematic presentation of the methods, (d) carefully explaining how the proposed model is similar / differs to the classical activator -inhibitor model proposed by Turing, and (e) discussing / showing if the fading patterns result from a turning instability.
Referees cross-commenting
I agree with the comments from Reviewer #1.
Significance
The work aims to bridge mathematical modelling to dermatological practice, which is much needed to enable the use of theoretical and computational tools to clinical decision-making. While some mathematical models of skin inflammation have been proposed in the past (refer to papers from the RJ Tanaka group in systems dermatology), most of these do not consider explicitly the spatial component, which is crucial for modelling the clinically visible spatial patterns. Potentially interested audience includes biomathematicians, systems biologists, systems dermatologists, and, if the validation of the model predictions is achieved (as suggested above), also dermatologists.
I am a systems biologists working on multi-scale mechanistic mathematical modelling of epithelial tissue diseases. The work I just reviewed falls exactly within my area of expertise.
-
-
www.biorxiv.org www.biorxiv.org
-
Note: This response was posted by the corresponding author to Review Commons. The content has not been altered except for formatting.
Learn more at Review Commons
Reply to the reviewers
Reviewer #1 (Evidence, reproducibility and clarity (Required)):*
The mechanisms that differentiate ER from the nuclear envelope (NE) remain to be fully elucidated but likely depend at least in part on junctions between the ER and NE. How such junctions are formed and maintained is the subject of this manuscript where extensive correlative light and electron microscopy is used to observe and characterize ER-nuclear envelope (ER-NE) junctions at distinct phases of the cell cycle. The authors make use of their own electron tomography data as well as publicly available focused-ion beam scanning electron microscopy (FIB-SEM) datasets to compare the morphology of these junctions in different human cell types as well as in budding yeast. The major finding is that ER-NE junctions in human cell lines are more constricted than ER-ER junctions, often to the point of excluding lumen. The examination of mitotic cells suggests that this constriction likely occurs at the end of mitosis as the NE is completing its maturation from ER to NE. The implications of these morphological changes are discussed but there are no mechanistic or functional studies. Overall, the data are well presented, are of high quality and are rigorously evaluated. The manuscript is well written and scholarly, and the speculations as to the function of the constrictions are reasonable. I only have minor comments. *We thank the reviewer for the positive evaluation on our work and for the useful suggestions on how to further improve the manuscript.
- * In Figure 2D, the authors present evidence to demonstrate that an hourglass-like constriction occurs at ER-NE junctions. From the side view, it is difficult to interpret this on the plot, particularly for the ER-NE junctions with a lumen. Perhaps, in the supplemental data, the authors could plot both with and without lumen data separately, and color-code individual traces? I believe this would convey the hourglass nature of these constrictions more clearly.* To make it easier to see individual membrane profiles, we will plot the profiles with and without lumen separately and labelled each profile with distinct colour, as the reviewer suggested.
* In the Methods section, the authors should describe how carbon-coating of sapphire discs was achieved. If these were provided from the manufacturer precoated, this should be specified.*
We coated the sapphire discs with carbon by ourselves. We will specify how the carbon-coating was done in the revised manuscript.
* On page 10, Figure 5F callout 9 lines from the bottom likely should be 5E. We will correct this error.
Reviewer #1 (Significance (Required)):
Overall, this work provides an important new morphological perspective on the nature of ER-NE junctions in human cells. As the authors describe in their introduction, such junctions have been noted previously in the literature but not in a dedicated study using modern imaging techniques in human cell lines. In describing the morphology of these junctions, the authors lay the groundwork for future mechanistic, functional, and structural studies. We thank the reviewer for appreciating the significance and the impact of our work.
*
-
*
-
*
Reviewer #2 (Evidence, reproducibility and clarity (Required)):*
Summary: In this manuscript, Bragulat-Teixidor et al., use correlative live-cell imaging and electron tomography to study the structure of the endoplasmic reticulum-nuclear envelope (ER-NE) junction in HeLa cells (and also in S. cerevisiae). The authors also make use of publicly available whole-cell FIB-SEM datasets to study ER-NE junctions in mouse pancreatic islet, HeLa, and human macrophage cells to corroborate their findings in other cell types.
The authors show that the structure of the ER-NE junction in interphase cells adopts an hourglass shape with a constricted neck. Comparing the ER-NE junction to the ER tubule-sheet junction, the authors show that these structures are different: the ER tubule-sheet junction is not constricted. Because the NE forms from the ER during postmitotic NE assembly, the authors compare the structure of the ER-NE junctions in anaphase, telophase, and interphase cells, and find that the junction becomes constricted in telophase. The number of ER-NE junctions increase going from telophase to interphase.
While the authors do not provide any direct evidence for this, they propose a functional model where the ER-NE junction is constricted because it regulates the supply of certain lipids and proteins from the ER to the NE. One proposed example is that the constriction of the ER-NE junction might prevent the passage of large protein aggregates from entering the NE.
The general question of how the structure of the ER-NE junction might regulate the passage of lipids and proteins from the ER to the NE is interesting and potentially important. However, the authors should address the following issues to improve the accuracy and completeness of this manuscript for it to be considered for publication. *We thank the reviewer for the appreciation of our work and the thoughtful suggestions for further improvements.
* Major comments: 1. The authors compare the structure of the ER-NE junction to the structure of the ER tubule-sheet junction in interphase cells. They should instead or in addition be comparing the ER-NE junction to ER sheet-sheet junctions. This is likely a better comparison for two reasons:
i) The NE is similar to an ER sheet due to its flat and extended structure. The ER membranes surrounding the NE consists mostly of a dense network of sheet-like ER (Zheng et al., 2022, PMID: 34912111). Therefore, the ER-NE junction should be compared to these NE-adjacent ER sheet-sheet junctions and not ER tubule-sheet junctions which are likely to be found in the cell periphery.
ii) In HeLa cells, the NE assembles from large ER sheets and not ER tubules (Zhao et al., 2023, PMID: 37098350; Otsuka et al., 2018, PMID: 29323269; Lu et al., 2011, PMID: 21825076). Therefore, the ER-ER junctions the authors are already studying in anaphase cells are likely to be ER sheet-sheet junctions, which should be kept the same in their analysis of the ER-ER junctions in interphase cells.
Related to this point, comparing the side view panels in Figure 2D with 2H, it seems that the width of the ER membranes on either side of the neck region of the ER-NE junction is in fact getting wider (more sheet-like). This is in contrast to the ER-ER junction where the width stays constant for the ER tubule that is fusing onto the ER sheet. This suggests that indeed, the ER-NE junction is more similar to an ER sheet-sheet junction. *It is a very interesting possibility that the ER-NE junction might be similar to the ER sheet-sheet junction. We will inspect whether the ER that forms the ER-NE junction consists of sheet or tubular ER in our EM tomograms, and describe the outcome in the revised manuscript.
* The authors claim that in late anaphase cells, the ER-ER/NE (written like this because the ER and NE cannot be distinguished like the authors also point out) junctions are not constricted and had a similar morphology to ER-ER junctions in interphase. However, this claim is only qualitative at the moment, as the authors do not provide any quantification of the width of the ER-ER/NE junctions in late anaphase cells. To make the current claim that the ER-NE junction only becomes constricted in telophase, the authors should report the width of the ER-ER/NE junctions in late anaphase cells.
In late anaphase cells, large ER sheets initially wrap around chromatin at the periphery of the chromosome mass (Zhao et al., 2023, PMID: 37098350; Otsuka et al., 2018, PMID: 29323269; Lu et al., 2011, PMID: 21825076). Therefore, the authors might find it easier to identify ER-ER/NE junctions in the so-called "non-core" regions, instead of in the current regions shown in Figure 3A. *As the reviewer pointed out, we did not provide quantification of the width of ER-ER/NE junctions in late anaphase cells. We will measure them and show the quantification in the revised manuscript.
* Minor comments: 1. In the Supplementary Figures 1 A-D, make the scale bars white. Currently, the black scale bars are especially difficult to see in the top panels in Supplementary Figure 1C. *We will change the colour of some scale bars to make them more visible in the Supplementary Figure 1.
* In the Results section entitled "The number of ER-NE junctions per cell increases from telophase to interphase", the authors should tone down this claim because the number of telophase cells examined is low (only 2 telophase versus 9 interphase cells). It would be better to include the word "slightly" in the title to change it to "slightly increases". *We will modify the text accordingly. * In the Results section entitled "The number of ER-NE junctions per cell increases from telophase to interphase", the authors state "These densities were much lower than those of ER-ER junctions...". For sure this is true for ER tubule-tubule junctions in the periphery of the cell as ER tubules form an intricate network by constantly fusing to each other, but it's not clear if this is also the case for ER tubule-sheet or ER sheet-sheet junctions. For clarity, the authors should state that they mean ER tubule-tubule junctions.
Same comment also for the statement "...although their abundance remains considerably lower than that of ER-ER junctions or nuclear pores at both cell cycle stages". The authors should state that they mean ER tubule-tubule junctions. We will clarify what we mean by ER-ER junctions in the revised manuscript. * In the Results section entitled "The constricted morphology of ER-NE junctions is observed in different mammalian cells, but not in budding yeast", the authors state "...pancreatic islet cells (Figure 5A), HeLa (Figure 5B), and macrophage (Figure 5C) were significantly smaller than most ER-ER junctions (Figure 5F)". The last figure reference here is wrong and should be changed to Figures 5D-E. We will correct this error. * In Discussion, the authors state "Proteins known to form and stabilize junctions in the ER, including Atlastins and Lunapark...". The authors should specify that they mean ER tubule-tubule three-way junctions. Also more generally throughout the manuscript, the authors should be more careful in specifying which ER-ER junctions they mean in each case.*
As pointed out in the Major comment 3 above, we will clarify this point in the revised manuscript.*
- In Discussion, the authors state "Thus, we favour a second scenario in which ER-NE junctions are generated from ER tubules that contact and eventually fuse with the ONM". Given that the ER membranes adjacent to the NE are mostly sheet-like (as pointed out in Major comment 1 above), the authors need to explain how they think an ER tubule (mostly found in the cell periphery) could access and fuse to the NE. As mentioned in the response to Major comment 1 above, we will examine if the ER that forms ER-NE junctions is tubule or sheet in our EM tomograms. Depending on the outcome of the examination, we will rephrase the text.
*
* Reviewer #2 (Significance (Required)):
Although the ER-NE junction has been studied in other organisms before, this study represents the first structural characterisation of the ER-NE junction in mammalian cells. Therefore, this study represents an advance for the field in gaining a better understanding of different ER structures and morphologies. How the ER is remodelled during the cell cycle is also an interesting question and an active field of research (Merta et al., 2021 PMID: 34853314; Zhao et al., 2023, PMID: 37098350) which this study further contributes to. This study would therefore be interesting for anyone interested in ER structure/morphology, ER-NE connections, and cell cycle regulation of such ER-NE connections.
My field expertise is in ER and NE. I do not have sufficient expertise to evaluate the methodology for the EM tomography part of this paper. We thank the reviewer for appreciating the novelty and the impact of our work.
*
*
*
* Reviewer #3 (Evidence, reproducibility and clarity (Required)):
The manuscript by Bragulat-Teixidor et al. is a study of the connection of the ER with the nuclear envelope. It uses advanced ultrastructural techniques: high pressure freezing instead of chemical fixation and EM tomography instead of serial sectioning. Synchronized HeLa cell cultures were examined during interphase, late anaphase (4-6 min after anaphase onset) and early telophase (8-10 minutes after anaphase onset).
The investigators find an unexpected, unusual structure - a constricted neck 7-20 wide and about 10 nm long where the ER connects to the nuclear envelope. The 7 nm connections had no apparent lumen. These are not seen in late anaphase when the NE has not yet formed, but they are seen a few minutes later during early telophase when there is a newly formed NE surrounding the chromosomes. A quantitation was made of their abundance, more was found later during interphase, and with wider lumens.
It is very nice to show the EM images as uncolored and segmented (colored). The images shown in the figures are presumably the best that were obtained during the study. Heavy metals do not stain membranes uniformly or exclusively, and identification of structures doesn't always seem unambiguous. The three dimensional information can certainly make this easier though this information is difficult or not possible to show in journal format. In the end, the reader must depend on the judgment of the person who did the analysis. Overall, the analysis seems trustworthy. *We thank the reviewer for the comment. To better present the three-dimensional structure of ER-NE junctions, we will provide movies of the EM sub-tomograms containing the junctions. In this way, the readers will be able to inspect the three-dimensional structure of six ER-NE junctions.
* HeLa cells are very convenient for getting information on cell cycle dependence. However, they are cancer cells in culture, so it is important to look at other cell types as well. The same methodology was used on budding yeast and they saw a wide tentlike connection, which reproduces an earlier study. This seems more consistent with what is known or expected from ER membranes. It is not less interesting but perhaps less puzzling. To get evidence on other mammalian cells, the authors did an analysis of data from OpenOrganelle. These are high pressure frozen cells / tissue imaged by FIB-SEM. The voxels are 4 nm, which is significantly larger than those in EM tomography. Unfortunately, the difficulty of identifying structures is correspondingly more significant. The images shown do not contradict the HeLa results but by themselves (without the HeLa cell data), a convincing case for narrow connections probably couldn't be made. *The reviewer raises a very good point about a limitation of the FIB-SEM datasets in OpenOrganelle. We agree with the reviewer that, as we had mentioned in the manuscript (line 6–11, page 10), the spatial resolution of the FIB-SEM datasets are not enough to gain insights into the exact morphology of the 7–20 nm wide ER-NE junctions because the voxel size is 4 nm. However, the resolution is good enough to examine if ER-NE junctions are narrower than ER-ER junctions, as shown in Figure 5A–E. The fact that we rarely found non-constricted ER-NE junctions in FIB-SEM datasets confirms the tiny nature of ER-NE junctions. To clarify this point, we will modify the text (line 24–25 on page 10) as below:
Previous: This analysis of FIB-SEM images confirms the hourglass morphology that distinguishes ER–NE from ER–ER junctions as seen in our EM tomograms…
Revised: This analysis of FIB-SEM images confirms that ER-NE junctions are narrower than ER-ER junctions as seen in our EM tomograms…
* The work in this manuscript seems to have been done well. Assuming that this structure is confirmed in other mammalian cells, another kind of question comes to mind: is this the final word on ER to NE connections? The lumenless neck does not seem like it would be a stable structure, somehow it seems like a transient one. In the future, it would help if a new structural protein was identified or some theoretical analysis to help explain the shape. *Certainly, this will not be the final word on ER-NE junctions, which are crucial for the ER-to-NE transport of lipids and transmembrane proteins. In the future, it will be important to identify structural proteins regulating the junctions and reveal how their constricted morphology affects the ER-to-NE transport. We believe that, as you kindly mentioned in the last paragraph of your comments, our observations “serve as a starting point for further structural and functional work” for this unique yet fundamental junctions that connect the ER to nucleus.
* It is generally now assumed that high pressure freezing preserves structure perfectly. However, in this reviewer's mind, there is a possibility that some structures are not. The sample is brought to 2000 atmospheres within a few milliseconds, frozen, then the high pressure is released after a second. Although many intracellular structures do seem well preserved, could the junction be susceptible to high pressure? A second source of uncertainty is that in order to embed the samples in resin, the water was removed by freeze substitution. This is known to cause a small amount of tissue shrinkage and possibly could alter a delicate structure. Another way to look at this kind of structure is cryo-EM tomography on hydrated lamellae from plunge frozen cells. I don't recommend that the authors do another arduous, possibly too arduous set of experiments with a completely different technique, but perhaps another group has data which could support their findings. *We think it is very unlikely that ER-NE junctions were deformed due to the high-pressure freezing. In general, high-pressure freezing allows vitrification of specimens up to 0.5 mm in thickness and the vitrification works better for thinner specimens. Our specimens are only 0.02 mm thick monolayer cells frozen in a chamber with 0.03 mm depth. Thus, the vitrification is expected to occur fast and the ER-NE junctions must have been frozen in the same way as in other regions of the cell.
However, as the reviewer pointed out, it is possible that the dehydration of the samples due to freeze substitution might cause deformation in ER-NE junctions. To verify the structural preservation of ER-NE junctions in our protocol, we will compare the morphology of the ER and NE in cryo-EM datasets that are available in public databases with ours. We will describe the outcome in the revised manuscript.
We think that our conclusion from the EM analysis is solid, because we observed significant structural difference between ER-NE junctions and ER-ER junctions in the same cells (Figure 2). In addition, we found the morphology change of ER-NE junctions in late-anaphase, early-telophase, and interphase cells that were high-pressure frozen and freeze-substituted on the same sapphire disc, and found that the ER-NE junctions became progressively constricted from telophase to interphase (Figure 3).
* The following are suggestions for the Discussion:
Yeast have many of the same biochemical processes as mammalian cells. Perhaps their lack of narrow connections can be used as a clue to the function of the narrow necks seen in HeLa cells. For instance, the authors speculate that the narrow connection serves to keep phosphatidylserine in the nuclear envelope low. If the yeast nucleus has the same concentration of phosphatidylserine as the ER, it would provide good evidence for this idea. Yes, it is indeed the case. It was shown that the yeast outer nuclear membrane has the same concentration of phosphatidylserine as the ER (Tsuji et al., Proc. Natl. Acad. Sci. U. S. A.*, 2019). We had described this in the discussion on page 14 “this phosphatidylserine enrichment occurs in mammalian cells and not in budding yeast (Tsuji et al., 2019)”, which was probably overlooked by the reviewer. In the revised manuscript, we will rephrase the text to make this point clearer.
* There might be other instances of lumenless neck structures. Dynamin mutants can cause a stable constricted tubule - are the dimensions of this tubule similar to that of the ER / NE connections? Or possibly some ESCRT related structure? These are very interesting questions. As shown in Figure 2A-D and Supplementary Figure 1B, the inner diameter (an inner leaflet distance) of the lumenless ER-NE junctions is below 1 nm. In contrast, the inner diameter of most constricted membrane tubules that the dynamin mutant K44A Dynamin 1 generates is 3.7 nm (Antonny et al., EMBO J., 2016, doi: 10.15252/embj.201694613). The inner diameter of membrane tubules that ESCRT-III subunits CHMP1B and IST1 form is 4.4 nm (Nguyen et al., Nat. Struct. Mol. Biol.*, 2020, doi: 10.1038/s41594-020-0404-x). Thus, the lumenless ER-NE junctions is unique in their highly-constricted nature and might be regulated by proteins other than dynamin or ESCRT proteins. We will discuss this point in the revised manuscript.
* There do not seem to be any recent studies of the ER / nuclear membrane connection in fixed cells. However, there is serial section data online which can be inspected. There are connections in mouse brain cortex in the data of Kasthuri et al., 2015 (https://neurodata.io/project/ocp/). Instead of a tubule connection, there seems to be a narrow sheet of ER that connects to the nuclear envelope. But there is something odd about these too. The authors may like to mention something about this or similar work in their manuscript. This reviewer has looked at chemically fixed data from several cell types from his own unpublished data and connections are surprisingly hard to find. Possibly, the connection is particularly sensitive to chemical fixation.* We inspected the serial section data of mouse brain cortex that was chemically fixed. The nuclear envelope in this dataset is deformed and does not seem well preserved. We do not think that we can extract useful information on the ultrastructure of ER-NE junctions from this dataset, and thus will not mention this work in our manuscript.
It is great to hear that the reviewer tried to look for ER-NE junctions in their own EM data. The frequency of ER-NE junctions is rare (only 0.1 junction per square micrometer, Figure 4). Thus, we think that the reason why it was hard to find the junctions in the reviewer’s data is due to the low-frequent nature of this junction and not due to the chemical fixation.
- *
* Reviewer #3 (Significance (Required)):
This is a careful and thorough study of the connection between the ER and the nuclear envelope. The discovery of reticulons and similar proteins, along with biophysical modeling, made the form of the ER accessible to analysis. The factors that govern ER structure are now much better understood. This is particularly true of sheets versus tubules, the three way tubule junctions and to some extent, the junction of ER tubules coming out of the edge of a sheet. However, with all this activity, the subject of the connection of the ER to the nucleus has not been examined in detail. What makes it different is that the tubule is connected perpendicular to the plane of a sheet.*
We thank the reviewer for appreciating the quality and novelty of our work.
* The manuscript uses the best ultrastructural techniques and provides strong evidence for a narrow neck at this connection in HeLa cells. With the same methodology, yeast cells (S. cerevisiae) have a wider connection. OpenOrganelle data from other mammalian cell types was examined. This data has less resolution and although it does not contradict the HeLa cell data, it does not support it strongly. *As mentioned in the response to one of this reviewer’s comments above, the spatial resolution of FIB-SEM datasets is good enough to examine if ER-NE junctions are narrower than ER-ER junctions. We think that our observation of several mammalian cells in FIB-SEM datasets strongly supports the conclusion that ER-NE junctions are narrower than ER-ER junctions and extends our findings in HeLa cells to two other mammalian cell types.
* This work is of interest to cell biologists specializing in membranous organelles or those interested in nuclear physiology. The connection of ER to nuclear envelope is an interesting problem that has not been studied recently. This manuscript could very well serve as a starting point for further structural or functional work by the authors or other groups. *We thank the reviewer for appreciating the significance and impact of our work.
*
Reviewer #4 (Evidence, reproducibility and clarity (Required)):
Summary: Membrane bound ribosomes and ER exit sites are present in the cytosolic side of nuclear envelope (NE), suggesting that NE shares protein translocation, folding and quality control functions with the endoplasmic reticulum (ER). Moreover, membrane continuity between the ER and outer NE membrane is evident, and, thus, NE is considered as a subdomain of the ER. To support this, during cell division, NE loses its identity, and participates to daughter cells as part of the ER. However, NE has also membrane proteins and luminal proteins that are enriched to NE and absent from ER during interface, and the segregation of NE specific proteins/lipids occurs concomitantly with NE formation during late anaphase/telophase. In this study, the ultrastructure of the ER-NE junctions is described using high resolution electron tomography. Results show convincingly a specific constriction at the ER-NE neck during interface in several mammalian cell types. This structure is absent during metaphase, and also from the budding yeast. Authors present a model for the formation of ER-NE junctions in higher eukaryotes and speculate about their functional role. *We thank the reviewer for the appreciation of our work and the valuable suggestions for further improvements.
* Major comments: The main conclusion of the paper is that although the ER and outer NE membranes are continuous, a specific hourglass shaped constriction at the neck is found in higher mammalian cells during interphase. The structure is specific to ER-NE necks, as it is absent during metaphase and ER-ER junctions. For the analysis, authors used high pressure freezing to ensure best structural preservation. Unfortunately, fixation is not the only potential source of artifacts; during tomography at ambient temperature, the thinning of the plastic sections under the beam can be up to 30%. In evaluation of the results, authors should consider how this thinning could affect the measurements of membrane distances and luminal width, and what type of distortions may happen as a consequence of asymmetric shrinkage.*
In addition to analysis of own samples, authors took advantage of the publicly available whole-cell datasets in OpenOrganelle and used these datasets to expand the number of cell types analyzed. Moreover, the 3D-datasets were generated with different imaging technique, FIB-SEM. Although this technique provides lower resolution in general, it provides isotropic resolution, and the data could be used to eliminate the shortcomings of the tomography, thinning of the sections and the missing wedge. The authors could expand the comparison of the data from these different sources from this perspective, especially since HeLa cells were used in their own tomography studies and FIB-SEM datasets in OpenOrganelle. Similarly, it would be interesting to see if similar approach could be used to compare their results to those obtained by cryo-EM by utilizing the cryo-EM database. Have authors checked if any suitable datasets for analysis of ER-NE junctions could be found from public archives? For the analysis of mitotic cells, double thymidine block was used to synchronize the cell culture. It is not clear, why synchronization was necessary, as CLEM was used to select the cells, and their number was rather low. Do cells continue growing and synthesizing new proteins during thymidine blocks? As one way to control potential artifacts due to the synchronization treatment, authors could compare the average thickness of ER and NE in naturally occurring interphase and mitotic cells vs. synchronized cells. We agree with the reviewer that it is important to clarify the degree of shrinkage and deformation of the sample that our EM protocol might introduce. To access the degree of sample shrinkage and deformation in the plastic sections, we will compare the ONM-INM distance measured in our plastic sections with the one in cryo-EM tomograms of rapidly-frozen and FIB-milled mammalian cells that are publically available (EMPIAR, the Electron Microscopy Public Image Archive, https://www.ebi.ac.uk/pdbe/emdb/empiar/), and describe the outcome in the revised manuscript.
The reason why we synchronized the cell cycle is to enrich cells in late anaphase and early telophase in the same plastic sections, so that we can compare their ultrastructure side-by-side. In the revised manuscript, we will examine if the double thymidine block affects the ER-NE junction morphology by comparing the morphology of the ER and NE between the synchronised and non-synchronised cells.
As we described in the response to Reviewer 3, we think that our conclusion from the EM analysis is solid because of the following reasons. (i) We observed a significant structural difference between ER-NE junctions and ER-ER junctions in the same cells (Figure 2). (ii) We discovered a morphology change of ER-NE junctions in late-anaphase, early-telophase, and interphase cells that were freeze-substituted on the same sapphire disc; the ER-NE junctions became progressively constricted from telophase to interphase (Figure 3).
Minor comments: On page 5, last chapter (+ Fig.1 legend and materials and methods): "the quick tomograms covered the entire NE" is misleading, as the imaging covered a thin layer of the entire NE only. - Authors could have analyzed the entire NE from the FIB-SEM datasets but chose to use stereological approach to minimize their work.
We will modify the text to make it clear that the quick tomograms covered the NE in a section and not the entire NE of the cell in the revised manuscript.
* To save time from the readers to follow the reference, authors could describe how the specimens used in OpenOrganelle datasets were fixed and processed, especially as they emphasize the importance of high pressure freezing in their own sample prep. Similarly, in Fig.4 legend, authors refer to measurements done in the previous study without explaining how and from what type of data. *We thank the reviewer for pointing these out. We will describe how the OpenOrganelle datasets were generated and how the nuclear surface area measurement was done.
- *
Is there a difference between mesh generation and segmentation, or is it just two different terms used for the same thing by different programs? We apologize our short description of these terms. We will clarify these terms in the revised manuscript.
*
Reviewer #4 (Significance (Required)):
General assessment: ER-NE gates were described earlier in the literature for specific cell types using standard thin-section TEM imaging, and in this study, the analysis was done with modern technology at 3D. The text is fluent and clear, and the quality of the images was excellent. The analysis of the data was thorough, and materials and methods including image analysis part were presented accurately and clearly. Ultrastructural analysis was done systematically, and generated models are beautiful and informative. Much thought has put into planning of the experiments and experimental approach. The shortcoming of the study is its limitation to ultrastructural analysis only without attempts to connect to any mechanism. The discussion part contains lot of speculation of the factors that might be needed for the formation and maintenance of the constriction and present several hypotheses for the function of the constriction. The paper would be much stronger if one of few of the leads would be followed, and if there would be any explanation for the role of these structures, or factors affecting them. *We thank the reviewer for the appreciation of the clarity and quality of our work. The molecular mechanism that regulates the function, shape and biogenesis of ER-NE junctions will be the subject of future studies, for which our discovery of a highly-constricted morphology of the ER-NE junctions lays the groundwork.
* Advance: The paper provides a very nice example for the reuse of publicly archived imaging datasets to complement own experimental work. Hopefully this paper encourages others to the same path, as the large volumeEM datasets require significant investments and contain wealth of potential for reuse. *We strongly agree with the reviewer. The volume EM datasets that are publically available contain wealth of potential for new discoveries. We also hope that our paper encourages other scientists to make good use of those datasets and also to deposit their own data to the public databases. We will deposit our EM tomograms to EMPIAR, the Electron Microscopy Public Image Archive.
* The paper strengthens the description of the ER-NE junction structure significantly and convincingly but does not further our understanding of the mechanisms behind the structure nor the function of them and raises more questions than provides answers. For structural analysis of this kind, the state-of-the-art technology is cryo-EM (e.g., preparation of lamella with cryo-FIB-SEM followed by cryo-tomography), and in this study, the technical limitations come from plastic embedding and ambient temperature imaging. The used techniques would be more adequate for cell biological study, where the described structure is somehow connected to the function in cell, or the factor(s) needed to the formation or maintenance are identified. *Indeed, a limitation of our current study is that we did not reveal the underlying molecular mechanism and the functions of the constricted morphology of ER-NE junctions. We do not think that cryo-EM is necessarily required because we have collected evidence that the ER-NE connections are distinct from the ER-ER junctions in not only our EM tomography data (Fig. 2) but also in the EM datasets deposited in public databases (Fig. 5).
* Audience: This study will be of special interest to cell biology community. The study could be an opening to several lines of research, e.g., identification of the factors forming or maintaining the structure, the potential function of the structure, how the structure affects the dynamics of the NE/ER membrane and luminal proteins. *We thank the reviewer for appreciating the impact of our work.
* Reviewer's expertise: The reviewer has long experience in electron microscopy, volumeEM techniques and image analysis, and operates mainly in the field of cell biology.*
-
Note: This preprint has been reviewed by subject experts for Review Commons. Content has not been altered except for formatting.
Learn more at Review Commons
Referee #1
Evidence, reproducibility and clarity
The mechanisms that differentiate ER from the nuclear envelope (NE) remain to be fully elucidated but likely depend at least in part on junctions between the ER and NE. How such junctions are formed and maintained is the subject of this manuscript where extensive correlative light and electron microscopy is used to observe and characterize ER-nuclear envelope (ER-NE) junctions at distinct phases of the cell cycle. The authors make use of their own electron tomography data as well as publicly available focused-ion beam scanning electron microscopy (FIB-SEM) datasets to compare the morphology of these junctions in different human cell types as well as in budding yeast. The major finding is that ER-NE junctions in human cell lines are more constricted than ER-ER junctions, often to the point of excluding lumen. The examination of mitotic cells suggests that this constriction likely occurs at the end of mitosis as the NE is completing its maturation from ER to NE. The implications of these morphological changes are discussed but there are no mechanistic or functional studies. Overall, the data are well presented, are of high quality and are rigorously evaluated. The manuscript is well written and scholarly, and the speculations as to the function of the constrictions are reasonable. I only have minor comments.
- In Figure 2D, the authors present evidence to demonstrate that an hourglass-like constriction occurs at ER-NE junctions. From the side view, it is difficult to interpret this on the plot, particularly for the ER-NE junctions with a lumen. Perhaps, in the supplemental data, the authors could plot both with and without lumen data separately, and color-code individual traces? I believe this would convey the hourglass nature of these constrictions more clearly.
- In the Methods section, the authors should describe how carbon-coating of sapphire discs was achieved. If these were provided from the manufacturer precoated, this should be specified.
- On page 10, Figure 5F callout 9 lines from the bottom likely should be 5E.
Significance
Overall, this work provides an important new morphological perspective on the nature of ER-NE junctions in human cells. As the authors describe in their introduction, such junctions have been noted previously in the literature but not in a dedicated study using modern imaging techniques in human cell lines. In describing the morphology of these junctions, the authors lay the groundwork for future mechanistic, functional, and structural studies.
-
-
www.biorxiv.org www.biorxiv.org
-
Note: This preprint has been reviewed by subject experts for Review Commons. Content has not been altered except for formatting.
Learn more at Review Commons
Referee #3
Evidence, reproducibility and clarity
Summary
In their manuscript, Tetzlaff et al. report a substantially improved protocol for the isolation of mitochondria from the parasitic apicomplexan Toxoplasma gondii, which allowed improved sequencing and in-depth analyses of the organism's peculiarly complex mitochondrial genome. Follow-up small RNA-sequencing made it then possible to confirm the expression of fragmented mitochondrial ribosomal RNAs (mt-rRNAs) and to identify a dozen new RNA species of unknown function. The authors document not only multiple Toxoplasma mitochondrial genes that overlap one another-including rRNA and protein-coding genes, otherwise a rare occurrence-but also show that some fragmented rRNA genes recombine, effectively leading to multifunctional sequence segments, another rare feature and consequence of the peculiar architecture of the organism's mitochondrial genome. Lastly, the authors confirm that products of three genes presumed to encode pieces of the highly fragmented mitochondrial large subunit (mtLSU) rRNA do indeed assemble-presumably with additional components-into large molecular-weight complex(es).
Major comments
Key conclusions of the manuscript are that Toxoplasma's mitogenome encodes overlapping rRNA and protein-coding genes, divergent and chimeric rRNA pieces, and several small RNAs (sRNAs) of unknown function. Provided evidence is very solid for certain aspects of the study, but objectionable for the others as detailed below.
- The extent of the presented analysis of rRNAs and unassigned sRNAs seems lacking. In several places of the manuscript, the authors wonder about potential implications of divergent rRNA sequences, but their analyses appear to have been limited to sequence similarity searches. Had modelling of secondary structure interactions been attempted, this conundrum could potentially be solved. Importantly, similarity searches (to conventional rRNAs) were performed using BLASTN, which is a rather crude tool for the purpose, instead of covariance models/HMMs. It is therefore not entirely surprising that some sRNAs remained unassigned. Admittedly, recognizing rRNA motifs in divergent RNAs is a challenging issue. However, it is important to not conflate similarity to conventional rRNA and the molecule's functionality as an rRNA, i.e., sequence divergence does not necessarily disqualify the unassigned sRNAs as potential rRNAs. Mitochondrial rRNA sequences are among the most divergent, often constrained only by base-pairing, if at all, as has shown the research on kinetoplastid and diplonemid mt-rRNAs, which contain very few conserved elements and very few base pairs (e.g., Ramrath,2018,Science & Valach,2023,NAR). Even in generally less divergent cases such as green algae, the fragment encoding a highly divergent and derived 5S-like rRNA has only been recognized as such only after the mitoribosome structures were determined (Waltz,2021,Nature Comm & Tobiasson,2022,Nature Comm). It would not be surprising if the same was the case for Toxoplasma's fairly quickly evolving mitochondrial genome.
- The discovery of overlapping protein-coding and rRNA genes is intriguing, but the authors do not explain why it should be considered as fundamentally groundbreaking as the 'Abstract' and 'Discussion' make it sound. Gene overlaps are found in mitochondria of many organisms (e.g., fungi, animals, various protists), especially of tRNA and protein-coding genes. Even in Plasmodium, a rather close relative of Toxoplasma studied in the presented work, LSUB (rRNA) gene overlaps cob (protein) gene in the antisense orientation. Admittedly, the extent of the overlaps in Toxoplasma does seem fairly high at a first glance, but it is necessary to provide more data and, importantly, broader context to make the case that Toxoplasma overlaps are in any way special. For instance, what is the average size of the overlaps? What is their cumulative size? How does their extent (i.e., the size of overlapping coding sequences compared to the total length of coding sequences) compare to gene overlaps in other (mitochondrial) genomes? Certain additional aspects of the analysis and interpretation of protein- and/or rRNA-coding sequence overlaps are somewhat underdeveloped. For example, are the RNA-coding regions that overlap protein-coding sequences more divergent in those three conserved proteins compared to other organisms, i.e., does their function as rRNA take precedence, or is the converse the truth, i.e., are the rRNA sections more divergent? RNA19 (overlapping coxIII and cob) is the only example discussed in depth, but at least a short sentence summarizing the overall picture would be useful. As for the authors' interpretations, proposed formation of sRNA:mRNA hybrids, through which sRNAs could by implicated in facilitating mRNA recognition by the mitoribosome, is an interesting hypothesis, but a simpler scenario, which is given very little space, is that the genes happen to overlap by chance and that the overlaps are merely a consequence of genome compaction (a phenomenon that the authors rightly highlight). Without a comprehensive analysis, it is impossible to conclude which possibility is more likely. For instance, if both protein-coding and non-protein-coding sequences are divergent, this would indicate that there are few evolutionary constraints and so the fact that these sequences overlap means very little and might be just due to neutral drift, an effect of genome compaction without much consequence for the organism. Lastly, considerable significance is attributed in the study to the presence of antisense overlaps, especially between rRNA- (or sRNA-) and protein-coding genes. Yet, the overall extent of sense and antisense overlaps in the Toxoplasma mitogenome is quite similar, which-again-seems to point to a neutral evolutionary process. Can the authors elaborate if this aspect of the genome architecture was taken into account and if they regard it as of lesser relevance (and why, if so)?
- Another controversial issue concerns prevalent sequence block combinations and their impact on mitochondrial gene expression regulation. The authors postulate that 5′-terminal blocks of protein-coding genes always occurring near other protein-coding blocks has some functional significance. However, concluding this from just two cases (even if out of two) is quite speculative and seems like reading too much into a pattern that could very well be due to chance alone. The authors argue that the fact that 5′ ends of coxI & coxIII genes overlap is another indication of potential gene expression coordination. While it is possible to envisage such a regulation because of the 5′ termini proximity, the overlap between these genes means that their connection is hardwired into the genome, making it difficult to compare this particular case to the other sequence blocks. Arguably, it is tempting to speculate that an evolutionary pressure exists to coordinate protein expression and such a coordination does not indeed seem implausible, but the presented data and arguments are not convincing. The authors should at least expand on their ideas in the 'Discussion' and indicate potential experiments and/or which additional data could support (or refute) their speculation.
- My last major point concerns the experimental examination of large-molecular weight complexes and the interpretation of its results. To prove incorporation of the sRNAs into the mitoribosome, i.e., confirm that they do indeed represent rRNAs, the authors opted to investigate their distribution across a sucrose velocity gradient. This is a relatively simple and powerful approach and although it does not provide an irrevocable proof, it can be used to gain very useful insights. However, the presented design has critical flaws: 1) all sRNAs selected for Northern blot were mtLSU components, so only the mtLSU would be detected; 2) a single cytosolic LSU component was used as the control, so the distribution of cyto-SSU subunit, cyto-ribosome, and cyto-polysomes is actually unclear; 3) the authors' interpretation relies on the assumption that both mitochondrial and cytosolic ribosomes preserve their association as polysomes, but no relevant control is provided for this. For example, in Figure 6, fractions 6-14 clearly contain cyto-LSU, but polysomes (e.g., disomes) might just as well start in fractions 12-14; without additional controls, or at least continuous monitoring of UV absorbance across the gradient (to show a typical polysomal pattern), it is not guaranteed that what was detected actually included cyto-polysomes. The main concern, however, is the migration of mitoribosomes. First, the authors presume that the fractions 7-8 contain the mitochondrial monosomes because they are the fractions closest to the gradient top. This is not guaranteed. In fact, based on the experience of our and our colleagues' labs and taking into consideration the conditions used for the described experiment (more precisely, the use of Triton and deoxycholate, which in many organisms lead to mitoribosome subunit dissociation), it seems quite likely that fractions 7-9 actually contain separated mtLSU, not monosomes. Fractions in higher sucrose concentration would then represent monosomes and possibly assembly intermediates, though perhaps also a minor polysomal fraction (if the interactions are preserved in the conditions used). In particular, if the assembly process in Apicomplexa is as complex as in Euglenozoa (e.g., see papers on kinetoplastid mitoribosomes Saurer,2019,Science & Tobiasson,2021,EMBO Journal), which does not seem unlikely in Toxoplasma given the necessity to incorporate ~15 distinct rRNA pieces per mitoribosomal subunit, then the assembly intermediates might form ribonucleoprotein complexes that migrate quite far into a sucrose gradient (e.g., as in kinetoplastid mtSSU, Maslov,2007,Mol Biol Parasit). Thus, while it can be reasonably well argued that the detected RNAs co-migrate with the mtLSU (and possibly mito-monosome), the claim that they associate with mito-polysomes is open to question. More critically, investigating only sRNAs that are clearly identifiable as rRNA pieces-and all from the mtLSU at that-does not automatically prove that all sRNAs associate with the mitoribosome. To argue that the unassigned sRNAs are associated with mitoribosomes, northern blots of as many as possible (but at the very least one) unassigned sRNAs are absolutely necessary. However, I encourage the authors to consider performing additional experiments to address the issues raised in the preceding paragraph: for example, a western blot of mitochondrial ribosomal protein(s), a northern blot with at least one mtSSU rRNA fragment (since all three shown are from mtLSU), as well as a test that would examine the influence of detergents on mitoribosome stability (e.g., use milder detergents such as digitonin or dodecylmaltoside). Furthermore, if experimental conditions are identified allowing subunit dissociation, it would be possible to discern to which subunit which sRNA belongs and, importantly, whether the unassigned sRNAs are just "disguised" rRNAs (simplest explanation) or something completely different (speculative explanation seemingly favoured by the authors). All this would substantially boost the significance of the presented work.
Minor comments
General comments
The word "novel" is rather overused in the manuscript. At several places, it is inappropriate, as the presented results are not as unprecedented as the manuscript makes them sound; at other places, it might be acceptable, but as the word's meaning is vague, the text would benefit from using more informative term(s) instead. The former case is exemplified by the sentence at the lane 102 "Here, we present a novel method for enriching organellar nucleic acids" - "novel" does not simply mean "new", but alludes to "unprecedented"; yet, the devised method, albeit clever, is a modification of existing approaches. The sentence at the lane 182 illustrates the latter case where "novel blocks" are mentioned, but "previously not detected blocks" would be more appropriate and to the point. The labelling of 5′ and 3′ is inconsistent throughout the manuscript - sometimes the prime is used, sometimes the apostrophe, sometimes it is the single quotation mark.
Abstract
In light of the raised concerns, the authors should consider carefully rewording this section, as some of the formulations are mis-representing the data and lead to unjustified generalizations.
Introduction
lanes 72-73: "How rRNA fragments are assembled into functional ribosomes remains an enigma." - Without proper context, this statement feels like an exaggeration. Fragmented rRNAs are known from other species and their mitoribosome structures were determined in the past few years (i.e., Tetrahymena, Polytomella, Chlamydomonas). Arguably, these mt-rRNAs are not as fragmented as in Toxoplasma, but at the very least, it is clear that base-pairing of rRNA pieces and RNA-binding proteins play significant roles in the process. If the authors think that this is not the case in apicomplexans, this should be at least alluded to, if not explained. l. 80-83: The paragraph mixes information on Plasmodium and Toxoplasma. To a non-initiated reader, this can be quite confusing. It would be useful to specify which species the authors refer to. l. 83-86: The information on the atovaquone impact lacks reference(s). l. 105: "demonstrated that they are incorporated into polysomes" - In light of the issues raised above and if the authors opt not to expand the work as suggested above, this claim (and similar throughout the text) should be emended. l. 106-108: "allowed us to identify novel transcripts, many of which originate from block boundaries and contain mixed origins from coding and noncoding regions." - This sentence would benefit from rephrasing because it is difficult to comprehend (the sequences overlap protein-coding and non-protein-coding regions, but do not contain any origins).
Results
l. 115-117: "cell fractionation method that takes advantage of the differential cholesterol content in plasma membranes" - Does Toxoplasma contain cholesterol? Perhaps it might be more practical to refer to sterols (since the effect of digitonin is not limited to cholesterol). l. 147: "significant increase" - It might be useful to specify that the increase was ~42-fold, so that readers can see the extent of improvement; it has the advantage of really highlighting the achievement. l. 180: "have been lettered from A-V" - Rewording to "designated by letters from A to V" works better. l. 213-218: This section is essentially a discussion so should be moved the corresponding section of the manuscript. l. 262-265: cotranscripts/transcript isoforms - It is a matter of nomenclature, but it seems more appropriate to refer to "a transcript containing LSUF and LSUG regions" instead of a co-transcript, because in the latter case, one then expects that these two will be separated in a following processing step, which-as the authors demonstrate-is clearly not the case for the vast majority of the population of these rRNA pieces. Given the prevalence of the larger pieces, it seems more appropriate to refer to the "smaller transcript isoforms" as possible degradation products and not isoforms, which implies some kind of functional relevance. l. 281: In the section "Discovery of novel rRNA fragments", it might be useful to provide a graphical representation or at least a sentence summarizing all different categories of sRNAs. For instance, what is missing from the text is that there are 11 species for which homologous sequences in "conventional" rRNAs were not identified and out of these only 4 seem to have sequence homologs in other Apicomplexa. In addition, in Table S5, the authors could indicate where these homologs are located in Plasmodium, since these appear to be newly identified candidates for Plasmodium sRNA species/rRNA pieces. l. 313-314: "In general, block combinations lead to the expression of novel RNAs in T. gondii that are not found in apicomplexan species with a simpler genome organization. " - It is not clear where this generalization comes from: Fig.S5A shows that RNA5, RNA7, RNA23t extend across block borders (but based on Table S5 are not unique to Toxoplasma), while only RNA31 and RNA34 are both absent from other Apicomplexa and extend across block borders - yet, this is still less than half of all newly identified sRNAs. In addition, the novelty claim is not clear either: based on the presented data, several sRNAs that overlap are clearly present in other apicomplexans (e.g., RNA1 and RNA2) and thus are not completely new, but merely more divergent in Toxoplasma, because parts of their sequence have been replaced by the shared sequence segment. l. 319-320: "None of the three RNAs had detectable homologies to rRNA." - Specify to which rRNAs were the sequences compared to make the inference. l. 320-321: "For all five coding-noncoding RNAs, homologs are present in the mitochondrial genome of P. falciparum." - Does this mean that they remain unassigned in Plasmodium as well or that they have not been previously recognized in Plasmodium? Confusingly, RNA34 is labeled as not having homologs in Apicomplexa in Table S5. In addition, mentioning "coding-noncoding RNAs" is somewhat misleading because some of the sRNAs clearly code for mt-rRNA pieces. l. 335-338: This section contains contradictory statements that should be reformulated. A couple of sentences prior, the authors experimentally determined that RNA19 actually overlaps only a single protein-coding sequence (coxI), but then refer to the original and demonstrably incorrect annotation of RNA19 overlapping also the cob gene. l. 341: The authors mention similarity to rRNA, but do not specify which rRNA. Referring to similarity to known or conserved rRNA sequences or segments would work better. Still, the region of the block S (i.e., 5′ proximal segment of RNA19) falls into the region between helices H51 and H60 of the domain III in the LSU secondary structure, which is sequence-wise relatively poorly conserved-especially in mitochondrial rRNAs-so sequence divergence is not unexpected. l. 366: "Note that RNA1 and RNA2 are registered according to their shared sequence" - Unclear what "registered" means here. l. 416-421: Specifying when reference is made to cytosolic vs. mitochondrial monosomes and polysomes would make this section and the related parts of the 'Discussion' clearer. Also, the authors clearly state here that there might be technical reasons for what they observed, but ignore this possibility in the 'Discussion' and assume that they did indeed separate polysomes.
Discussion
l. 444: "the reshuffling appears limited to specific block borders and is not random" - How many biological replicates of nanopore sequencing were performed? Did the authors test other T. gondii strains? What about other apicomplexan species? Unless this has been done, there is no demonstration that the block order and block-joining frequencies documented here are (more or less) constant and that block order is under some kind of purifying selection. Hence, the conclusion that the block borders are not random is debatable. Arguably, it is not random in this particular experiment, but neither is it limited to specific blocks because most combinations have been detected (even if at low frequency; Figure S1). l. 450: "One intriguing finding is the obligate linkage of coding sequences" - Presuming this sentence is about protein-coding sequences, this should be reformulated because it mis-represents the actual data. Figure 2 clearly shows that protein-coding blocks are often linked to rRNA-coding blocks. l. 454: "balancing the expression of coxI and coxIII" - Not clear where this information comes from, as it is not from the cited papers. l. 460-461: "Our small RNA sequencing results revealed another potential advantage of the block organization of the T. gondii mitochondrial genome" - This should be reformulated. Clearly, the discovery of the 15 sRNAs was facilitated by the recognition of block order, but the presented argument is a bit confusing: how does the organization into blocks provide an "advantage" and what kind of advantage do the authors mean? (An evolutionary advantage or an advantage related to gene expression regulation or an advantage for their sRNA-Seq data mapping?) l. 462-478: Multiple explanations are provided for the existence of sRNAs at block borders and what these sRNAs represent. While I agree that it is important to consider all options, even the more debatable ones, the authors seem to forget the simplest possibility: the identified unassigned sRNAs could well be rRNA pieces and them being encoded across block borders is not any more, nor any less surprising than the fact that protein-coding genes are encoded across (several) gene blocks. l. 485: "antisense RNA surveillance" - In contrast to the nuclei, the existence of a genuine antisense RNA "surveillance" mechanism in mitochondria is uncertain. Given what is known from mitochondria of other organisms (especially plants and kinetoplastids), it seems more likely that certain regions of sense and antisense transcripts are protected from exonucleases by RNA-binding proteins (RBPs such as PPR and related helix-turn-helix repeat proteins, e.g., Toxoplasma's homologs HPRs discovered in Plasmodium [Hillebrand,2018,NAR]), leading to RNAs that partially overlap, but are actually protected from base-pairing by these RBPs. This is not taken into account in any presented explanation of the phenomenon of antisense gene overlaps. l. 490: "start codon. while also " - Typo: should be a comma, not a dot. l. 500: "discovery of block-border sRNAs highlights the complex regulatory mechanisms at play" - This should be reformulated: the claim is very speculative, since no hard data are provided on such regulatory mechanisms in the presented work. l. 504: "sRNAs are incorporated into polysome-size structures" - In light of the concerns raised in the preceding section, this should be reformulated. l. 539-540: The closing sentence should be reformulated. The mitogenome organization in blocks per se does not "allow" the sequences to function as both mRNA and rRNA. Rather, it seems to be a combination of 1) the compactness of the genome that seems to lead to the re-use of certain segments in both mRNA and rRNA or in two distinct rRNAs, and 2) the apparently dynamic nature of the genome (due to recombination among gene blocks) that brings together certain combinations of gene blocks.
Methods
l. 607: Only agarose gel separation is mentioned, but most experiments shown are of denaturing PAGE separations (which is actually mentioned in several figure legends). l. 636: "Paste your materials and methods section here." - To be removed. l. 662: "NUMTS" - This should be "NUMTs"; the same typo occurs at multiple places in the 'Methods' section. l. 704: "Homology search for novel transcript annotation" - Somewhat confusing title; it is possible to guess what the authors likely mean, but it is unclear. l. 715: "New block annotations can be found in GenBank." - 1) The whole community would very likely appreciate if the GenBank entries were properly annotated (i.e., genes added), not just showed sequences as is currently the case for all Namasivayam,2021,Genome Res entries (not sure about the authors' own entries because they were inaccessible). If impossible to update the entries of the Namasivayam,2021,Genome Res study, then just submitting anew properly annotated GenBank entries would be appropriate. 2) It was not possible to properly assess some of the claims in the manuscript because access to the files was not provided to reviewers, nor have been the newly submitted GenBank entries made public by the authors.
Figures
Figure 1B - The load of total proteins into each well is unclear. Ponceau stain does not show identical loads, so it is unclear what the reader should take as the reference. Figure 1D -The phrasing "fragments found in the pellet fractions of the protocol" is a bit awkward. The fragments are in the pellet fractions after plasma membrane permeabilization and benzonase incubation, not in the "fractions of the protocol". Figure 2 - The chosen hues of red and green (for coxI and coxIII) are of such similar intensity that they are virtually indistinguishable to ~2% of the readers. A colourblind-friendly palette would be very much appreciated. For guidelines, see for example: https://www.nature.com/articles/nmeth.1618 . Figure 3 - The use of lowercase letters to indicate the probes (instead of the full probe names) is a nice idea and simplifies the reading experience, but the use of the same letter 'a' in different figures for different probes is confusing. Labeling each probe with a unique ID/letter and indicating this ID in the Table S6 (e.g., by adding an additional column) would work much better. Figure 4A - The wiggle lines for rRNAs are coloured in purple shades, which contrast with the grey colour that is assigned to them in the Figure 2. Keeping a consistent colour palette across figures would be preferable. Figure 4C - If the E.coli sequence was on the outer lines, the Toxoplasma sequences could be closer to one another, which would make it easier for the reader to understand the alignment. Figure 5 - Purple shades for rRNA are somewhat difficult to discern from the blue cob. Also, the 'reference' wiggles would work better if demarcated as a key because this would make it visually clearer that they are shared by the A and B panels.
Supplementary Information
Figure S1 - An explanation what the A and B panels show is missing. Figure S5 - It is difficult to appreciate the extent of overlaps with protein-coding sequences if these are missing from the figure (unlike in Fig.5). Table S4 - Nuclear genome accession number is missing. Add "mitochondrial" to the label of the column "sequence blocks". Table S5 - 1) It is unclear what the 'rRNA homology' refers to. (It does not seem to be the nomenclature used by Feagin et al.,2012, PLoS One.) 2) An extension of the table (or perhaps a separate table) with the cumulative size of mtLSU and mtSSU rRNA pieces, as well as unassigned sRNAs, would be useful. 3) It should also be stated somewhere if homologs of any of the rRNA pieces known from Plasmodium are missing in Toxoplasma. (If so, they could be among the newly identified short RNAs.)
Referees cross-commenting
Referee #2 rightly pointed out that basic statistics on nanopore reads, as well as omitted methodological details (e.g., minimap2 and SAMtools settings) would be welcome. Similarly, Figure 2 should indicate the upstream/downstream block orientation. If the authors intend to position their work as a major achievement in mitochondrial enrichment for Toxoplasma (as the text currently indicates), I also agree that a comparison with previously published protocols would not be out of place.
Significance
Speaking from personal experience, devising a protocol for such a substantial mitochondrial enrichment, as the study presents, is a great technical achievement, which cannot be understated, especially for a protist or any somewhat unconventional model organism. The mitoribosomal community will certainly take notice of the improved catalogue of mitochondrial rRNA pieces, while the discovery of overlapping protein-coding and rRNA genes will be of interest to those working in the field of mitochondrial evolutionary biology. The study already provides a significant upgrade from the previous attempts to understand the nature of the mitochondrial genome in Toxoplasma (and in Apicomplexa in general), and is well positioned to become a source of inspiration for future studies in the field. However, being at a crossroad of genomics, evolution, and molecular biology, it has certain limitations in its current form, mainly because the evolutionary and molecular biology aspects would benefit from further development (see 'Major concerns'). The text is generally well written and accompanying figures well designed, but clarifications, broader context, and less speculative interpretation would be welcome (as detailed mostly in 'Minor concerns'). To justify publication in a journal with a broad readership, the authors should provide additional experimental evidence to strengthen their case and generalize their findings.
-
Note: This preprint has been reviewed by subject experts for Review Commons. Content has not been altered except for formatting.
Learn more at Review Commons
Referee #2
Evidence, reproducibility and clarity
In this article, the authors delve into an intriguing topic, aiming to enhance our understanding of the organization of the mitochondrial genome of T. gondii, a parasite of significant importance in both human and animal health contexts.
In essence, their approach involves enriching mitochondrial material, followed by genome sequencing and the analysis of mitochondrial short RNAs. They achieve a remarkable depth of mitochondrial sequencing and generate valuable RNA data. Furthermore, their efforts lead to the discovery and annotation of new short RNAs.
Overall, the article is well-crafted and presents compelling results. However, it's worth noting that, at times, the authors appear somewhat self-congratulatory, and certain results might be perceived as overly ambitious. Nevertheless, the discussion is aptly constructed.
Major comment:
They assert certain discoveries that had already been reported. Notably, they adapt an existing protocol for mitochondrial enrichment and describe it as 'We developed a protocol to enrich T. gondii mitochondria.' It's worth noting that they neither reference a more recently described protocol (PMC6851545) nor compare the performance of their modified protocol with the original.
The protocol they employ does not seem to yield exceptionally high success rates, as mitochondrial DNA constitutes less than 10% of the total sequenced DNA.
Additionally, they frequently mention the identification of specific combinations of sequence blocks previously identified by Namasivayam et al. (PMC8092004), which was also discussed in Namasivayam et al. 2021."
Missing in the supplementary material are basic details on the sequences performed. Distribution of mitochondrial reads length, depth, etc.
Further clarification is needed for Figure 2. Specifically, the frequency units or combinations of frequency (A, B, and C) are not clearly explained. While the matrix's asymmetry suggests a 5'- 3' orientation difference, this orientation difference is not explicitly specified (B). Additionally, the fragment Mp does not appear in the block combination figure (C).
Some points to improve the introduction:
Provide an evolutionary context for the following phrase: 'An idiosyncratic feature of Apicomplexa is a highly derived mitochondrial genome.' Specify what you intend to emphasize.
Line56: The sentence must begin with a capital letter
In line 58 "Nuclear genes encoding proteins with functions in mitochondria contribute strongly to P. falciparum and T. gondii cell fitness" Although it is mentioned later, it would be more effective to introduce the fact that all but three genes are encoded in the nucleus.
Line68: "Apicomplexan mitogenomes usually code only for three proteins" It seems to me that 'usually' should not be included.
Line 65-67: The sentence should include that the mitochondrial genome is composed of a total of 20 blocks of repeating sequences organized in multiple DNA molecules of varying length and non-random combinations
At the end of the introduction, the authors state that they have developed a protocol for mitochondrial enrichment. The text should be modified taking into account that: 1- The new protocol is an adaptation of another existing protocol. In fact, the Methods the authors say the protocol was "slightly" modified. 2- There is already existing mitochondrial enrichment protocol available [Reference: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6851545/#mmi14357-bib-0074]. In any case, they should consider performing a comparative analysis between the proposed protocol and existing ones to determine its relative effectiveness. It should be noted that the proposed protocol enriches in organelles (including the nucleus and apicoplast), but when sequencing DNA, mitochondrial DNA accounts for only 5% of the total reads, which may raise doubts about its overall efficacy.
Some points related to Results section:
Lines 113-115: 'To distinguish between NUMTs (nuclear DNA sequences that originated from mitochondria) and true mitochondrial sequences, it is necessary to enrich mitochondrial DNA.' I disagree with this sentence. NUMTs, in general, consist of very short sequences. With long reads, it is relatively straightforward to differentiate mitochondrial sequences from those nuclear sequences that have small mitochondrial fractions. In my opinion, even many Illumina reads can be confidently identified as belonging solely to the mitochondria. I found this article that supports this argument, indicating that the majority of NUMTs are less than 100 nucleotides in length [Reference: https://pubmed.ncbi.nlm.nih.gov/37293002/].
Lines 166-168: 'A previous sequencing study used Oxford Nanopore sequencing technology (ONT) to identify combinations of sequence blocks in T. gondii mitochondria (Namasivayam et al. 2021).' However, it's important to note that Namasivayam's group did not merely use ONT to identify combinations of blocks; rather, they discovered, identified, and defined these combinations based on sequencing with long reads.
Line 177: "The length of mitochondrial reads ranged from 87 nt to 17,424 nt" It would be beneficial to include a histogram depicting the length distribution of the obtained reads. It's worth noting that nanopore reads tend to be shorter than Illumina reads
Line 194-195 "we found that only a small fraction of all possible block combinations are prevalent within the genome" this has been previously described (PMC8092004)
Line 201. "This indicates that the genome's flexibility is limited and that not all block combinations are realized". This is consistent with the findings published by Namasivayam et al. in 2021, which have already established that the combination of the 21 blocks is non-random.
Line 205: "All combinations are well covered in our ONT results and helped to refine block borders relative to previous annotations (Fig. S2)" In the supplementary materials the authors say: "However, the blocks Fp, Kp, and Mp frequently occur separately in the mitochondrial genome We therefore treated Fp, Kp and Mp as separate blocks and have shortened the blocks F, K and M accordingly". As far as I understand, for this very reason, Namasivayam and collaborators annotate them as partial fragments, which may appear in other regions but are, in turn, parts of larger F, K, and M fragments. To redefine the segments F, K, and M without the sequences corresponding to Fp, Kp, and Mp, as shown in Figure S2, these fragments should be distinct from the 'partials.' In other words, segments of the type (F minus Fp), (K minus Kp), and (M minus Mp) should appear in the reads, and should be distinguishable from Fp, Kp, and Mp. If this distinction is made, I am satisfied with the new definition.However, if such a separation is not evident, it seems important to clarify it in the text or to reconsider this new definition.
Lines 221-223: "This suggests that there is no need to postulate mechanisms of genomic or posttranscriptional block shuffling to arrive at full-length open reading frames." The authors argue that invoking mechanisms of genomic or post-transcriptional block shuffling is unnecessary to explain the presence of full-length open reading frames, given that genes represent 2-3% of mitochondrial sequences. However, there is a missing estimate regarding the probability of encountering all three genes within a single molecule or mitochondrial genome, as well as the total number of sequenced mitochondria. Consequently, the statement appears overly assertive. In the absence of alternative mechanisms for generating complete genes, this would mean that at most only 1646 mitochondrial genomes would have been sequenced. To comprehensively address this issue, the authors should consider discussing this scenario further. They should also provide information about how many reads they found containing all three genes and how many contained two of the genes.
Lines 249-250 "using the block combinations identified here by ONT sequencing " which is the difference between blocks identified here with those on Namasivayam ? The division of M, K and F fragments?
Line 287: "The six remaining small RNA fragments are specific to T. gondii" I would suggest being more cautious in this sentence by stating that they were not found in other organisms. Given the similarity of the mitochondrial genome between T. gondii, N. caninum, and other coccidians, it would be expected to find them in these organisms as well.
Line 300 "Among the novel small RNAs identified, there is also a class that was only detectable due to our insights into genome block combinations." A valid strategy is to map the small RNAs to the generated nanopore reads or to an assembly made with these reads, rather than solely relying on the single blocks or combinations of blocks, as this approach would yield the same result.
Line 444: "Upon closer scrutiny, however, the reshuffling appears limited to specific block borders and is not random" This was already established by Namasivayam et al 2021.
I would like to highlight the potential for a more comprehensive examination of the mitochondrial genome in the discussion. While the proposed explanations for the presence of sRNAs at the 'block borders' appear plausible, it's worth noting that the definition of these blocks is artificial rather than biological. I think it is interesting to discuss without the concept of block sequences, but of sequences existing in the mitochondrial genome. Therefore, it's important to discuss whether these sequences (the block borders) are consistently present in all mitochondrial genomes. The total cumulative length of the blocks is 5.9 Kb, which is relatively small and comparable to one of the smallest mitochondrial genomes on record. It is conceivable that recombination and the generation of new sequences play a role in expanding genomic space for encoding, such as RNAs.
Line 535-536 "We developed a protocol to enrich T. gondii mitochondria and used Nanopore sequencing to comprehensively map the genome with its repeated sequence blocks." I find this sentence to be somewhat assertive, especially considering that they modified an existing protocol and obtained results that may not be optimal. Additionally, they have not compared their protocol with other available methods for mitochondrial enrichment.
Some points related to Method section: In none of the experiments is it specified how many parasites were initially used as a starting point
"Masking NUMTs in the T. gondii nuclear genome" it's unclear whether the authors utilize all hits or filter the results of BLASTN. It would be helpful if they specify the criteria for filtering, such as identity percentage or query coverage. Additionally, it's not clear how they generate the GFF3 file from the BLAST results, or whether they instead create a BED file. Providing clarification on this process would enhance the reproducibility of their methods. Moreover, it would be beneficial if the authors include information regarding the number of sequences they intend to mask, the average length of the NUMTs, and the total percentage of the genome these masked sequences represent.
Line 657 "Mapping results were filtered using SAMtools"<br /> The text does not specify the filtering criteria or the parameters used for this process.
Line 673 establish "No matching reads were found" in the "Sequence comparisons of ONT reads found here with published ONT reads for the T. gondii mitochondrial genome" but in the results the authors say: "While smaller reads of our dataset are found in full within longer reads in the published datasets, we do not find any examples for reads that would be full matches between the dataset. Could you provide a more detailed explanation? Specifically, I would like to know how many reads from the dataset (including their length) are also present in other datasets, and at what minimum length do they cease to coincide?
689 - The text does not specify the filtering criteria or the parameters used for Samtools filtering process.
Lines 689-693 Please describe better the methodology used.
Line 696: the program is fastp not fastq (Chen et al. 2018)
Line 697: what do you mean only the ends of the reads were mapped? how many bases? Or do they mean that they map the reads fowrards and reverse reads?
Significance
In this article, the authors delve into an intriguing topic, aiming to enhance our understanding of the organization of the mitochondrial genome of T. gondii, a parasite of significant importance in both human and animal health contexts.
In essence, their approach involves enriching mitochondrial material, followed by genome sequencing and the analysis of mitochondrial short RNAs. They achieve a remarkable depth of mitochondrial sequencing and generate valuable RNA data. Furthermore, their efforts lead to the discovery and annotation of new short RNAs.
Overall, the article is well-crafted and presents compelling results. However, it's worth noting that, at times, the authors appear somewhat self-congratulatory, and certain results might be perceived as overly ambitious. Nevertheless, the discussion is aptly constructed.
-
-
www.biorxiv.org www.biorxiv.org
-
Note: This response was posted by the corresponding author to Review Commons. The content has not been altered except for formatting.
Learn more at Review Commons
Reply to the reviewers
Manuscript number: RC-2023-02224R
Corresponding author(s): Austin Smith
1. General Statements [optional]
This section is optional. Insert here any general statements you wish to make about the goal of the study or about the reviews.
We thank the reviewers for constructive comments and helpful suggestions which we have adopted to clarify and improve the manuscript. In addition, we have added a link to a web portal that will allow readers to visualise gene expression profiles and create their own plots using our early human embryo UMAP embedding (https://bioinformatics.crick.ac.uk/shiny/users/boeings/radley2024umap_app/). Stefan Boeing created this tool and is added to the author list with agreement of other authors.
2. Point-by-point description of the revisions
Reviewer #1 (Evidence, reproducibility and clarity (Required)):
Summary In this manuscript, Arthur Radley and Austin Smith designed a new feature selection method for scRNA-Seq, which is a successor to ESFW previously proposed by the same authors. As an evolution of this earlier framework, cESFW is also based on the idea that informative genes share information with other genes, whereas non-informative genes have a more random relative expression. The authors emphasize the key importance of feature selection in the scRNA-Seq workflow and assess the current state of the art for this step. They also propose that better feature selection leads to less data transformation. They show that cESFW outperforms Scran and Seurat feature selection in most cases of synthetic datasets. cESFW is then used in the context of early human development, re-analysing data from several published datasets where they show that they do not require batch correction. They also further strengthen the conclusion that a "2-step" model for TE-ICM and EPI-Hyp differentiation is also present in human embyros. Finally, they map several types of in vitro pluripotent stem cells, in particular primed and naive, to their manifold and study the evolution of the gene signatures during early human development. Overall, the manuscript is well written and presents a solid methodology. The re-analysis of human early development is convincing and justified. The main critic is that the quality of figures can be greatly improved: their resolution is too low and they are hard to read. For instance, more contrasted color schemes could be used to improve clarity, and given the high number of clusters for some UMAPs, indicating the name of some cluster near their centroids should improve clarity.
We agree that the resolution of the figures should be improved. We had to compress the images to satisfy the size limit for uploaded documents to bioRxiv. Our final submission will be of higher quality (original figures are at 900dpi). With regards to colour schemes, this is a surprisingly difficult problem. We tried multiple colour palettes but could not achieve greater contrast. The suggestion to add key cluster names near to their centroids on the UMAPs is an excellent idea, which we have implemented.
Comments: Page 2 I think the criticism of PCA is unfair because it is not a true feature selection method, and it is mainly used for computational purposes. I believe that for most workflows, between 30 and 50 PCs are retained, which do not significantly change the results in the downstream analyses. The citation (Yeung and Ruzzo 2001) does not seem appropriate, as they examine cases where only a small number of PCs are retained, outside the context of scRNA-seq.
We agree that the criticism of PCA is insufficiently justified by the citation. We thank the reviewer for pointing this out and have removed the comment.
"Furthermore, HVG selection has been found to be biased toward selecting highly expressed genes over low expressed genes." Could the author justify or remove this statement, as the Seurat and Scran methods are specifically designed to consider average expression to determine HVG? The cited article (Yip, Sham, and Wang 2019) raises this issue for methods other than Seurat and scran.
The reviewer is correct that the provided citation highlights Seurat and Scran HVG selection as relatively insensitive to the average gene expression levels compared with other HVG selection methods. We again thank the reviewer and have deleted the comment.
More generally, we have shortened the introduction, focusing on cESFW as a new approach to feature selection rather than critiquing alternative methods.
Page 6 I might have missed it, but I do not understand the number of cells in the early human development dataset also shown in Figure S2B. The Petropoulos et al. dataset alone is larger than the sum of cells from different cell types. Is there some filtering step that is not described?
We have added text in the data availability section to clarify the cells used in our analysis:
“The pre-implantation raw counts scRNA-seq data from Yan et al. 2013, Petropoulos et al. 2016, Fogarty et al. 2017, and Meistermann et al. 2021, were compiled into a single gene expression matrix by Meistermann et al. 2021. For information regarding quality control and cell filtering of these 4 datasets, please refer to Meistermann et al. 2021.”
The unsupervised clustering used to annotate cell types is unconventional (especially with the high number of clusters chosen), which is not a problem, but should be clarified. Improving the figure 3D to make it clearer and providing a cell cluster correlation plot might help to better appreciate the relationship between cell types.
We agree that the gene expression heatmap in figure 3D contributed little to the interpretation of the data/results. As suggested, we have replaced this heatmap with a cell cluster correlation plot to help appreciate cell state similarities. (Changes in figure 3.)
It could be emphasized that the ICM/TE branch cell type is a major difference with the mouse topology, as the readers might not be aware that the ICM/TE is an unspecified blastocyst state that only exists in humans.
There appears to be some misunderstanding around the use of “ICM/TE branch”. The cluster comprises an uncommitted population at the branching point from morula to either ICM or TE, as also described in the mouse embryo. We have adjusted the discussion to make more clear that the two branching point clusters are heterogeneous populations, not unitary cell types or states:
“The branching populations reside at critical junctures in blastocyst formation, the partitioning of extraembryonic and embryonic lineages. These branchpoint clusters do not define unitary states. On the contrary, cells in these clusters are heterogeneous and may become specified to alternative fates. For example, PDGFRA, a hypoblast marker (Corujo-Simon et al. 2023), and NANOG, an epiblast marker (Allegre et al. 2022), are heterogeneously distributed in the Epi/Hyp branching population. Furthermore, branch cluster boundaries extend beyond the topological bifurcation, potentially indicating that cells remain plastic and may be redirected. This would be consistent with the demonstration in mouse embryos that cells expressing ICM genes remain capable of generating TE up to the late 32-cell stage (Posfai et al. 2017).”
Page 9 To further substantiate the stepwise ICM/TE and EPI/PrE specification events, authors could project cells from each embryo on the UMAP, and analyze what are the co-occurrence of cells (as performed for instance in Meistermann et al 2021). This should show as reported (and cited by the authors) that some GATA3 positive cells (TE fated) start appearing from late morula stage and that ICM cells almost never co-exist with EPI nor Hyp in embryos.
We appreciate this suggestion. We have generated the requested plots showing where cells from individual embryos at different developmental timepoints are positioned on our UMAP embedding. (new supplemental figure (New figure, Figure S6). We present a summary heatmap of cell co-occurrence in revised Figure 4. These results offer greater insight than the RNA velocity analysis, which we have moved to supplemental Figure S6. We have added discussion of these analyses in the “Lineage branching blastocyst development” Results section.
Reviewer #1 (Significance (Required)):
The presented methodology shows significant value especially in the field of scRNA-Seq, where the critical step of feature selection is often inadequately addressed. Furthermore, this field is characterized by a limited set of feature selection methodologies. cESFW appears to be an important alternative to HVG methods that could improve scRNA-Seq analysis in certain contexts.
The new findings on early human development are somehow incremental, but a welcome addition to solidify the two-step model and refine the concept of reject cells. The audience for this early development context is specialized, but cESFW will most likely have an impact to the entire field of scRNA-Seq analysis.
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
Here, Radley and Austin present a novel approach for feature weighting in scRNAseq data based on entropy sorting. Feature selection is a central part of scRNAseq analysis, and it is most likely the case that there is no single approach that outperforms all others across all datasets. Hence, innovation in this space is needed for the field. The cESFW method presented here has several appealing properties from a theoretical point of view, and it also performs well on the synthetic and real datasets considered. Nevertheless, there are several major issues that need to be addressed before I can recommend the manuscript for publication:
1 The original entropy sorting (eq 1 in SI 1) is based on only two discrete states. However, calculating entropy for continuous distributions can be more tricky and it is unclear to me what assumptions are made regarding the gene expression. Could the authors clarify what properties of the distribution are required for the updated ESE equation to be valid? Is the only assumption that values are drawn from the [0, 1] interval? What happens if values are highly skewed, ie forming a bimodal or power-law distribution rather than something close to a uniform distribution?
We agree that it is beneficial to clarify these points. We have added a section titled “Assumed properties of underlying sample distributions” to the supplemental information. Briefly, we show that the ESS correlation metric is directly linked to the commonly used correlation metric, Mutual Information (MI). A desirable properly of MI is that it is able to capture non-linear/skewed relationships between features. The ES framework and ESS share this property with MI, allowing the ES framework to be relatively robust to presence of non-uniform distributions.
The main assumption for applying ES is that the features can be meaningfully scaled between values of 0 and 1. For gene expression, an intuitive way of achieving this is to inspect each gene and designate 0 count values as having 0 expression activity, and the maximum counts as having activities of 1, and all values in between existing within the [0,1] interval. A useful property of ES is that we do not need to assume a particular shape or distribution of the samples within the [0, 1] interval. The ES framework is non-parametric and does not require an assumed distribution to calculate the conditional entropy (CE), even in the continuous form. This is possible because the ES framework is formulated by turning the probabilistic form of CE into an ordinary differential equation (ODE), where the only dependent variable, x, is the overlap between the minority state activities of each individual sample. This calculation is explicitly identifiable/calculable, and is permutation invariant, meaning the shape of the distributions of a reference feature (RF) and query feature (QF) does not need to be assumed/defined. In other words, the ES framework quantifies to what degree active expression states enrich/overlap with one another in a manner that is robust to different distribution shapes.
2 How robust is the procedure for the choice of percentile for normalizing the gene expression scores? Does one get roughly the same results for 90-99th percentile or is it sensitive to this choice?
We have carried out a sensitivity analysis on the choice of percentile for each of the synthetic datasets and added it to the manuscript. (New figure, Figure S11). We find that on each of our 4 synthetic datasets the final results of cESFW are robust to a wide range of normalisation percentiles.
3 Similarly, I am concerned about the procedure for how to choose the number of significant genes. How robust is this process? Also, it is not altogether clear how to generalize the procedure outlined on p19. Most potential users would benefit from more quantitative guidelines. In particular, having to rely on interpretation of GO terms typically requires a considerable amount of understanding about the system at hand which could make it challenging to apply the procedure for others. For most users it would be helpful to know how robust the procedure is to this step and also if there could be more stringent guidelines for how to decide which genes to include.
We understand the reviewers concern regarding the robustness of feature selection on real scRNA-seq datasets. We have now applied our cESFW workflow to peripheral blood mononuclear cells (PBMC) scRNA-seq data, and found cESFW feature selection to be comparable, and by one metric more robust, than Seurat and Scran HVG selection (New Figure S2).
As cESFW is applied to more scRNA-seq data, we will learn more about how results compare to highly variable gene selection, and how workflows may be adapted to optimise results in different scenarios. For example, we have found that supervising the selection of gene clusters using a small set of markers known to be important in the system of study can help identify which clusters of genes should be retained during gene selection. We have added this to the materials and methods with the following paragraph:
“Furthermore, we suggest supervising the selection of gene clusters using a small set of markers known to be important in the system of study. In this work, we found that genes known to be important during early human embryo development (FigS4) are enriched in the dark blue cluster of genes, further suggesting that this cluster of genes is more likely to separate cell type identities in downstream analysis.”
While gene cluster selection supervision in this manner requires a degree of domain expertise, we believe this is not unreasonable for most applications, and is the case for many scRNA-seq analysis pipelines.
Our primary software contribution is the cESFW algorithm which calculates the ESS and EP matrices. With this manuscript we provide 6 commented workflows for applying cESFW to different datasets (4 synthetic data, human embryo data, PBMC data). We believe these workflows provide a good balance of documented use cases and user flexibility for cESFW usage. This is important because it is advantageous to be able easily to adapt workflows to incorporate domain expertise and different methodologies. Although workflows such as Seurat and Scran are user-friendly, their rigidity can be difficult when wanting to deviate from their standard workflows. In summary, we believe that our provided workflows are suitable for users to implement cESFW, while providing the flexibility to apply adapted pipelines.
4 The comparison of the clusterings on p6 is not really fair is it? If I understand it correctly, the 3,012 genes identified by cESFW was used to define clusters in fig 3c through unsupervised clustering. The authors then use HVG methods to identify 3,012 genes and then carries out clustering based on those. To evaluate the methods the silhouette score is used, but the labels from the cESFW clustering is used as ground truth. This does not sound like a fair way to compare. Could the authors please clarify, and if needed come up with an approach where the three methods have a more level playing field if needed.
The reviewer raises a fair point regarding the comparison of cluster identities and ranked gene lists. This issue is a chicken and egg problem, in that we require a baseline to benchmark different methodologies but lack an explicitly defined ground truth. For that reason we used synthetic datasets for initial comparison.
For the human embryo data, we have presented substantial evidence that our cluster annotations are biologically coherent and consistent with prior knowledge. We therefore consider it legitimate to compare the ranked lists of Seurat, Scran and cESFW. However, we acknowledge the potential bias and have mentioned this in the “Limitations of the study” section.
In addition, we have now analysed the peripheral blood mononuclear cells (PBMC) scRNA-seq dataset that is used in the tutorial workflows of Seurat and Scran. This PBMC dataset is arguably better defined since it has more discrete populations of cells, and by using the Seurat generated cell type labels we bias the analysis towards Seurat rather than cESFW. The results show that cESFW performs comparably to Seurat and Scran, and that the cESFW ranked gene list may be more stable than Seurat and Scran. These results suggest that cESFW can be widely applicable as a suitable alternative for feature selection. We have included this analysis in the Results and as a supplemental figure (New figure, Figure S2).
5 The main cESFW.py file in the github repository is clearly well structured and commented. However, I would like to see a much better documentation so that one does not have to go through the source code to understand what functions there are and what they do. In particular, I would like to see a vignette to make it easier for others to incorporate cESFW into their workflows.
We thank the reviewer for the positive comments regarding our cESFW.py commenting. We accept that our initial submission failed to point the reader directly towards our example workflows that provide step by step, well commented vignettes for using cESFW to analyse scRNA-seq data. In our initial submission we provided 5 workflows (4 synthetic data and the human embryo data), and in the re-submission we have added a workflow for analysing PBMC data. We have updated our cESFW Github to guide users to these example workflows (https://github.com/aradley/cESFW/tree/main).
Please note, the embryo workflow will be easily accessible through GitHub, whereas the synthetic data and PBMC workflows will be provided through a Mendeley data link (referenced in the manuscript and on our GitHub). However, the content of the Mendeley link cannot be made public until the paper is finalised, as it cannot be changed after publication. We provide a temporary public Dropbox link for the reviewers so that they may access the additional workflows (https://www.dropbox.com/scl/fo/xr5o9xm6490ftjsa55wxg/h?rlkey=maindrxwdqnirsw1en3my5qsr&dl=0).
Minor:
Why are the figures not always in order? For example, fig S10 is mentioned before fig S2 on p 6
Thank you for pointing this out; we have amended the text.
I am not sure if the indexing in eq 1 (p 18) is correct. j is both on the LHS and it is also being summed over on the RHS. Should one of these be i instead?
The indexing is correct. Each column j of a matrix refers to gene/feature on the RHS, and in the calculation on the RHS we take the column averages, leading to vector on the LHS that is still indexed by genes/features j. We have clarified this in the text.
Reviewer #2 (Significance (Required)):
The work presents a new method for feature selection in scRNAseq. Feature selection is a very important step and can have a big impact on findings. The method presented here is theoretically sound and it seems to provide interesting result when applied to early embryo development. However, as cESFW is only tested for one dataset it is unclear how well the method generalizes to other problems and datasets.
Appreciation of the utility of cESFW will grow as it is applied to more datasets. However, we would like to highlight that the human embryo dataset consists of 6 independent scRNA-seq datasets from different laboratories, and that cESFW was able to identify common and differing structure between them without any batch correction, smoothing or feature extraction. We have added to our summary that we propose cESFW may be best suited to analysis of transcriptome trajectories in time course and developmental data. However, we have also now performed comparison of Seurat, Scran and cESFW feature selection in a different context, using a reference PMBC scRNA-seq dataset. The results demonstrate that cESFW is a viable alternative for feature selection in that static system also (New figure, Figure S2).
-
Note: This preprint has been reviewed by subject experts for Review Commons. Content has not been altered except for formatting.
Learn more at Review Commons
Referee #2
Evidence, reproducibility and clarity
Here, Radley and Austin present a novel approach for feature weighting in scRNAseq data based on entropy sorting. Feature selection is a central part of scRNAseq analysis, and it is most likely the case that there is no single approach that outperforms all others across all datasets. Hence, innovation in this space is needed for the field. The cESFW method presented here has several appealing properties from a theoretical point of view, and it also performs well on the synthetic and real datasets considered. Nevertheless, there are several major issues that need to be addressed before I can recommend the manuscript for publication:
- The original entropy sorting (eq 1 in SI 1) is based on only two discrete states. However, calculating entropy for continuous distributions can be more tricky and it is unclear to me what assumptions are made regarding the gene expression. Could the authors clarify what properties of the distribution are required for the updated ESE equation to be valid? Is the only assumption that values are drawn from the [0, 1] interval? What happens if values are highly skewed, ie forming a bimodal or power-law distribution rather than something close to a uniform distribution?
- How robust is the procedure for the choice of percentile for normalizing the gene expression scores? Does one get roughly the same results for 90-99th percentile or is it sensitive to this choice?
- Similarly, I am concerned about the procedure for how to choose the number of significant genes. How robust is this process? Also, it is not altogether clear how to generalize the procedure outlined on p19. Most potential users would benefit from more quantitative guidelines. In particular, having to rely on interpretation of GO terms typically requires a considerable amount of understanding about the system at hand which could make it challenging to apply the procedure for others. For most users it would be helpful to know how robust the procedure is to this step and also if there could be more stringent guidelines for how to decide which genes to include.
- The comparison of the clusterings on p6 is not really fair is it? If I understand it correctly, the 3,012 genes identified by cESFW was used to define clusters in fig 3c through unsupervised clustering. The authors then use HVG methods to identify 3,012 genes and then carries out clustering based on those. To evaluate the methods the silhouette score is used, but the labels from the cESFW clustering is used as ground truth. This does not sound like a fair way to compare. Could the authors please clarify, and if needed come up with an approach where the three methods have a more level playing field if needed.
- The main cESFW.py file in the github repository is clearly well structured and commented. However, I would like to see a much better documentation so that one does not have to go through the source code to understand what functions there are and what they do. In particular, I would like to see a vignette to make it easier for others to incorporate cESFW into their workflows.
Minor:
Why are the figures not always in order? For example, fig S10 is mentioned before fig S2 on p 6
I am not sure if the indexing in eq 1 (p 18) is correct. j is both on the LHS and it is also being summed over on the RHS. Should one of these be i instead?
Significance
The work presents a new method for feature selection in scRNAseq. Feature selection is a very important step and can have a big impact on findings. The method presented here is theoretically sound and it seems to provide interesting result when applied to early embryo development. However, as cESFW is only tested for one dataset it is unclear how well the method generalizes to other problems and datasets.
My expertise is in computational genomics.
-
-
www.biorxiv.org www.biorxiv.org
-
Note: This response was posted by the corresponding author to Review Commons. The content has not been altered except for formatting.
Learn more at Review Commons
Reply to the reviewers
Reviewer #1 (Evidence, reproducibility and clarity (Required)):
This is an excellent paper experimentally exploring variations in carboxylation rates of form I rubisco.
We thank the Reviewer for this positive feedback on our study.
I have three comments
Q. Did the authors also measure the oxygenate activity of the enzyme? This is relevant to the evolution of carboxysomes and CCMs in general.
We agree that this is relevant but there are technical limitations for achieving this with the current framework. This pipeline’s emphasis on the carboxylation rate allows for the screening of a high number of rubisco variants covering a wide genetic diversity. It provides a way to approach the complexity of the kinetic constraints of this enzyme with a realizable and reproducible method. However, we agree that the measurements of oxygenase activity, as well as affinities to CO2 and O2 , would enrich our understanding of the evolution of rubiscos in the context of CCMs, and thus expanding our pipeline in the future to cover the other kinetic dimension would be worthwhile but cannot be achieved currently due to various technical constraints (other dimensions to explore, like the temperature effect, could also be included). In line also with the comment of reviewer #3, we have expanded our discussion in the manuscript to clarify this matter more thoroughly:
“By centering our analysis on the carboxylation rate, this pipeline systematically shows the particularity of carboxysome-associated rubiscos which are characterized by a poor affinity to CO2 (Badger et al, 1998; Falkowski & Raven, 2007) alongside a relatively high carboxylation rate (our data). This likely reflects the aforementioned catalytic tradeoff, suggesting that higher local concentrations of CO2 within CCMs probably allowed rubiscos to evolve towards higher kcat and KM. High-throughput measurements of other kinetic parameters beyond what was achieved here, such as the KM for both gasses or the oxygenation rate, would be valuable. It could provide values of the carboxylation efficiency, or even the enzyme specificity, which would enrich our understanding of this enzyme and of its adaptation to the atmospheric composition over geological timescales.”
- How do predicted structures (e.g., using Alpha fold) vary with catalytic efficient?
We generated Alpha Fold structures of the 98 active variants revealed in this study and performed preliminary structural analysis of the active site. There was no strong correlation between measured rates and the active site structure. The RMSD of generated structures has a median RMSD of 2.7 Å for the large and small subunit together, and 1.3 Å for the active site, suggesting high conservation of the structure of rubisco, and probably explaining the difficulty to associate rate variation with any clear structural feature (especially coming from prediction algorithm already showing uncertainties on the order of magnitude of an angstrom (Jumper et al, 2021; Terwilliger et al, 2024)).
We added a paragraph about this new analysis in the Results and in the Materials and Methods sections of the manuscript, and we present the results in new Supplementary Fig. 12. We made these structures available on the gitlab folder associated with this paper.
- The authors should note the paper by Tortell (not this reviewer) https://aslopubs.onlinelibrary.wiley.com/doi/pdfdirect/10.4319/lo.2000.45.3.0744
Thank you. We added a reference to this paper in the following sentence: “Another strategy consists of the evolution of rubisco towards stronger affinity for CO2 (Tortell 2000).”
Reviewer #1 (Significance (Required)):
This is an excellent paper experimentally exploring variations in carboxylation rates of form I rubisco.
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
I very much enjoyed reviewing this manuscript. de Pins et al provide a timely report on the catalytic turnover rate of a large number of Rubisco enzymes within the FormI group. These data provide novel insights into generalities of Rubisco function, specifically within certain phylogenies, and extend our understanding of carbon acquisition in these systems. In particular, the data presented by de Pins et al provide new insights into the relative carbon fixation rates of alpha-cyanobacteria, for which there are very few studies reporting catalytic turnover. It is apparent that the CO2 concentrating mechanisms (CCM) of cyanobacteria, especially the alpha-cyanobacteria (containing FormIA Rubisco) are a globally important contributor to CO2 capture into the biosphere via their carboxysomal Rubisco enzymes. This report provides then first broad selection of FormI Rubiscos to enable comparisons of catalytic turnover across this dominant enzyme family and shows that FormIA Rubiscos from phototrophic systems, and encapsulated in carboxysomes, are on average the fastest enzymes.
We thank the Reviewer for the positive remark on the novelty of the study.
de Pins et al use a high throughput screening technique that provides a highly correlative estimate of Rubisco turnover compared with traditional assays. This screen is based upon bulk expression of enzymes within E. coli from synthesised genes, and in some cases the co-expression of chaperonin factors to boost expression and solubility of holoenzymes. The assay process is sound and of high quality and the interpretations clear and uncomplicated.
The conclusions are sound and I only have a number of minor issues for consideration.
Minor comments: Temperature effects on 'true' Rubisco turnover rates. The authors quite reasonably note that a single measurement temperature was used in the assay and that this may not necessarily reflect the catalytic turnover of Rubiscos from thermophiles. Suppl. Fig 5b indicates a relatively large number of 'hot spring' species that have, generally, a low median kcat compared with, for example, both cyanobacterial classes. Can the authors comment on whether or not the thermophile set is not highly represented by one group (e.g. phototrophic alpha-cyanobacteria). Does this thermophile dataset have the potential to influence the generalities presented? Fig 2 would suggest this is not the case but it is not possible for the reader to know if all or any thermophiles are represented in Fig2 (as opposed to Suppl. Fig 4).
We indeed identify 3 groups of rubiscos that are either expressed by thermophilic bacteria (Supplementary Figure 10), and/or are associated with hot environments (hot spring and hydrothermal vent; see Supplementary Figure 5). These rubiscos show relatively lower rates. We grouped them together and reproduced our main analysis (from Figure 2) with and without rubiscos from this group to create a new Figure (Supplementary Fig. 11). Interestingly, alpha-cyanobacteria had no representatives among this group of thermophilic and hot-environments-associated rubsicos. However, this is unlikely to explain the result observed as removing them does not influence the obtained tendencies. We add the following sentences in the Results section:
“Additionally, the slightly lower carboxylation rate of rubiscos originating from thermophilic bacteria and isolated from hot environments (Supplementary Fig. 5 and 10) aligns with expectations, considering that these rubiscos naturally work at higher temperatures than in our in vitro assay (30°C). However, this is unlikely to explain the observed trends as the main results of this study are not affected by the removal of these rubiscos (Supplementary Fig. 11).”
We also noticed, while reviewing the study, that the code generating Supplementary Fig. 8 and 10 incorrectly duplicated some dots for a few rubiscos (less than 5), although this did not affect the overall results. We have fixed this issue and have now updated the figures accordingly.
Line 98: "in spite of" should be "despite"
Done.
Lines 171-173: There is an additional alpha carboxysomal Rubisco for which there are catalytic parameters described (Chapter 11 Engineering Photosynthetic CO2 Assimilation to Develop New Crop Varieties to Cope with Future Climates. RE Sharwood, BM Long - Photosynthesis, respiration, and climate change, 2021). This book chapter reports a kcat of 11.9 s-1 for the alpha carboxysomal Rubisco from Synechococcus WH8102, very much in line with the authors conclusions.
We added this rubisco to the list of previously characterized rubisco and updated Figure 1 accordingly. We also updated the following sentence:
“However, such statements were made based on scarce measurements, with only three kcat,C values currently available for both rubisco groups (Shih et al, 2016; Long et al, 2018; Sharwood & Long, 2021; Wilson et al, 2018; Aguiló-Nicolau et al, 2023).”
Lines 232-234: I note that ref 30 posits that low CO2 was the more likely driver of carboxysome evolution than high O2.
We agree that our phrasing did not point clearly enough that CO2 was more likely the driver of carboxysome evolution, rather than high O2. We rephrase the sentence to: “Carboxysomes likely evolved during the Proterozoic eon - in the context of the continuous decrease of carbon dioxide in Earth’s atmosphere (Flamholz & Shih, 2020)”
Line 235: The preferred term is either "CO2 concentrating mechanism" or "inorganic carbon concentrating mechanism"
We rephrased into “CO2 concentrating mechanisms”.
Lines 254-256: The relative saturation of carboxysomes with Rubisco is still somewhat undecided, although relatively new datasets enable more accurate comparisons. A number of papers from the Liu Lab (Liverpool) enable estimates of Rubisco active site concentrations for alpha and beta carboxysomes in the range of 2-6 mM. It appears at this stage that Rubisco active site concentrations may be highest in alpha-carboxysomes.
We thank the reviewer for drawing our attention to the work of the Liu lab which, while acknowledging potential inaccuracies in the estimates, tends to show a higher concentration of rubiscos in alpha-carboxysomes than in beta-carboxysomes (Sun et al, 2019; Sun et al, 2022). We removed this statement from the text.
Lines 312-318: That genes were codon optimized for E. coli expression raises an interesting question about the effect of Raf1 on Rubisco solubility. Assuming expression rates were not constrained, can any conclusions be made as to the amino acid sequence differences that led to lower solubility? One assumes that the Rubisco sequences had a high degree of identity?
We indeed note that the fact that every rubisco gene from this study was codon optimized for E. coli expression suggests that the solubility issues met here were post-translational. This suggests a post-translational role for Raf1 in rubisco folding/assembly that is in line with the mechanism proposed by Xia et al. of an interaction of Raf1 with rubisco large subunits dimer, further mediating the assembly of an octameric core and the recruitment of rubisco small subunits (Xia et al, 2020). We also note that insoluble rubiscos were met in all form I clades, suggesting that this property was not linked to a specific group of rubiscos sequences with a high identity degree.
We add that we also tested the effect of not performing codon optimization on 8 rubiscos that were insoluble following codon optimization (to assess whether the codon optimization could negatively affect the folding, for instance by making the translation too fast). This did not yield any improvement in solubility.
Lines 333-344: Was there an attempt to use acRAF (Raf2?) for FormIA Rubiscos that did not fold successfully in E. coli?
While only 33% of β-carboxysome-associated (IB) rubiscos were originally soluble (i. e. without the coexpression of Raf1 from E. natronophila), 85% of the tested α-carboxysome-associated (IAc) rubiscos were soluble in our experimental conditions. We therefore decided that testing for the effect of co-expressing them with the chaperone acRAF would be less cost-effective.
Reviewer #2 (Significance (Required)):
This manuscript presents a significant advance in our broader understanding of the major enzyme involved in carbon input into the biosphere, Rubisco. It will be of key interest to those studying carbon biogeochemistry, global CO2 modelling, cyanobacterial and proteobacterial CCMs, and those interested in using these systems to improve plant-based carbon capture for food security and global carbon abatement systems. It provides, for the first time, a large dataset of hitherto unknown Rubisco kinetics in a globally important group of organisms. The study is extremely well carried out and will likely form the basis of future Rubisco screens to provide greater clarity to our knowledge base of this globally important enzyme.
My expertise is in the study and application of CCMs as CO2 acquisition systems that can be used for Synbio applications.
Reviewer #3 (Evidence, reproducibility and clarity (Required)):
Summary: The authors have presented a tremendous study into the diversity of carboxylation speed (kcatc) from bacterial Form I Rubisco enzymes. The authors identified some nice diversity in kcatc which resulted in the finding that Rubisco's originating from within a CCM were faster, which confirms what has been previously observed in the literature. The authors provided information on a pipeline to screen large numbers of Rubisco variants. In this manuscript, the authors tested 144 different enzymes with 112 of these successfully expressed in E.coli and of these 98 showed substantial catalytic activity. The authors showed that alpha cyanobacterial Rubisco possessed the fastest kcatc when compared to beta cyanobacterial counterparts which is contrary to that published in the literature so far. The authors have provided some nice insight into how they improved expression of soluble Rubisco with expressing bacterial chaperonin and Rubisco assembly factors such as Raf1 and rbcX. All which have been previously discovered plant and cyanobacteria. The authors also presented some nice correlations as shown in figure 2 and some weaker and non-correlations to various environmental parameters in the supplementary data.
Overall, the field will learn something from this large body of work that has characterized only one Rubisco catalytic parameter.
We thank the Reviewer for these positive comments.
Major points: 1) The authors only measured carboxylation speed using a spec assay. The Michaelis constant for CO2 measured in N2 and 21% Oxygen is also valuable to understand the diversity in Rubisco catalysis. The authors should perhaps mention this and that the carboxylation efficiency is also an important measure for comparing Rubisco enzymes.
In line with this and with the comment of reviewer #1, we have expanded the Discussion to include the following:
“By centering our analysis on the carboxylation rate, this pipeline systematically shows the particularity of carboxysome-associated rubiscos which are characterized by a poor affinity to CO2 (Badger et al, 1998; Falkowski & Raven, 2007) alongside a relatively high carboxylation rate (our data). This likely reflects the aforementioned catalytic tradeoff, suggesting that higher local concentrations of CO2 within CCMs probably allowed rubiscos to evolve towards higher kcat and KM. High-throughput measurements of other kinetic parameters beyond what was achieved here, such as the KM for both gasses or the oxygenation rate, would be valuable. It could provide values of the carboxylation efficiency, or even the enzyme specificity, which would enrich our understanding of this enzyme and of its adaptation to the atmospheric composition over geological timescales.”
2) The authors mentioned that they used E.coli lysates. Did the authors test for background activity due NADH dehydrogenases which are present in bacterial lysates? This could impact the catalytic rates measured.
The background activity due to dehydrogenases from the lysate is negligible compared to the measured reaction (see as an example, in Supplementary Fig. 16A, the gray curve, corresponding to a CABP concentration of 90 nM, fully inhibiting rubisco in the reaction). Moreover, the use of CABP in the spectroscopic assay allows to take into account any “background activity” that would come from an element independent of rubisco because this background would be identical at every CABP concentration. We modified Supplementary Note 1 to make this point clearer:
“We note that any NADH dehydrogenation due to other native E. coli proteins, while low (see the [CABP] = 90 nM gray curve of Supplementary Fig. 16A), is not influencing the measurement of the carboxylation rate which relies on the differential Vmax values at changing CABP concentrations (which should not affect the rates of these dehydrogenases).”
3) For the microtitre plate assay, did the authors correct for the different pathlength? This is crucial for the Beer-Lambert law which is used to calculate the consumption of NADH.
Because our assay is performed in a microwell plate instead of a standard 1 cm quartz cuvette, we indeed calibrated the assay to empirically determine the optical path length in our conditions. We add the following sentence in the Material and Methods section to better explain the measurement of NADH concentration in our assay:
“Knowing the NADH extinction coefficient at 340 nm (ε340 = 6220 M-1cm-1), and after measuring the optical path length (𝑙 = 0.26 cm) with an NADH calibration curve in our setting, we used Beer-Lambert law (𝐴340 = ε340 . 𝑙 . 𝑐) to measure the NADH concentration 𝑐.”
4) Did the authors consider studying the temperature response of kcatc for these enzymes? This could also reveal some interesting insight into their data.
We indeed considered this. With a high-throughput pipeline involving >100 enzymes to express and test in parallel, we had to limit the experimental testing conditions to be realistic both in terms of time and budget. However, the finding that rubisco variants from thermophiles tend to have lower carboxylation rates in our standardized conditions (30°C) suggest that they will probably show faster rates at temperatures closer to their optimal growth temperature. We therefore agree that the study of the temperature response of the carboxylation rate in further works could validate these hypotheses and bring more insight into the catalytic characteristics of rubisco. We further emphasize this point in the following modified sentence:
“Investigating the temperature response of rubisco carboxylation rate in further work could shed light on the importance of this parameter, especially among thermophilic or psychrophilic associated enzymes.”
5) With this new catalytic knowledge, what can the field now do with this data to inform new research directions?
We believe this new knowledge can inform new research directions in the field of microbial ecology, metabolic engineering, and machine learning in the context of kinetic parameters prediction. We modified a paragraph in the discussion to elaborate on this point:
“This study provides a systematic exploration of bacterial form I rubisco maximal rates and its relationship with various contextual factors that could have shaped the evolution of this most abundant enzyme on Earth. It holds potential for future metabolic and ecological studies about specific bacterial species – for instance among cyanobacteria for which 40 rubiscos have been characterized here. By enriching our knowledge on carboxylation rates and their connection to environmental factors, it can also contribute to more accurately modeling global carbon fluxes. Additionally, this dataset of rubisco sequences and their associated rates can facilitate linking sequence motifs to catalytic function. Ultimately, it can improve our understanding, and possible harnessing, of bacterial CCMs for the potential development of plant-based carbon capture strategies, the increase of agricultural yields and the support of sustainable food production in the face of a changing climate.”
Minor comments: The figures are of outstanding quality and easy to follow. This will set the bar high in the literature. I have no other minor comments.
We thank the Reviewer for noting the care taken in the graphic presentation of our results.
Reviewer #3 (Significance (Required)):
Overall, the authors have presented an excellent study into bacterial Form I Rubisco's that will further enhance our understanding of Rubisco evolution. The pipeline for expression of bacterial Rubisco's in E.coli is developed nicely by the authors and the next step will be to determine how other important catalytic parameters can be determined to have more detailed understanding of Rubisco catalysis.
-
-
www.biorxiv.org www.biorxiv.org
-
Note: This response was posted by the corresponding author to Review Commons. The content has not been altered except for formatting.
Learn more at Review Commons
Reply to the reviewers
We thank the three reviewers for their thoughtful and constructive comments. The changes to the text and figures made in response to the questions raised have made this a clearer and stronger manuscript. The additional citations suggested by the reviewers helped to further anchor our study within the growing literature on facultative parthenogenesis. Below we have responded to each comment in blue. We have added new data to the manuscript (Fig. 4C, Fig. S10B and Fig. S10D).
Point-by-point description of the revisions
This section is mandatory. *Please insert a point-by-point reply describing the revisions that were already carried out and included in the transferred manuscript. *
Reviewer #1 (Evidence, reproducibility and clarity (Required)):
- Summary: Here Ho et al. provide strong molecular evidence for the production of facultatively parthenogenetic whiptail lizards, through a gametic duplication. As evidenced through multiple routes, including microsatellites, WGS, RADseq, and RBC ploidy, and lines of evidence from multiple specimens, this study is timely in furthering our understanding of the mechanisms underlying FP. The findings are conclusive.
That said, I have several comments that should be addressed prior to publication. The introduction which addresses FP in other systems fails to cite several key studies that provide strongly molecular support for terminal fusion automixis. Similarly, the study pushes the idea that this is an adaptive trait, however without proving that the parthenogens can themselves reproduce, this is a moot point at this stage.
That said, my comments are minor. I found this to be an excellent study, well written, comprehensive in methodology, and one that I strongly advocate for publication.
We thank reviewer 1 for referring to our manuscript as an excellent study and strongly advocating for its publication. We concur with his/her points that evidence for automixis in other systems was not sufficiently referenced and that the adaptive trait hypothesis for FP is somewhat speculative. The text has been modified accordingly (see below).
Major comments - None.
Minor Comments: Should be addressed.
Line 36 - However, data that supports terminal fusion are no longer restricted to microsat data. Studies utilizing RADseq and whole-genome sequencing in snakes and crocodiles have now provided further evidence supporting terminal fusion.
See: Booth et al. 2023. Discovery of facultative parthenogenesis in a new world crocodile. Biology Letters. 19, 20230129.
Card et al. 2021. Genome-wide data implicate terminal fusion automixis in king cobra facultative parthenogenesis. Scientific Reports. 11, 1-9
Allen et al. 2018. Molecular evidence for the first records of facultative parthenogenesis in elapid snakes. R. Soc. Open. Sci. 5, 171901.
We have now included that automixis in other systems is supported by both microsatellite and NGS data in the abstract of our manuscript. The references have been included in the main text.
Ln 42 - Evidence suggesting that isolation from males was not a pre-requisite for FP has previously been reported in snakes.
See: Booth et al. 2011. Evidence for viable, non-clonal but fatherless Boa constrictors. Biology Letters. 7, 253-256.
Booth et al. Facultative parthenogenesis discovered in wild vertebrates. Biology Letters. 8, 983-985.
Booth et al. 2014. New insights on facultative parthenogenesis in pythons. Biol J Linn Soc. 112, 461-468.
Despite the prior evidence to the contrary cited by the reviewer, it is still a commonly held belief among scientists and science journalists that isolation from males promotes or triggers FP. We have placed our findings in the context of other studies, including those mentioned above, that came to the same conclusion that isolation from mating partners is not a requirement for FP. We thank the reviewer for the additional citations, which are now included in the discussion section.
Ln 48 - Is this really an argument. While an immediate transition to homozygosity will purge some deleterious alleles, given the genome-wide nature of this, there will also conversely have been strong selection for mildly deleterious alleles.
Even though many FP animals have congenital defects, our data, combined with that of others, show that seemingly healthy animals arise as well. Even if these healthy animals harbor slightly deleterious alleles, the most detrimental alleles would have therefore been purged especially for subsequent generations. We have modified the abstract to be clearer: “Conversely, for animals that develop normally, FP exerts strong purifying selection as all lethal recessive alleles are purged in one generation.”
Ln 56 - I would recommend the inclusion of both Allen et al. 2018. R. Soc. Open Sci, and Card et al. 2021. Sci Reports, here, as they are members of the elapids, not represented in the other examples.
These two citations have been added.
Ln 60 - Recent studies have highlighted the significance of sperm storage in reptiles. For example, Levine et al. 2021. Exceptional long-term sperm storage by a female vertebrate. PLos ONE. 16(6).e0252049, describe the storage of sperm by a female rattlesnake for ~70 months, with two instances of its utilization to produce healthy offspring during that period. Clearly, molecular tools are providing both support for long-term sperm storage, and an understanding of its utilization.
Recent work has indeed provided new evidence for instances of long-term sperm storage and the two mechanisms are no longer competing hypotheses, but it is clear that both mechanisms exist in nature. We have modified the text accordingly to include “Nevertheless, clear examples of long-term sperm storage have also been documented in the recent literature (29), underscoring the need for molecular methods such as MS analysis or sequencing data to elucidate the underlying mechanisms.”
Ln 68 - American Crocodile would also be suitable to include here.
This has now been included in the list of examples of endangered species.
Ln71 - The problem with this hypothesis is that parthenogens produced through FP tend to have very low viability. For example, Adams et al. 2023. Endangered Species Research, follow a cohort of sharks produced through FP and all survive. Similarly low levels of survival are reported across other systems for which FP was reported. More likely, FP is simply a neutral trait. The mother is not negatively impacted through producing parthenogens and can go on to produce sexual offspring. Few instances report successful reproduction of a parthenogen. See pers. Comm in Card et al. 2021. And Straube et al. 2016.
We thank the reviewer for the comment and agree that more data on the successful reproduction of parthenotes are needed to claim that FP is an adaptive trait. We have modified the text to include that studies on “the successful reproduction by FP offspring” are needed to support this hypothesis and have included the Straube et al. 2016 citation. We decided to omit the Card et al. 2021 citation as the reports of second-generation FP was through personal communication mentioned in this study and the results themselves have not yet been published.
Ln 79 - I doubt that there is a desperate need for this for conservation. However, I think there is a need to simply further our understanding of basic biological function, given that it is not uncommon, and is phylogenetically widespread in species lacking genomic imprinting.
We agree that understanding FP as a basic biological function is important in light of the realization that it occurs more commonly than previously thought. We have added this aspect to the text: “A better understanding of the triggers and molecular mechanisms underlying FP and the fitness of the resulting offspring are therefore needed in a variety of contexts. These include: to understand a fundamental biological mechanism and its significance in vertebrate evolution, to aid in conservation efforts including captive breeding programs, and to possibly harness FP in an agricultural context (28).”
Ln 85 - It would be worth citing Card et al. 2021., here given that they used genome-wide ddRAD markers to show support for terminal fusion.
The citation has been added.
Ln 91 - Better citations here are Card et al. 2021. Allen et al. 2018, and Booth et al. 2023, which all utilize either RADseq or WGS.
These citations have been added.
Ln 95 - The conclusion of genome duplication here was supported only by a small number of microsatellite loci. As such, given that terminal fusion has been supported through genome-wide markers in other species of snakes and crocodiles, the conclusion of genome duplication is likely incorrect.
In light of the other examples that show terminal fusion in snakes, we have removed this sentence.
Ln 96 - I would strongly disagree with this statement. Allen et al. 2018, Card et al. 2021, Booth et al. 2023, all provide evidence of heterozygous loci and thus support terminal fusion. While no species-specific chromosome level reference genome is available for any of these species, the fact that levels of heterozygosity are below 33% percent supports terminal fusion. Rates over 33% support central fusion, but have not been reported in any vertebrate to date. AS such, I would recommend the removal of this statement.
We agree that the studies listed by the reviewer all support terminal fusion in snakes and crocodiles and therefore, we have removed the statement.
Ln 121 - Recent work in Drosophila mercatorum and D. melanogaster suggest that three genes play a role in the activation of FP in unfertilized eggs. In this case, through the fusion of meiotic products. That said, it is plausible to assume that FP in these lizards has an underlying genomic mechanism that is not related to isolation from males. See Sperling et al. 2023. Current Biology. 33, P3545-P3560.E13.
Clearly isolation from males is not a key trigger in FP in whiptail lizards and other vertebrate species. With recent work from Sperling et al. 2023 and the fact that selection has led to increases in parthenogenesis in birds, an underlying genetic mechanism may well be at play. We have cited and addressed this in the discussion and propose identifying the genetic basis for FP in whiptail lizards in future studies.
“Recent work identifying key cell cycle genes inducing FP in two species of Drosophila (71) and selection resulting in higher incidences of parthenogenesis in birds (24, 33) suggest a genetic basis for the initiation of FP. [...] Additional whole-genome sequencing data for species with documented FP will aid in the understanding the genetic basis, propensity, and evolutionary significance of FP.”
Ln 126 - While these data strongly support FP of the two unusual A. marmoratus appearing offspring, can long term sperm storage be ruled out. Either through captive history or allelic exclusion of other males in the group?
We have added the following sentence to the text: “Given that all of these offspring are female, inherited only maternal alleles, and animal 122 had no history of being housed with a conspecific male during its lifetime, both interspecific hybridization and long-term sperm storage are all but ruled out and FP is strongly supported.”
Ln 171 - 191 - Given that the topic of this manuscript is the genomic mechanism underlying FP in this species, are these data necessary? These are not discussed later and as such I would recommend that they are moved supplemental material. Otherwise, they simply clutter that manuscript and detract from the key question. Indeed, they are important to show that the genome constructed is of high quality, but online Supp Mat is the place for that here.
We chose to keep this section in the main text for the following reasons: There is still a lack of published reference quality genomes for many reptile species and therefore we want to highlight that this A. marmoratus reference adds not only to the understanding of FP, but also expands the small list of reptile genomes and makes the first Aspidoscelis genome available to the community. The high quality and contiguity of the genome (as indicated by the high N50 value and BUSCO score) is important to emphasize in the main text because the absence of any heterozygous regions in FP animals supports a mechanism of post-meiotic genome duplication. We would not want to bury these key points in the supplement.
Ln 296 - Comparable estimates were made for parthenogenetic production in wild populations of two North American pitviper species. See Booth et al. 2012. Biology Letters.
In Booth et al. 2012, 2 out of 59 litters of the two pitvipers (3.39%) were identified to contain FP offspring and these results are very similar to our reported rate of FP in whiptail lizards. We have now included this similarity in our discussion. “Interestingly, these rates are similar to what has been reported for wild populations of two North American pitviper species (10)”.
Ln 312 - Again, can this really be suggested? Above, the authors state that most FP animals that hatched had congenital defects, and a large number failed to hatch. This does not sound like strong support for generating individuals that counter the effects of population bottlenecks and inbreeding depression. The authors need to take this study further and monitor the long-term viability of the FP individuals that survive.
We agree with the reviewer that the adaptive advantages of FP reproduction are dependent on the fitness and reproductive potential of FP offspring and present data is insufficient to clearly support this notion. We have modified the text to include that long-term studies are needed to support or refute this hypothesis: “However, support for this hypothesis is predicated on the fitness and reproduction of FP offspring and therefore more long-term studies on seemingly healthy individuals of FP origin are needed.”
Ln 348 - To be able to provide support for this, you need to track animals long term to understand their reproductive competence, and that of their offspring.
We have added the text: “To assess whether the co-occurrence of sexual and FP reproduction in vertebrates can indeed be considered a reproductive strategy rather than biological noise will require further studies to assess the reproductive competence and fecundity of offspring produced by either mode of reproduction.”
Ln 358 - But, the caveat is that the parthenogens must themselves reproduce. This must me stated.
The statement that parthenogens must be able to reproduce to support a hypothesis of FP as an adaptive trait has been added: “One must now consider the possibility that FP is an adaptive trait and that low rates of successful FP could contribute significantly to genome purification. Such a role for FP hinges on further studies demonstrating the ability of parthenogens to reproduce themselves either through further FP or sexually.”
Ln 359 - Note that FP can also fix mildly deleterious alleles. Only if it is strongly deleterious will it be lost.
We now make it clearer that selection only applies to strongly deleterious alleles.
Ln 361 - See above comments.
We have modified the text to include that “FP offspring will have low genetic load and only pass on neutral and mildly-deleterious alleles to the next generation.”
Reviewer #1 (Significance (Required)):
- Significance:
While reports of parthenogenesis have been reported as far back as the early 1900's, it has only been over the last decade that reports are become common. Such that facultative parthenogenesis is no longer considered a rarity, but is recognized now as being relatively common and phylogenetically widespread in species that lack genomic imprinting - particularly reptiles, birds, and sharks. Reasons for this are both an increased understanding that the trait can occur, hence recognizing it as an alternative mechanism to long-term sperm storage, and the ease of using molecular approaches.
The fundamental questions of recent times have been understanding the mechanisms driving FP. Recent papers utilizing whole genome sequencing and ddRADseq have provided support for terminal fusion automixis in snakes and sharks. Here, this study provides evidence of gametic duplication in whiptails, a mechanism with an alternative outcome in regards to the levels of retained heterozygosity. As such, this study compares to the recent work of Card et al. 2021 (Scientific Reports), and Booth et al. 2023 (Biology Letters), in providing substantive advances in the field.
The audience for this will be broad. Parthenogenesis is a fascinating topic that attracts significant media attention. See the Altmetric score of recent papers on the topic, particularly Booth et al. 2023 (Altmetric score - ~3100). As such, the study will be of interest to both a broad readership, but will also be of great significance to a specialized group working on parthenogenesis. All round, an excellent paper that has promise to advance the field.
We thank reviewer 1 for this positive assessment and for putting our work into context.
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
Summary: The researchers bring together microsatellite and whole-genome sequencing data from long-term laboratory cultures of lizards to discover occasional production of parthenogenetic offspring by several species of otherwise sexually producing whiptail lizards ("facultative parthenogenesis, "FP") and to show that these FP-produced lizards have patterns of genomic homozygosity that are incompatible with currently held assumptions about mechanisms of FP. Instead, the FP lizards seem to have been produced by a mechanism that results in almost complete homozygosity, likely a consequence of post-meiotic duplication of genomes from haploid unfertilized oocytes. They also show that FP offspring were produced by females housed with males and along with sexually produced offspring, counter to prevailing assumptions that FP offspring are only produced in situations where mates are not available. Many of the FP-produced offspring did not survive to hatching or had major abnormalities, consistent with a situation where this high homozygosity exposes harmful alleles. Finally, the authors used reduced-representation sequencing (RAD-seq) to survey heterozygosity in 321 wild-collected whiptail lizards from 15 species, showing evidence for strikingly low homozygosity in at least one individual and perhaps up to 5, consistent with the potential for FP in nature. These data are of broad interest in demonstrating several exciting new possibilities. Most importantly, the data hint at a different mechanism of FP than previously assumed, and one that causes immediate near-complete homozygosity. This scenario would likely lead to immediate purging of harmful recessive alleles. If the selective load of this purging wasn't insurmountably high, a lineage with a history of purging could produce FP offspring of relatively high fitness. Other exciting possibilities suggested by the data include the existence of FP even in a setting where mating occurs and in natural populations, versus just captivity.
Major Comments:
I found it difficult to impossible to sort out exactly what the researchers did and with what lizards. For example, in line 107, they refer to a "systematic MS analysis" for all individuals of gonochoristic species in their laboratory, but where are these data? Indeed, at this early spot in the paper, the introduction from here on out suddenly reads like a discussion. What would be better here would be to summarize what was known and wasn't known about the system and questions involved, why gaps in knowledge were important, and what the researchers actually did for this paper. In my opinion, the paper would be a much easier read if the researchers left the results and interpretation for later in the paper.
As a consequence of the reviewers’ comments, the text of the manuscript has undergone major revision, and we trust that reviewer 2 will find this new version far more accessible. The MS data collection of more than 1000 individuals is the subject of another ongoing study and was only mentioned peripherally here to put the identification of FP into context. As most of the MS data relates to gonochoristic reproduction and interspecific hybridization, we are only presenting the data that are directly relevant to this manuscript as part of this study. To our knowledge, there is no common repository to upload raw MS data, but we have provided the data for the FP animals and controls discussed in this paper in the Github repository (see section “Data availability”).
Even with this suggested fix, however, the data are still too inaccessible and analyses too opaque. For example, in line 202, a critical definition is laid out regarding heterozygous sites as those having "equal support" for two alleles. What do the researchers mean by "equal support"? My presumption is that this is something about equal or close to equal numbers of reads, but this definition needs to be spelled out and justified because it underpins much of the downstream analyses. A similar problem occurs in line 208-209, where the authors make a statement about limiting further analysis to positions in the genome where the coverage is "equal" to the mean sequencing depth.
We have changed the text to “we defined heterozygous sites as those having two alleles supported by an equal number of reads. This stringent requirement was chosen to limit the search to apparent heterozygous sites with strong support, decreasing the chance of false positives.”. We further look at only sites where the coverage is equal to the average sequencing depth to exclude regions where over-assembly and collapse of repetitive elements would artificially increase the coverage.
Another data/analysis issue emerges with the components of the manuscript that deal with mixoploidy. As far as I can tell, these data come from one sexually produced lizard, one FP A. marmoratus, and one FP A. arizonae. While the reports of bimodality of nuclear size are certainly interesting, the data and discussion are no more than an anecdotal case study in the absence of careful replication across multiple FP lizards and comparison to sexually produced lizards. Without these data, the conclusion that “Animals produced by facultative parthenogenesis are characterized by mixoploidy” (Figure 4 caption; also see lines 324-331) is far too strong.
We have added animal IDs to figure legends 4 and S10 to clarify that these erythrocyte staining come from two FP A. marmoratus, and one FP A. arizonae. In addition, imaging from two sexually produced control animals (1 A. marmoratus and 1 A. arizonae) have now been included in S10 (as S10B and S10D). We also have included an extra panel of flow cytometry data (new Figure 4C) as a complementary methodology for ploidy determination. Both imaging and flow cytometry support similar amounts of haploid cells. With the additional data and clarification, we hope that the reviewer agrees that the observations of mixoploidy are well beyond “anecdotal”. Nevertheless, we have changed the title for Figure 4 to “Detection of mixoploidy associated with facultative parthenogenesis.” We hope that our observations here will indeed inspire future studies to see if mixoploidy is a widespread phenomenon in FP outside of whiptails as indicated by earlier work in birds.
I had a similar reaction to the discussion of developmental abnormalities and embryonic lethality of embryos of FP origin presented in lines 263-281 (also lines 307-309). What is the baseline level of such abnormalities and the frequency of lethality in sexually produced eggs/embryos/hatchlings, and especially those produced via inbreeding? These comparisons are needed to interpret the significance of the patterns observed in the FP eggs/embryos/hatchings. Analogously, the comparison of the ovaries and germinal vesicles from one FP individual relative to one sexual individual do not tell us anything nearly so definitive as the text in lines 279-281 (also see Fig. S12 title, which is too broad of a conclusion for N = 1). This overly ambitious conclusion also underpins the discussion regarding the potentially adaptive nature of FP with respect to genome purification (lines 341-363; also see lines 47-50). If FP does not actually increase the rate of purging in FP lizards relative to inbred sexual counterparts (sounds like inbreeding is common from line 339), it seems less likely that we can view FP as adaptive at least from this perspective.
We have now included a comparison between defects seen in sexually produced animals vs FP animals: “six out of 16 FP animals (37.5%) hatched with no discernable developmental defects (Fig. S11A-B). This is in stark contrast to sexually produced animals, where over 98% of hatchlings showed no abnormalities. Additionally, most of the defects noted in sexually produced animals were less severe than in FP animals including bulges in tails or truncated digits.”
We agree that our statement on the lack of differences between sexually produced and FP animals was too general. We have modified the title of Fig. S12 from “No differences between ovaries and germinal vesicles of Aspidoscelis marmoratus produced by facultative parthenogenesis or fertilization” to "Ovaries of Aspidoscelis marmoratus FP animal 8450 and germinal vesicles of FP sister 8449 revealed no differences in structure and anatomy compared to fertile sexually reproducing animals.” Due to instant complete homozygosity, FP would indeed have a higher rate of purging than inbreeding. While one hypothesis is that FP is adaptive (in large enough populations), our intentions were to highlight the alternative that FP could be detrimental in smaller populations (that already would likely experience high inbreeding rates). We would expect inbreeding to not be common in whiptails relative to other lizards given that they tend to have large population sizes and actively range across generalist habitats.
A final data concern is with the use of liver tissue for whole-genome sequencing and reference genome assembly (lines 389-390) and then using these data and the reference genome to make conclusions about ploidy/coverage. Liver tissue is very commonly endopolyploid, meaning that coverage could be artificially high for animals for which liver (vs. tail) tissue was used for DNA extraction. In particular, it would be helpful if the researchers consider whether endopolyploidy could have affected their ability to make accurate estimation of coverage and thus, heterozygosity, when libraries generated from diploid (tail) tissues are aligned to a reference genome generated from a polyploid tissue as was done here.
This is an interesting point and indeed hepatic cells in various organisms have been documented to be polyploid. The proportion of polyploid cells though vary and as far as we are aware, all published studies on polyploid hepatocytes are in mammals (DOI: 10.1016/j.tcb.2013.06.002). Reference genomes have been generated from a variety of tissue sources and liver is commonly used. As most assemblies are for haploid genomes, polyploidy (unlike aneuploidy) does not impact the assembly quality. The reference genome was also from an animal of FP origin and therefore has genome-wide homozygosity that aids in a more contiguous genome assembly by eliminating the phasing problem. For the 10 animals sequenced, genomic DNA was derived from liver for three animals and the rest from tail tissue. The sequencing data generated from either liver or tail resulted in similar coverage levels (Figure S6) and similar levels of heterozygosity (Figure 2A). Minor Comments:
Line 410: Please explain why the BLAST cutoff was changed from the default.
The BLAST cutoff was changed from the default 1e-03 to 1e-06 to be more stringent and thereby increase confidence in the BUSCO results.
Lines 441-443: Please explain why this dataset was seemingly larger than expected.
Animal 122 was sequenced on one flow cell without any multiplexing with other samples and therefore yielded more reads than other animals sequenced. We subsampled the reads from this animal for analysis, so it is directly comparable with the other WGS data.
Line 510: The link to the Github repository was broken, so I was unable to access the code and data denoted as available here.
We apologize for the unavailability of the link at the time of review. Review Commons did not request a reviewer token. The repository will be made public upon journal acceptance. We would be happy to provide a reviewer token in the meantime upon request by Review Commons.
Figure 1, and other figures featuring comparisons of MS data across parents and offspring: The authors need to engage here with the alleles that do not match either parent here (e.g., allele 282 at MS7), explaining the likelihood that these alleles indeed represent a binning error (or, perhaps, stepwise mutation from parental allele), and these alleles should be flagged. Instead, they bin these unique alleles with the most similar parental allele without any explanation or flagged. The authors do bring this point up in Figure S1, but this issue needs to be addressed in the main text (related point: the mix of red/green in MS16 offspring appear more green than red. Is this meant to denote a probability different than 50:50? If not, the authors should adjust the shading so that this shape is half green, half red).
We have added to the figure legend that single nucleotide differences are most likely binning errors and are therefore not considered “de novo” alleles. Instead, they are assigned it to the most similar parental allele, consistent with Figure S1. The shading at MS16 has been removed so that it is consistent with Figure 3.
Figure 3: Indicate that white background for alleles means that allelic inheritance is not determinable, or use the mix of colors applied in Fig. 1 to indicate as such. Unique offspring alleles should be flagged rather than just automatically assigned to the most similar parental allele. Finally, it would be helpful if the alleles were presented within loci from the shorter to the longer alleles.
We have included in the figure legend that non-shaded alleles are those for which multiple potential parents share the same allele and the inheritance therefore remains ambiguous for this locus. Single nucleotide differences are also now addressed, and sizes are ordered from smallest to largest.
Figure S7. Indicate visually which panels indicate FP animals.
We have now indicated which animals are FP and included this in Figure S6 as well.
Fig. S13. The 5 animals that had especially low heterozygosity should be flagged. The title of this figure should be toned down in light of the tentative nature of the conclusions regarding FP in nature: low heterozygosity could instead reflect, for example, a long history of inbreeding. My reaction to the data is also that the % heterozygosity distribution for many of the species looks continuous rather than the bimodality one might expect under FP vs. sexual reproduction.
Since FP has not been further confirmed in these animals, unlike those examples from our captive colony, there could indeed be other reasons for low heterozygosity. We have changed the title of the figure from “Facultative parthenogenesis in whiptail lizards collected in nature” to the more neutral “Heterozygosity estimates of whiptail lizards collected in nature.” Since there are so relatively few animals, one would not necessarily expect a bimodal distribution to be apparent in the current data. We did show that the animal with the lowest calculated level of heterozygosity (deppii LDOR30) was a statistical outlier when compared to other individuals of the same species though. Since these animals were sampled across different locations and habitats, the effective population sizes would be assumed to be different as well, reflecting the range of heterozygosity estimates seen here. This has been made clear in the text.
Reviewer #2 (Significance (Required)):
General assessment: strengths and limitations. The paper's strengths include the combination of data from lab and natural populations, the characterization of an unexpected means of achieving FP, with dramatic genetic consequences, and the data suggesting that this type of FP is fairly common and occurs even in the context of mating.
Audience: The biological questions of relevance to these discoveries are of broad interest, and the paper is likely to garner some attention from the life sciences community as whole and the popular press.
Advance: These data fill an important knowledge gap regarding the mechanisms potentially driving FP in vertebrates, how often FP is likely to occur, and its genetic consequences. The discoveries are potentially conceptual/fundamental, though the extent to which they are ground breaking is not clear in the absence of functional characterization of how FP occurs as well as the need for more rigorous comparisons and replication that I outlined above.
We thank reviewer 2 for summarizing the strengths of this manuscript, pointing out the broad interest and stating that this work fills an important knowledge gap.
Reviewer #3 (Evidence, reproducibility and clarity (Required)):
Summary: The occurrence of facultative parthenogenesis has been described in a number of vertebrate lineages but the underlying cytological mechanism(s) have remained largely speculative due to sparsity of data. Here, Ho & Tormey et al. provide a detailed analysis of facultative parthenogenesis in gonochoristic species of the lizard genus Aspidoscelis. They show that parthenogenesis leads to a complete loss of heterozygosity (LOH) within a single generation. They attribute the LOH to diploidization through duplication of the oocytes haploid genome after completion of meiosis. This mechanism is consistent with their finding of mixoploidy in erythrocytes of asexually produced offspring. Based on LOH the authors additionally show that facultative parthenogenesis in Aspidoscelis is not condition dependent (no developmental switch): it can occur in the presence of males, alongside with sexual reproduction in the same clutch, and both in captivity and the wild. Finally, the authors show that facultative parthenogenesis is associated with developmental aberrations, likely caused by expression of homozygous recessive deleterious mutations.
Major comments: In my opinion, this study presents a very comprehensive, careful documentation of mechanistic aspects and consequences of facultative parthenogenesis in a vertebrate. The genomic and microsatellite results leave little to no doubt that facultative parthenogenesis has led to complete LOH in Aspidoscelis. I am particularly impressed by the meticulous analysis of genomic coverage to exclude e.g. false positive heterozygosity due to merged paralogs in the assembly. I also follow the authors conclusion that a post-meiotic "gamete duplication"-like mechanism is likely causative for the LOH (and the mixoploidy of erythrocytes; but I am no expert on that). I was wondering if terminal fusion automixis together with a complete absence of recombination would be worth mentioning as an (probably very unlikely) alternative in the discussion. It would be exciting to corroborate the conclusion of diploidization by genome duplication in the future, e.g. via early embryonic DNA stainings to show the duplication "in action" (if that is practically possible)...? As for this manuscript, I suggest emphasizing the indirect nature of the evidence for the mechanism of parthenogenesis a little bit more.
We thank the reviewer for highlighting the effort that went into the genomic analysis that led us to our conclusions. In terms of terminal fusion without recombination, we argue that this is not an obvious alternative explanation as a large body of work has established that at least one crossover per homologous chromosome pair is required to advance into meiosis I in many organisms (e.g. see https://doi.org/10.3389/fcell.2021.681123) and therefore the absence of recombination would likely not produce the polar bodies necessary for automixis.
We have added to the text: “In whiptail lizards, we have not been able to examine post-meiotic oocytes as locating the post-meiotic nucleus within a large yolked egg is inherently difficult. The difficulty is compounded by the unpredictability of which eggs will undergo FP development and the need to sacrifice animals to remove eggs.”
While the genome duplication mechanism we propose is indeed indirect because we are unable to visualize developing FP embryos, the most parsimonious explanation from the whole-genome sequencing analysis is genome duplication because of the lack of heterozygous regions associated with automixis. In the text, we have made sure to state genome-wide homozygosity as the basis for our conclusion.
I agree that facultative parthenogenesis in the presence of males hints at a baseline rate of parthenogenesis without requiring a developmental switch. However, this makes it difficult to rule out that sperm played a role in activation of embryonal development (gynogenesis; however I am only aware of gynogenesis in fishes and amphibians)... maybe, the authors want to take this up in the discussion. Were the five parthenogenetic individuals for whole genome sequencing actually produced in the presence of males, too?
FP has been reported to occur in isolated females for other reptile and bird species, suggesting that sperm activation is at least not a general requirement in FP of amniotes. (Watts, et al. 2006, W. W. Olsen, S. J. Marsden 1954). In all cases in this study, the female mothers were housed with conspecific or heterospecific males. While we cannot completely rule out a non-genetic contribution of sperm in these cases, it would seem to be an unlikely explanation in light of the sperm-independent reproduction by obligate parthenogenesis in other species of whiptail lizards (unlike the sperm-dependence of all unisexual reproduction in amphibians and fish). We decided to not include speculation on sperm-dependence in this manuscript as we have no evidence in favor of it, nor is there any evidence for this in the literature relating to other amniotes. In fact, most examples of FP were reported from isolated females, most likely because offspring were not expected in those cases and prompted further analysis as to their origin.
I agree with the interpretation of the LOH in the RADseq data as a likely case of facultative parthenogenesis in the wild. However, when looking at figure S13 I noticed some bimodal looking distributions (e.g. in A. guttatus). It may be interesting for future studies to look into what factors influence heterozygosity in natural populations of Aspidoscelis (e.g. inbreeding vs parthenogenesis). Could there be different mechanisms of facultative parthenogenesis in different Aspidoscelis species explaining different LOH intensities?
The continuous nature of the data may reflect natural variation between individuals and collection at various locations with possibly different effective population sizes and levels of hybridization. Low levels of heterozygosity could be indicative of inbreeding or FP in some cases. This is important to note in future studies and we have added this to the manuscript (“Further fieldwork and analysis will be required to assess the level of FP in natural populations of gonochoristic Aspidoscelis species (and other factors that could influence the observed heterozygosity such as population size, levels of hybridization, and inbreeding) …”). While there are different mechanisms of FP in other vertebrate groups, the most parsimonious hypothesis is that within a genus, the mechanism would be the same.
The manuscript is well written, the introduction nicely explains the significance of the study, the methods are fully appropriate and the results (and supplementary results) displayed comprehensibly and in great detail. The discussion might benefit from going a bit more generally into the occurrence and mechanism of obligate asexuality in Aspidoscelis. One might e.g. speculate on whether the ability for facultative parthenogenesis in gonochoristic species has facilitated the transitions to obligate parthenogenesis in the hybrid lineages and what peculiarities might predispose Aspidoscelis to parthenogenesis (e.g. are centrioles contributed by sperm required?). In addition, I think the occurrence of LOH due to gamete duplication (facultative and obligate) in invertebrates (e.g. due to Wolbachia) is worth mentioning in the discussion: e.g. there is a similar case in facultative asexual Bacillus rossius stick insects, where the early dividing cells are haploid. Some of them diploidize via duplication later and form the embryo.
Thank you for complimenting each section of the manuscript and referring to it as well-written. Our lab has a long-standing interest in obligate parthenogenesis. While it is interesting that both obligate and facultative parthenogenesis occur alongside each other in this genus, the mechanisms appear to be fundamentally different, and we would like to focus the discussion on FP in a variety of systems and its potential implications in conservation and evolution. Parthenogenesis in general is a fascinating topic for a broad audience and not discussing another form of parthenogenesis (obligate in this case), the focus remains on FP and keeps the manuscript more accessible for non-specialists. We have included the stick insect as another example of diploid restoration through genome duplication in the discussion.
Minor comments:
39-41: I am a bit puzzled by the usage of the term "post-meiotic" to contrast the diploidization through duplication with automixis. Wouldn't one consider polar body fusion after completion of meiosis II also post-meiotic? Maybe I am just not aware of how the term is usually used in this context here...
We use the term “post-meiotic” because the restoration of an entirely homozygous diploid cell can only occur after the completion of both meiotic divisions. It is our understanding that polar body fusion and meiotic restitution after meiosis I or meiosis II are generally considered meiotic mechanisms in the specialized literature, even though polar body fusion would also occur after the meiotic divisions.
65: isn't that gynogenesis (sperm-dependent parthenogenesis) in the amazon molly?
While sperm is required for parthenogenesis in the Amazon Molly, it is an all-female species that exclusively reproduces through gynogenesis. In this case, it is considered an example of obligate parthenogenesis rather than FP.
78: the term "economically viable" may be a bit puzzling for a biologist's audience. "Economically sustainable" could be an alternative.
This has been changed.
129: the Arizona male was referred to as ID 4272 above. Here it is ID 4238?
This has been corrected. The correct ID is 4272.
218: please define over-assembly (see line 207)
The definition of “over-assembly” is collapsing paralogous loci into a single representative sequence. This is now explained in the text.
263-281: please, indicate a hatching rate/ rate of malformations of sexually produced offspring for comparison.
A comparison has been added: “This is in stark contrast to sexually produced animals, where over 98% of hatchlings had no abnormalities noted.”
333: in the haploid cells recessive deleterious mutations would be exposed in the hemizygous state but in the diploid cells in the homozygous state.
The text has been modified to reflect the difference between haploid and diploid cells.
470: please, provide more detail for the RADseq analyses (variant calling, calculation of heterozygosity etc.)
We have elaborated on the analysis in the methods.
Figure 1B: please, mention in the legend that the shown mechanisms are not exhaustive, e.g. first polar body fusion could occur right after meiosis 1 or polar body formation could be skipped completely.
This has been added.
Figure 1C: it may be interesting for non-specialists to name the distinctive morphological characters setting apart the three species in the figure legend and highlight them e.g. with arrows in the figure.
We have now included in the figure legend characteristic color patterns for each species: “(C) Photographs of Aspidoscelis arizonae with characteristic blue ventral coloration (top), A. gularis with light spots in dark fields that separate light stripes on dorsum (middle), and A. marmoratus with light and dark reticulated pattern on dorsum (bottom).” Since the descriptions are specific and apparent, we did not add arrows to the pictures.
Reviewer #3 (Significance (Required)):
Significance: The study by Ho & Tormey et al. substantially enhances the understanding of (facultative) asexuality in vertebrates. In particular, while most reports of facultative parthenogenesis in vertebrates have been attributed to a form of automixis, the authors conclusively show an instance of diploidization through genome duplication, a mechanism functionally similar to "gamete duplication". The study is novel, very comprehensive and of interest for a general audience within the field of evolutionary biology.
We thank reviewer 3 for pointing out that our study substantially enhances the understanding of asexuality in vertebrates, is very comprehensive and of interest for a general audience within
-
Note: This preprint has been reviewed by subject experts for Review Commons. Content has not been altered except for formatting.
Learn more at Review Commons
Referee #2
Evidence, reproducibility and clarity
Summary:
The researchers bring together microsatellite and whole-genome sequencing data from long-term laboratory cultures of lizards to discover occasional production of parthenogenetic offspring by several species of otherwise sexually producing whiptail lizards ("facultative parthenogenesis, "FP") and to show that these FP-produced lizards have patterns of genomic homozygosity that are incompatible with currently held assumptions about mechanisms of FP. Instead, the FP lizards seem to have been produced by a mechanism that results in almost complete homozygosity, likely a consequence of post-meiotic duplication of genomes from haploid unfertilized oocytes. They also show that FP offspring were produced by females housed with males and along with sexually produced offspring, counter to prevailing assumptions that FP offspring are only produced in situations where mates are not available. Many of the FP-produced offspring did not survive to hatching or had major abnormalities, consistent with a situation where this high homozygosity exposes harmful alleles. Finally, the authors used reduced-representation sequencing (RAD-seq) to survey heterozygosity in 321 wild-collected whiptail lizards from 15 species, showing evidence for strikingly low homozygosity in at least one individual and perhaps up to 5, consistent with the potential for FP in nature. These data are of broad interest in demonstrating several exciting new possibilities. Most importantly, the data hint at a different mechanism of FP than previously assumed, and one that causes immediate near-complete homozygosity. This scenario would likely lead to immediate purging of harmful recessive alleles. If the selective load of this purging wasn't insurmountably high, a lineage with a history of purging could produce FP offspring of relatively high fitness. Other exciting possibilities suggested by the data include the existence of FP even in a setting where mating occurs and in natural populations, versus just captivity.
Major Comments:
-
I found it difficult to impossible to sort out exactly what the researchers did and with what lizards. For example, in line 107, they refer to a "systematic MS analysis" for all individuals of gonochoristic species in their laboratory, but where are these data? Indeed, at this early spot in the paper, the introduction from here on out suddenly reads like a discussion. What would be better here would be to summarize what was known and wasn't known about the system and questions involved, why gaps in knowledge were important, and what the researchers actually did for this paper. In my opinion, the paper would be a much easier read if the researchers left the results and interpretation for later in the paper.
-
Even with this suggested fix, however, the data are still too inaccessible and analyses too opaque. For example, in line 202, a critical definition is laid out regarding heterozygous sites as those having "equal support" for two alleles. What do the researchers mean by "equal support"? My presumption is that this is something about equal or close to equal numbers of reads, but this definition needs to be spelled out and justified because it underpins much of the downstream analyses. A similar problem occurs in line 208-209, where the authors make a statement about limiting further analysis to positions in the genome where the coverage is "equal" to the mean sequencing depth.
-
Another data/analysis issue emerges with the components of the manuscript that deal with mixoploidy. As far as I can tell, these data come from one sexually produced lizard, one FP A. marmoratus, and one FP A. arizonae. While the reports of bimodality of nuclear size are certainly interesting, the data and discussion are no more than an anecdotal case study in the absence of careful replication across multiple FP lizards and comparison to sexually produced lizards. Without these data, the conclusion that "Animals produced by facultative parthenogenesis are characterized by mixoploidy" (Figure 4 caption; also see lines 324-331) is far too strong.
-
I had a similar reaction to the discussion of developmental abnormalities and embryonic lethality of embryos of FP origin presented in lines 263-281 (also lines 307-309). What is the baseline level of such abnormalities and the frequency of lethality in sexually produced eggs/embryos/hatchlings, and especially those produced via inbreeding? These comparisons are needed to interpret the significance of the patterns observed in the FP eggs/embryos/hatchings. Analogously, the comparison of the ovaries and germinal vesicles from one FP individual relative to one sexual individual do not tell us anything nearly so definitive as the text in lines 279-281 (also see Fig. S12 title, which is too broad of a conclusion for N = 1). This overly ambitious conclusion also underpins the discussion regarding the potentially adaptive nature of FP with respect to genome purification (lines 341-363; also see lines 47-50). If FP does not actually increase the rate of purging in FP lizards relative to inbred sexual counterparts (sounds like inbreeding is common from line 339), it seems less likely that we can view FP as adaptive at least from this perspective.
-
A final data concern is with the use of liver tissue for whole-genome sequencing and reference genome assembly (lines 389-390) and then using these data and the reference genome to make conclusions about ploidy/coverage. Liver tissue is very commonly endopolyploid, meaning that coverage could be artificially high for animals for which liver (vs. tail) tissue was used for DNA extraction. In particular, it would be helpful if the researchers consider whether endopolyploidy could have affected their ability to make accurate estimation of coverage and thus, heterozygosity, when libraries generated from diploid (tail) tissues are aligned to a reference genome generated from a polyploid tissue as was done here.
Minor Comments:
-
Line 410: Please explain why the BLAST cutoff was changed from the default.
-
Lines 441-443: Please explain why this dataset was seemingly larger than expected.
-
Line 510: The link to the Github repository was broken, so I was unable to access the code and data denoted as available here.
-
Figure 1, and other figures featuring comparisons of MS data across parents and offspring: The authors need to engage here with the alleles that do not match either parent here (e.g., allele 282 at MS7), explaining the likelihood that these alleles indeed represent a binning error (or, perhaps, stepwise mutation from parental allele), and these alleles should be flagged. Instead, they bin these unique alleles with the most similar parental allele without any explanation or flagged. The authors do bring this point up in Figure S1, but this issue needs to be addressed in the main text (related point: the mix of red/green in MS16 offspring appear more green than red. Is this meant to denote a probability different than 50:50? If not, the authors should adjust the shading so that this shape is half green, half red).
-
Figure 3: Indicate that white background for alleles means that allelic inheritance is not determinable, or use the mix of colors applied in Fig. 1 to indicate as such. Unique offspring alleles should be flagged rather than just automatically assigned to the most similar parental allele. Finally, it would be helpful if the alleles were presented within loci from the shorter to the longer alleles.
-
Figure S7. Indicate visually which panels indicate FP animals.
-
Fig. S13. The 5 animals that had especially low heterozygosity should be flagged. The title of this figure should be toned down in light of the tentative nature of the conclusions regarding FP in nature: low heterozygosity could instead reflect, for example, a long history of inbreeding. My reaction to the data is also that the % heterozygosity distribution for many of the species looks continuous rather than the bimodality one might expect under FP vs. sexual reproduction.
Significance
General assessment: strengths and limitations. The paper's strengths include the combination of data from lab and natural populations, the characterization of an unexpected means of achieving FP, with dramatic genetic consequences, and the data suggesting that this type of FP is fairly common and occurs even in the context of mating.
Audience: The biological questions of relevance to these discoveries are of broad interest, and the paper is likely to garner some attention from the life sciences community as whole and the popular press.
Advance: These data fill an important knowledge gap regarding the mechanisms potentially driving FP in vertebrates, how often FP is likely to occur, and its genetic consequences. The discoveries are potentially conceptual/fundamental, though the extent to which they are ground breaking is not clear in the absence of functional characterization of how FP occurs as well as the need for more rigorous comparisons and replication that I outlined above.
-
-
-
www.biorxiv.org www.biorxiv.org
-
Note: This response was posted by the corresponding author to Review Commons. The content has not been altered except for formatting.
Learn more at Review Commons
Reply to the reviewers
Reviewer #1 (Evidence, reproducibility, and clarity (Required)):
- In this manuscript, Imoto et al. analyze the specific role of the Dynamin1 splice variant Dyn1xA in so-called ultrafast endocytosis, an important mechanism of synaptic vesicle recycling at synapses. In a previous publication (Imoto et al. Neuron 2022), some of the authors had shown that Dyn1xA, and not the other splice variant Dyn1xB, is essential for ultrafast endocytosis. Moreover, Dyn1xA forms clusters around the active zone for exocytosis and interacts with Syndapin 1 in a phosphorylation dependent manner. However, it was unclear which molecular interactions underlie the specific role of Dyn1xA. Here, the authors provide convincing evidence with pull down assays and CSP that Dyn1xA PRR interacts with EndophilinA1/2 with two binding sites. The first binding site lies in the part common to xA and xB, was previously characterized. The second site was previously uncharacterized, is specific for Dyn1xA, and is regulated by phosphorylation (phosphobox 2). The location of these splice variants and mutated forms at presynaptic sites correlate with the prediction made by the biochemical assays. Finally, the authors perform rescue experiments ('flash and freeze' and VGLUT1-pHluorin imaging experiments) to show that Dyn1xA-EndophilinA1/2 binding is important for ultrafast endocytosis. I find the results interesting, providing an important step in the understanding of the interplay between dynamin and the endocytic proteins interacting with it (endophilin, syndapin, amphiphysin) in the context of synaptic vesicle recycling. The manuscript is clearly written and for the most part the data supports the authors' conclusions (see specific comments below). However, there are some issues which need to be clarified before this manuscript is fully suitable for publication.
We thank the reviewer for noting the importance of our study. Indeed, our previous study has raised the question as to why only the Dyn1xA splice variant mediates ultrafast endocytosis, and our current manuscript now resolves this issue.
Introduction: the dynx1B Calcineurin binding motif is written PxIxIT consensus but actual sequence is PRITISDP. Is this a typo?
The sequence is correct. One thing we failed to mention is that the last amino acid in this motif can be either threonine or serine for calcineurin binding, as we demonstrated previously [Jing, et al., 2011 JBC; PMC3162388]. We have amended the text as follows.
- calcineurin-binding motif (PxIxI[T/S]) 19.
Figure 1: the difference between the constructs used in panels C and D is not clear. In D, is it a truncation without residues 796 and 845? If so, it should be labelled clearly in the Western blots. In Panel E, Dyn1xA 746-798 should be labeled Dyn1x 746-798 because it is common to both splice variants.
We thank the reviewer for pointing this out. Both C and D used the full-length PRRs of Dyn1xA-746 to 864 and xB-746 to 851. To make the labeling clear, we changed Dyn1xA PRR to “Dyn1xA PRR (746-864)” and Dyn1xB PRR to “Dyn1xB PRR 746-851” in Figure 1. In the main text, we made the following changes.
- 4: “To identify the potential isoform-selective binding partners, the full-length PRRs of Dyn1xA746-864 and xB746-851 (hereafter, Dyn1xA-PRR and Dyn1xB-PRR, respectively).”
Figure 1: For amphiphysin binding the authors write that "No difference in binding to Amphiphysin 1 was observed among these peptides (Figure1D-F)." They should write that Dyn1x 746-798 does not bind Amphiphysin1 SH3 domain, confirming the specificity of binding to the 833-838 motif.
We edited the sentence as suggested.
- “Dyn1x 746-798 does not bind Amphiphysin1 SH3 domain (Figure 1G), confirming the specificity of binding to the 833-838 motif as reported in previous studies 29,30. (Figure 1D-F).”
Figure S2. The panels are way too small to see the shifts and the labelling. Please provide bigger panels
As suggested, we have now provided bigger panels in Figure S2, and amended the text and Figure legend accordingly.
We also removed Figure S2B as it was not referred to in the text in any way. (It was the reverse experiment – HSQCs of 15N-labelled SH3 titrated with unlabelled dynamin).l
Figure 2 panel B. There is a typo in the connecting line between the sequence and the CSP peaks. It is 846 instead of 864 (after 839).
Corrected.
Figure 3 panel E. In the text, the authors write that "Western blotting of the bound proteins from the R838A pull-down experiment showed that R838A almost abolished both Endophilin and Amphiphysin binding in xA806-864 (Figure 3D), and reduced Endophilin binding to xA-PRR (Figure 3E)." I think they should write "only slightly reduced Endophilin binding..." it is more faithful to the result and consistent with the conclusion that Endophilin A1 has two binding sites on Dyn1xA PRR.
We have now provided quantitative data for R838A and R846A (Fig. 3F and G). Endophilin binding is significantly reduced with R846A.
It is unclear why the R846A mutant affects binding of Dyn1xA 806-864 but not Dyn1xA-PRR-.
The reviewer asks why the R846A mutant affects binding of Dyn1xA 806-864, but not so much of Dyn1xA-PRR. The explanation is simply that there are two endophilin binding sites in Dyn1xA-PRR. The first is not present in the xA806-864 peptide, while both are present in Dyn1xA-PRR (the full length tail). When doing pull-down experiments, the binding tends to saturate – even when the second site is blocked by R846A. The first site is still able to bind, and the binding appears as normal. The same applies to the R838A mutant.
Moreover, it affects binding to endophilin as well as amphiphysin, and therefore it is not specific. It is thus not correct to write that "R846 is the only residue found to specifically regulate the Dyn1 interaction with Endophilin as a part of an SDE". In the Discussion (page 11), the authors refer to the R846A mutation as specifically affecting Endophilin binding. This should be toned down, as it also affects Amphiphysin binding. For this important point, the data on quantification of Endophilin binding should be presented.
The reviewer’s concern is about our claims of specificity of Endophilin A binding in Dyn1xA R846 mutation experiments. The reviewer is correct, and we have now defined specific parameters for those claims. Specifically, we have added new quantitative data from the Western blots in Fig 3E (full-length Dyn1aX-PRR) as Fig 3F-G. We used full-length Dyn1aX-PRR rather than the xA806-864 peptide because the subsequent transfection experiments use full length Dyn1xA. In the new figures 3F and 3G, we quantified Endophilin A, Amphiphysin and Syndapin1 amounts from the multiple Western blots such as Figure 3E (now n=14, 6 experiments, each in with 2-4 replicates for Dyn1xA PRR). R846A mutated in Dyn1xA-PRR significantly reduces the binding to Endophilin A, but it does not significantly affect the binding to Amphiphysin 1and Syndapin1 (Fig 3G). Therefore, this particular Dyn1xA-PRR mutation specifically affects Endophilin A binding, in the context of the full-length tail Dyn1aX-PRR. To make these results clear, we modified the text as below.
P7. “R838A and R846A caused smaller reductions in Endophilin binding compared to wild-type Dyn1xA-PRR, (Figure 3E, 3F, R838A, median 68.5 ; Figure 3G, R846A, median 59.3 % : R838A reduced the Dyn1/Amphiphysin interaction (Figure 3E, 3F, median 14.2 % binding compared to wild-type Dyn1xA-PRR). By contrast, R846A did not affect Amphiphysin and Syndapin binding to Dyn1xA-PRR (Figure 3E, 3G). Therefore, R846, being part of an SDE, is the only residue we found to specifically regulate the Dyn1 interaction with Endophilin in the context of the full length tail (DynxA-PRR)”.
Additionally, the reviewer notes that “the authors refer to the R846A mutation as specifically affecting Endophilin binding. This should be toned down, as it also affects Amphiphysin binding.” In the light of the above data and new quantitative analysis (Fig 3F-G), we have clarified the conclusion. However, to be clear that this statement is only correct in the context of the full-length DynxA-PRR, we amended texts as follows:
P7. “By contrast, R846A did not affect Amphiphysin and Syndapin binding to Dyn1xA-PRR (Figure 3E, 3G). Therefore, R846, being part of an SDE, is the only residue we found to specifically regulate the Dyn1 interaction with Endophilin in the context of the full length tail (DynxA-PRR)”.
New legends for Figure 3F and G have now been added as follows.
“(F) The binding of Endophilin A, and Amphiphysin 1 and Syndapin1 to Dyn1xA-PRR (wild type) or R838A mutant quantified from Western blots in (E). n=14 (6 experiments with 2-4 replicates in each). Median and 95% confidential intervals are shown. Kruskal-Wallis with Dunn’s multiple comparisons test (**p (G) The binding of Endophilin A, and Amphiphysin 1 and Syndapin1 to Dyn1xA-PRR (wild type) or R846A mutant quantified from Western blots in (E). n=14 (6 experiments with 2-4 replicates in each). Median and 95% confidential intervals are shown. Kruskal-Wallis with Dunn’s multiple comparisons test was applied (*p
Figure 3F-G (which are now 3H and 3I in the revised text): what do the star symbols represent in the graphs? I guess the abscissa represents retention time. Please write it clearly instead of a second ordinate for molecular mass, which does not make much sense if this reflects the estimate for the 3 conditions.
The “stars” are crosses (x) and represent individual data points. The figure legends have been updated for clarity. The reviewer is correct that the X-axis is retention time (min). The second Y-axis is needed to define the points in the curve marked with crosses (x’s). The legends for Figure 3H and I are now changed as follows.
“(H) SEC-MALS profiles for Dyn1xA alone (in green), Endophilin A SH3 alone (in red) and the complex of the two (in black) are plotted. The x-axis shows retention time. The left axis is the corresponding UV absorbance (280 nm) signals in solid lines, and the right axis shows the molar mass of each peak in crosses. The molecular weight of the complex was determined and tabulated in comparison with the predicted molecular weight. x represent individual data points.
(I) SEC-MALS profiles for a high concentration of Dyn1xA-PRR/Endophilin A SH3 complex (0.5 mg) (in dark blue) and a low concentration of Dyn1xA-PRR/endophilin A SH3 complex (0.167 mg) (in blue). The x-axis shows retention time. The left axis is the corresponding UV absorbance (280 nm) signals in solid lines, and the right axis shows the molar mass of each peak in crosses. The molecular weight of the complex was determined and tabulated in the table. x represent individual data points.”
Figure 4: The statement that "By contrast [to Dyn1xA], Endophilin A1 or A2 formed multiple clusters (1-5 clusters)" is not at all clear on the presented pictures. The authors should provide views of portions of axons with several varicosities, for the reader to appreciate the cases where there are more EndoA clusters than Dyn1 clusters.
In the revised Figure S4, we added additional STED images for a region of axons with more EndoA1/2 clusters than Dyn1xA clusters. The locations of Dyn1xA and EndoA1/2 clusters are annotated in each image based on the local maximum of intensity, which is determined using our custom Matlab analysis scripts (Imoto, et al., Neuron 2022; for the description of the methods, please refer to the Point #14 below). We also added Figure S3 to describe our analysis pipelines. In the Dyn1xA channel, outer contour indicates 50% of local maxima (boundary of Dyn1xA cluster) while inner contour indicates 70% of local maxima of the clusters. In the EndoA1/2 channel, local maxima of the clusters are indicated as points. To reflect these changes, we modified text as below.
P 9. “By contrast, Endophilin A1 or A2 formed multiple clusters (1-5 clusters) (Figure S4)”
The legends for Figure S4 are now as follows.
“Figure S4. Additional STED images for Figure 4.
(A) The top image shows an axon containing multiple boutons. Signals show overexpression of GFP-tagged Dyn1xA (Dyn1xA) and mCherry-tagged Endophilin A1 (EndoA1). The bottom images show magnifications of four boutons in the top image. Red hot look-up table (LUT) images on the right side of Dyn1xA and EndoA1 images are enhanced contrast images. Outer and inner contours represent 50% and 70% of local maxima of the Dyn1xA, respectively. Black circles represent local maxima of Endophilin A1. In these boutons, multiple EndophilinA1 puncta are present.
(B) The top image shows an axon congaing multiple boutons. Signals show overexpression of mCherry-tagged Dyn1xA (Dyn1xA) and GFP-tagged Endophilin A1 (EndoA1). The bottom images show magnifications of four boutons in the top image. Red hot LUT images on the right side of Dyn1xA and EndoA2 images are enhanced contrast images. Outer and inner contours represent 50% and 70% of local maxima of the Dyn1xA, respectively. Black circles represent local maxima of Endophilin A2. In these boutons, multiple EndophilinA2 puncta are present.
(C) STED micrographs of the same synapses as in Figure 4E with an active zone marker Bassoon (magenta) visualized by antibody staining. GFP-tagged Dyn1xA, Dyn1xA S851D/857D or Dyn1xA R846A (green) are additionally stained with GFP-antibodies. Local maxima of Dyn1xA, Dyn1xA S851D/857D or Dyn1xA R846A signals and minimum distance to the active zone boundary are indicated by dark blue lines.”
Moreover, overexpression of EndophilinA1/2-mCherry is not sufficient to assess its localization. Please consider either immunofluorescence or genome editing (e.g. Orange or TKIT techniques).
We agree with the reviewer that overexpression obscures the endogenous localization of proteins. To address this point in our previous publication, we titrated the amount of plasmids for Dyn1xA-GFP and transfected neurons just for 20 hours – this protocol allowed us to uncover the endogenous localization of Dyn1xA despite the fact that it was overexpressed in wild-type neurons (Imoto, et al., 2022). We also confirmed this localization by ORANGE-based CRISPR knock-in of GFP-tag in the endogenous locus of Dyn1 just after the exon 23 and confirm the true endogenous localization of Dyn1xA (Imoto, et al., 2022). Similar approaches were taken by the Chapman lab to localize Synaptotagmin-1 and Synaptobrevin 2 in axons (Watson et al, 2023, eLife, PMID: 36729040). We did not emphasize this in the first submission, but we took the same approach for the EndoA1/2 localization. This does not mean that they also unmask the endogenous localization, and the reviewer is correct that additional evidence would strengthen the data here. Thus, as suggested, we have looked at the endogenous EndophilinA1 localization by antibody staining. As the reviewer is likely aware, EndophilinA1 also localizes to other places including dendrites and postsynaptic terminals, making it difficult to analyze the data. However, we observe colocalization of Dyn1xA with endogenous EndoA1. Thus, we believe that our major conclusion here drawn based on EndoA1/2-mCherry overexpression is valid (Reviewer’s Figure 1). Since the Endophilin signals in neighboring processes obscures its localization in synapses-of-interest, repeating this localization experiments with ORANGE-based knock-in would be ideal. However, with the lead author starting his own group and many validations needed to confirm the knock-in results, this experiment would require us at least 4-6 months, and thus, it is beyond the scope of our current study. We will follow up on this localization in the near future, but given that endophilin is required for ultrafast endocytosis (Watanabe, et al., Neuron 2018, PMID: 29953872) and these proteins need to be in condensates at the endocytic sites for accelerating the kinetics of endocytosis (Imoto, et al., Neuron 2022, PMID: 35809574), we are confident that endogenous
EndoA1/2 are localized with Dyn1xA.
The analysis of the confocal microscopy data is not explained. How is the number of clusters determined? How far apart are they? Confocal microscopy may not have the resolution to distinguish clusters within a synapse.
We apologize for the insufficient description of the method. We had provided a more thorough description of the methods in our previous publication (Imoto, et al., Neuron 2022, PMID: 35809574). To make this more automated, we improved our custom Matlab scripts. Please note that all the analysis for the cluster location is performed on STED images, not on normal confocal images. To determine the cluster, first, presynaptic regions (based on Bassoon signals or Dyn1xA signals within boutons) in each STED image are cropped with 900 by 900 nm (regions-of-interest) ROIs. Then, our Matlab scripts calculate the local maxima of fluorescence intensity within the ROIs. To determine the distance between the active zone and the Dyn1xA or EndoA1/2 clusters, the Matlab scripts perform the same local maxima calculations in both channels and make contours at 50% intensity of the local maxima. The minimum distance reflects the shortest distance between the active zone and Dyn1xA/EndoA1/2 contours. To make these points clearer, we modified the main text and the Methods section. In addition, we have added workflow of these analysis as Figure S3.
P9. Main. “Signals of these proteins are acquired by STED microscopy and analyzed by custom MATLAB scripts, similarly to our previous work23.”
P20. Methods. “All the cluster distance measurements are performed on STED images. For the measurements, a custom MATLAB code package23 was modified using GPT-4 (OpenAI) to perform semi-automated image segmentation and analysis of the endocytic protein distribution relative to the active zone marked by Bassoon or relative to Dyn1xA cluster in STED images. First, the STED images were blurred with a Gaussian filter with radius of 1.2 pixels to reduce the Poisson noise and then deconvoluted twice using the built-in deconvblind function: the initial point spread function (PSF) input is measured from the unspecific antibodies in the STED images. The second PSF (enhanced PSF) input is chosen as the returned PSF from the initial run of blind deconvolution62. The enhanced PSF was used to deconvolute the STED images to be analyzed. Each time, 10 iterations were performed. All presynaptic boutons in each deconvoluted image were selected within 3030-pixel (0.81 mm2) ROIs based on the varicosity shape and bassoon or Dyn1xA signals. The boundary of active zone or Dyn1xA puncta was identified as the contour that represents half of the intensity of each local maxima in the Bassoon channel. The Dyn1xA clusters and Endophilin A clusters were picked by calculating pixels of local maxima. The distances between the Dyn1xA cluster and active zone boundary or Endophilin A clusters were automatically calculated correspondingly. For the distance measurement, MATLAB distance2curve function (John D'Errico 2024, MATLAB Central File Exchange) first calculated the distance between the local maxima pixel and all the points on the contour of the active zone or Dyn1xA cluster boundary. Next, the shortest distance was selected as the minimum distance. Signals over crossing the ROIs and the Bassoon signals outside of the transfected neurons were excluded from the analysis. The MATLAB scripts are available by request.”
In the legend of Figure S3,
“Protein localization in presynapses is determined by semi-automated MATLAB scripts (see Methods).
(A) Series of deconvoluted STED images are segmented to obtain 50-100 presynapse ROIs in each condition.
(B) Two representations of the MATLAB analysis interface are shown. The first channel (ch1, green) is processed to identify the pixels of local maxima within this channel. The second channel (ch2, magenta) is normally an active zone protein, Bassoon. Active zone boundary is determined by the contour generated at 50% intensity of the local maxima of ch2. The contours outside of the transfected neurons are manually selected on the interface and excluded from the analysis. Minimum distances from each pixel of the local maxima in ch1 to the contour in ch2 are calculated and shown in the composite image. The plot “Distance distribution” shows all the minimum distance identified in this presynapses ROI (unit of the y axis is nanometer). The plot “Accumulated distance distribution” shows the accumulated distance distribution from the initial to the current presynapses ROI. The plot “Histogram of total intensity” shows the intensity counts around individual local maxima pixels in ch1.”
For the STED microscopy, a representation of the processed image (after deconvolution) and the localization of the peaks would be important to assess the measurement of distances. If Dyn1xA S851/857D is more diffuse, are there still peaks to measure for every synapse?
We thank the reviewer for bringing up this important question. In Figure S4C, we have added the position of the local maxima of wild-type and mutant Dyn1xA shown in the main Figure 4E. As the reviewer pointed out, when a protein is more diffuse, it is difficult to find the peak intensity by STED. However, since these proteins are still found at a higher density within a very confined space of a presynapse and synapses are packed with organelles like synaptic vesicles and macromolecules, signals from even diffuse proteins can be detected as clusters, and local maxima can be detected in these images.
To illustrate this point better, we added Reviewer’s Figure 2 below. In this experiment, we transfected neurons with a typical amount of plasmids (2.0 µg/well) or ~10x lower amount (0.25 µg/well). When the density of cytosolic proteins is high (Reviewer’s Figure 2A), the depletion laser has to be strong enough to induce sufficient stimulated emission and resolve protein localization. Insufficient power would produce low resolution images, leading to inappropriate detection of the local maxima (Reviewer’s Figure 1A). Thus, we set our excitation and depletion laser powers to resolve the protein localization to ~40-80 nm at presynapses. Furthermore, to avoid mislocalization of proteins due to the overexpression, we use 0.25-0.5 ug/well (in 12-well plate) of plasmid DNA for transfection, which is around 10 times lower than the amount used in the typical lipofectamine neuronal transfection protocol (Imoto, et al., Neuron 2022). We also change the medium around 20 hours after the transfection instead of the typical 48 hours (Imoto, et al., Neuron 2022). With these modifications and settings, we can obtain the location of the local maxima of the diffuse signals (Reviewer’s Figure 1B and Figure 4E and Figure S4). We modified the Method section to make these points clearer.
P 17, “Briefly, plasmids were mixed well with 2 µl Lipofectamine in 100 µl Neurobasal media and incubated for 20 min. For Dyn1xA and Endophilin A expressions, 0.5 µg of constructs were used to reduce the overexpression artifacts23. The plasmid mixture was added to each well with 1 ml of fresh Neurobasal media supplemented with 2 mM GlutaMax and 2% B27. After 4 hours, the medium was replaced with the pre-warmed conditioned media. To prevent too much expression of proteins, neurons were transfected for less than 20 hours and fixed for imaging.”
P 20, “Quality of the STED images are examined by comparing the confocal and STED images and measuring the size of signals at synapses and PSF (non-specific signals from antibodies).”
Legends for Figure S4C,
“(C) STED micrographs of the synapses shown in Figure 4F with an active zone marker Bassoon (magenta). GFP-tagged Dyn1xA, Dyn1xA S851D/857D or Dyn1xA R846A are visualized by antibody staining of GFP (green). Local maxima of Dyn1xA, Dyn1xA S851D/857D or Dyn1xA R846A signals and minimum distance to the active zone boundary are overlaid.”
Figures 5 and 6: No specific comment. The data and its analysis are very nice and elegant. The comment on the lack of rescue of Dyn1xA on endosome maturation may be a bit overstated, because many "controls" (shRNA control Figure S5 or Dyn3 KO in Imoto et al. 2022) have a significant number of endosomes 10 s after stimulation.
We thank the reviewer for noting the strength of our data and pointing out this issue on endosomal resolution. In particular, the reviewer is concerned about our interpretation of the ferritin positive endosomes present at 10 s in time-resolved electron microscopy experiments. Indeed, the number of ferritin positive endosomes in Dyn1 KO, Dyn1xA OEx neurons (0.1/profile) is similar to the control conditions: scramble shRNA control (0.1/profile, Figure S5) and Dyn3KO neurons (0.2/profile) in our previous study (Imoto et al. 2022). Although we do not consider Dyn3 KO as a control, given the presence of abnormal endosomal structures, we agree with the reviewer that scramble shRNA control in Figure S5 does indicate that some ferritin-positive endosomes even at 10 s after stimulation. We would like to note that this result is in stark contrast to our previous studies where we observed the number of ferritin positive endosomes returning to the basal level in both wild-type neurons and many scramble shRNA controls (Watanabe et al. 2014, 2018, Imoto et al 2022). Thus, the majority of the data we have indicate that the number of ferritin positive endosomes returns to basal level by 10 s, suggesting that endosomes are typically resolved into synaptic vesicles by this time. However, given that we do not know the nature of the inconsistency here and we cannot exclude the possibility of overexpression artifact of Dyn1xA as an alternative, we changed the following lines.
P. 10, “Interestingly, the number of ferritin-positive endosomes did not return to the baseline (Figure 5E, F) as in previous studies3,35,36, suggesting that Dyn1xA may not fully rescue the knockout phenotypes or that overexpression of Dyn1xA causes abnormal endosomal morphology.”
By the way, why did the authors use Dyn1 KO in this study, and not Dyn1,3 DKO as in Imoto et al. 2022?
This is simply because Dyn3KO displayed an endosomal defect in our previous study (Imoto et al 2022), and we wanted to focus on endocytic phenotypes of Dyn1 KO and mutant rescues in this study.
In the Discussion, the authors present the binding sites (for endophilin and amphiphysin SH3 domains) as independent. However, these proteins form dimers or even multimers as they cluster around the neck of a forming vesicle. Even though they provide evidence in vitro (Figure 3) that in these conditions of high concentration one dyn1xA-PRR binds one SH3 domain, in cells multiple binding sites on the PRR to these proteins may involve avidity effects, as discussed for example in Rosendale et al. 2019 doi 10.1038/s41467-019-12434-9. For example, the high affinity binding of Dyn1-PRR to amphiphysin cannot be explained only by the sequence 830-838.
The reviewer suggests “In the Discussion, the authors present the binding sites (for endophilin and amphiphysin SH3 domains) as independent.” However, we do not claim these interactions are functionally independent, except in the context of in vitro experiments where they are sequence-independent.
They also suggest “However, these proteins form dimers or even multimers as they cluster around the neck of a forming vesicle”. However we do not agree with this in the context of our Discussion, because the evidence of multimers and clustering is convincing but is entirely in vitro data.
Thirdly they comment that “For example, the high affinity binding of Dyn1-PRR to amphiphysin cannot be explained only by the sequence 830-838.” We fully agree with the statement and felt we had addressed this in the manuscript. To explain, it’s important to point out our relatively new concept here and previously reported by us (Lin Luo et al 2016, PMID: 26893375) of the existence and importance of SDE and LDE for SH3 domains (Endophilin here, syndapin in our previous report). These elements act at a distance from the so-called core PxxP motifs and they provide much higher affinity and specificity than the core region alone. We had further mentioned this in the p11 discussion “Although this is a previously characterized binding site for Amphiphysin and is also present in Dyn1xB-PRR, the extended C-terminal tail of Dyn1xA contains short and long distance elements (SDE and LDE) essential for Endophilin binding, making it higher affinity for Endophilin.” Because the NMR identified F862 as a chemical shift for dynamin, we performed a pulldown with this mutant in the xA746-798 construct (which only contains the higher affinity site) and found that indeed “.F862A reduced Endophilin binding 29% (pOverall, the reviewer correctly points out that “multiple binding sites on the PRR to these proteins may involve avidity effects*” could play a role in vivo. We agree that avidity is an additional possibility, not examined in our study. Therefore, as suggested, we added the following sentence to the discussion on the SDE and LDE impacts.
P. 11. “Our pull-down results showed that R846A abolished endophilin binding to xA806-864 (which contains only the second and higher affinity binding site and the associated SDE (A839) and LDE (F862)) and reduced about 40% of endophilin binding to the Dyn1xA-PRR (which contains both binding sites) without affecting its interaction with Amphiphysin, providing important partner specificity, although we cannot exclude the possibility that avidity effects may additionally come in play in vivo 42
Reviewer #1 (Significance (Required)):
This study provides a significant advance on the mechanisms of dynamin recruitment to endocytic zones in presynaptic terminals. The work adds a significant step by experienced labs (Robinson, Watanabe) who have provided important insight in the mechanisms by many publications in the last years.
We thank the reviewer for the careful read of our manuscript and positive outlook of our work.
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
- This is a compelling study that reports a key discovery to understand the molecular mechanism of ultrafast endocytosis. The authors demonstrate that the Dynamin splice version 1xA (Dynamin 1xA) uniquely binds Endophilin A, in contrast to Dynamin splice version 1xB (Dynamin 1xB) that does not bind Endophilin A and it is not required for ultrafast endocytosis. In addition, the Endophilin A binding occurs in a dephosphorylation-regulated manner. The study is carefully carried out and it is based on high quality data obtained by means of advanced biochemical methodologies, state-of-art flash-freezing electron microscopy analysis, superresolution microscopy and dynamic imaging of exo-and endocytosis in neuronal cultures. The results convincingly support the conclusions.
We thank the reviewer for supporting the conclusions of our study.
- Although additional experiments are not essential to support the claims of the paper there is room, however, for improvement within the pHluorin experiments. These experiments, that are clearly informative and consistent with the rest of experimental data, do not apply the useful approach to separate endo- from exocytosis. The use of bafilomycin or folimycin to block the vesicular proton pump allows the unmasking the endocytosis that is occurring during the stimulus, that should correspond to ultrafast endocytosis. It would be very elegant to demonstrate that such a component, as expected according to the electron microscopy data, requires the binding of Endophilin A to Dynamin 1xA. If the authors have the pHluorin experiments running, the suggested experiments are very much doable because the reagents and the methodology is already in place and the new data could be generated in around six weeks.
We thank the reviewer for the suggestion. The reviewer is concerned that vGlut1 pHluorin experiment in Figure 6 may not correspond to ultrafast endocytosis. We agree that bafilomycin/folimycin treatment will reveal the amount of endocytosis that takes place while neurons are stimulated. However, we are not certain that endocytosis during this phase would fully correspond to ultrafast endocytosis because reacidification of endocytosed vesicles typically takes 3-4 s (Atluri and Ryan, 2006, PMID: 16495458; although see https://elifesciences.org/articles/36097) and thus, the nature of endocytosis cannot be fully determined by this assay. To claim that endocytosis measured by pHluorin assay during stimulation all correspond to ultrafast endocytosis, we would need to perform very careful work to track single pHluorin molecules at the ultrastructural level and corelate their internalization to pHluorin signals. Perhaps, a rapid acid quench technique used by the Haucke group would also be appropriate to estimate the amount of ultrafast endocytosis (Soykan et al. 2017 PMID: 28231467), but we are not set up to perform such experiments here. Also, our lead author, Yuuta Imoto, is leaving the lab to start up his own group, and it will take us months rather than weeks to get the requested experiments done. Since the point of this experiment was to test whether the interaction of Dyn1xA and EndoA is essential for protein retrieval regardless of the actual mechanisms and the reviewer acknowledges that this point is sufficiently supported by the experiments, we will set this experiment as the priority for the next paper.
Instead of the bafilomycin or rapid acid quenching experiments, we have now added data from vglut1-pHluorin experiment with a single action potential. With a single action potential, all synaptic vesicle recycling is mediated by ultrafast endocytosis in these neurons (Watanabe et al, 2013 PMID: 24305055; Watanabe et al. 2014, PMID: 25296249). Our electron microscopy experiments in Figure 5 is also performed with a single action potential. As with 10 action potentials, 20 Hz experiments, re-acidification of vglut1-pHluorin is blocked when Dyn1 and EndophilinA1 interaction is disrupted (Figure 6 F-I). We added a description of this result as below.
P 11. “Similar defects were observed when the experiments were repeated with a single action potential – synaptic vesicle recycling is mediated by ultrafast endocytosis with this stimulation paradigm25 (S851/857 recovery is 73.3% above the baseline; R846A, recovery is 30.0% above the baseline) (Figure S9 A-D). Together, these results suggest that the 20 amino acid extension of Dyn1xA is important for recycling of synaptic vesicle proteins mediated by specific phosphorylation and Endophilin binding sites within the extension.”
The methods are carefully explained. Some of the experiments are only replicated in two cultures and the authors should justify the reasons to convince the audience that the approaches used have enough low variability for not increasing the n number. The pHluorin experiments, however, are performed only in a single culture; they should replicate these experiments in at least 3 different cultures (three different mice).
The reviewer is correct. The variability is very low in our ultrastructural studies and STED imaging, and thus, in all our previous publications, two independent cultures are used. We do agree that in the ideal case, we would like to have three independent cultures, but given the nature of ultrastructural studies (control, mutants, and multiple time points), triplicating the data would add another year to our work. We are currently developing AI-based segmentation analysis, and once this pipeline is established, we will be able to increase N. However, please note that for these experiments, we examine around 200 synapses from each condition in electron microscopy studies (Table S2)– these numbers are far more than the gold standard in the field. Likewise, 50-100 synapses are examined for STED experiments (Table S2). To examine variability of our analysis results, we compared a significance between the dataset using cumulative curves and Kolmogorov–Smirnov test (Figure S11). As shown in the summarized data and p value in each condition, there are no significant difference between the datasets.
For pHluorin analysis, the reviewer is correct. We repeated the experiments twice to increase the N after the initial submission. The data are consistent, and the conclusions are not changed by the additional experiments (Figure 6 and Figure S9). We also changed the Statistical analysis section in Methods as below.
P. 19. “All electron microscopy data are pooled from multiple experiments after examined on a per-experiment basis (with all freezing on the same day); none of the pooled data show significant deviation from each replicate (Table S2).”
p 19, “All fluorescence microscopy data were first examined on a per-experiment basis. For Figure 4, the data were pooled; none of the pooled data show significant deviation from each replicate (Figure S11 and Table S2). Sample sizes were 2 independent cultures, at least 50-100 synapses from 4 different neurons in each condition..”
Legends for Figure S11
Figure S11. Data variability in Figure 4.
Cumulative curves are made from each dataset of (A) distance of Endophilin A1 puncta from the edge of Dyn1xA puncta, (B) distance of Endophilin A2 puncta from the edge of Dyn1xA puncta, distance distribution of Dyn1xA from active zone edge in (C) neurons expressing wild-type Dyn1xA-GFP, (D) Dyn1xA-S851/857-GFP and (E) Dyn1xA-R846-GFP. n > 4 coverslips from 2 independent cultures. Kolmogorov–Smirnov (KS) test, p values are indicated in each plot.
Minor comments: 4. Prior studies referenced appropriately and the text and figures are clear and accurate.
We thank the reviewer for the careful read of our manuscript.
The authors should discuss about the mediators (enzymes) responsible for dephosphorylation of phosphor-box 2 that is key for the Dynamin 1xa-Endophilin A interaction.
We thank the reviewer for the suggestion. We added a discussion on a potential mediator, Dyrk1, as below.
P. 12. ”What are the kinases that regulate Dyn1? The phosphorylation of phosphobox-1 is mediated by Glycogen synthase kinase-3 beta (GSK3ß) and Cyclin-dependent kinase 5 (CDK5)17, while phosphobox-2 is likely phosphorylated by Trisomy 21-linked dual-specificity tyrosine phosphorylation-regulated kinase 1A (Mnb/Dyrk1)44,45 since Ser851 in phosphobox-2 is shown to be phosphorylated by Mnb/Dyrk1 in vitro32. Furthermore, overexpression of Mnb/Dyrk1 in cultured hippocampal neurons causes slowing down the retrieval of a synaptic vesicle protein vGlut146. Consistently, our data showed that phosphomimetic mutations in phosphobox-2 results disruption of Dyn1xA localization, perturbation of ultrafast endocytosis, and slower kinetics of vGlut1 retrieval. However, how these kinases interplay to regulate the interaction of Dyn1xA, Syndapin1 and Endophilin A1 for ultrafast endocytosis is unknown.”
It would be very helpful to include a final cartoon depicting the key protein-protein interactions regulated by dephosphorylation (activity) and the sequence of molecular events that leads to ultrafast endocytosis
As suggested, we made a model figure, (new Figure 7) showing how Dyn1xA and its interaction with EndoA and Syndapin1 increases the kinetics of endocytosis at synapses. Regarding the sequence of molecular events, we think that there are already dephosphorylated fraction of Dyn1xA molecules sitting on the endocytic zone at the resting state and they mediate ultrafast endocytosis. However, it is equally possible that activity-dependent dephosphorylation of Dyn1xA also may play a role (Jing et al. 2011, PMID: 21730063). However, we have no evidence about the sequence of activity dependent modulation of Dyn1xA and its binding partners during ultrafast endocytosis yet. This is much beyond what we have reported in this work and therefore, excluded from the model figure. We added the following to the end of the discussion:
p13, “Nonetheless, these results suggest that Dyn1xA long C-terminal extension allows multivalent interaction with endocytic proteins and that the high affinity interaction with Endophilin A1 permits phospho-regulation of their interaction and defines its function at synapses (Figure S7)”.
Figure legend Figure 7,
“Figure 7. Schematics depicting how specific isoforms Dyn1xA and Endophilin A mediate ultrafast endocytosis.
A splice variant of dynamin 1, Dyn1xA, but not other isoforms/variants can mediate ultrafast endocytosis. (A) Dyn1xA has 20 amino acid extension which introduces a new high affinity Endophilin A1 binding site. Three amino acids, R846 at the splice site boundary, S851 and S857, act as long-distance element which can enhance affinity of proline rich motifs (PRM) to SH3 motif from outside of the PRM core sequence PxxP. (B) At a resting state, Dyn1xA accumulates at endocytic zone with SH3 containing BAR protein Syndapin 123 and Endophilin A1/2. When phosphobox-1 (Syndapin1 binding) and phosphobox-2 (Endophilin A1/2 binding, around S851/S857) within Dyn1xA PRD are phosphorylated, these proteins are diffuse within the cytoplasm. A dephosphorylated fraction of Dyn1xA molecules can interact with these BAR domain proteins. Loss of interactions including Dyn1xA-R846A or -S851/857D mutations, disrupts endocytic zone pre-accumulations. Consequently, ultrafast endocytosis fails.”
Reviewer #2 (Significance (Required)):
This is a remarkable and important advance in the field of endocytosis. The study reports a key discovery to understand the molecular mechanism of ultrafast endocytosis. Scientist interested in synaptic function and the general audience of cell biologist interested in membrane trafficking will very much value this study. The mechanism reported will potentially be included in textbooks in the near future.
My field of expertise includes molecular mechanisms of presynaptic function and membrane trafficking.
I have not enough experience to evaluate the quality of the NMR experiments, however, I do not have any problem at all with, in my opinion, elegant results reported.
We thank the reviewer for the positive outlook of our manuscript.
-
-
www.biorxiv.org www.biorxiv.org
-
AbstractComputational drug discovery is intrinsically interdisciplinary and has to deal with the multifarious factors which are often dependent on the type of disease. Molecular Property Diagnostic Suite (MPDS) is a Galaxy based web portal which was conceived and developed as a disease specific web portal, originally developed for tuberculosis (MPDSTB). As specific computational tools are often required for a given disease, developing a disease specific web portal is highly desirable. This paper emphasises on the development of the customised web portal for COVID-19 infection and is referred to as MPDSCOVID-19. Expectedly, the MPDS suites of programs have modules which are essentially independent of a given disease, whereas some modules are specific to a particular disease. In the MPDSCOVID-19 portal, there are modules which are specific to COVID-19, and these are clubbed in SARS-COV-2 disease library. Further, the new additions and/or significant improvements were made to the disease independent modules, besides the addition of tools from galaxy toolshed. This manuscript provides a latest update on the disease independent modules of MPDS after almost 6 years, as well as provide the contemporary information and tool-shed necessary to engage in the drug discovery research of COVID-19. The disease independent modules include file format converter and descriptor calculation under the data processing module; QSAR, pharmacophore, scaffold analysis, active site analysis, docking, screening, drug repurposing tool, virtual screening, visualisation, sequence alignment, phylogenetic analysis under the data analysis module; and various machine learning packages, algorithms and in-house developed machine learning antiviral prediction model are available. The MPDS suite of programs are expected to bring a paradigm shift in computational drug discovery, especially in the academic community, guided through a transparent and open innovation approach. The MPDSCOVID-19 can be accessed at http://mpds.neist.res.in:8085.
This work has been published in GigaByte Journal under a CC-BY 4.0 license (https://doi.org/10.46471/gigabyte.114), and has published the reviews under the same license. These are as follows.
Reviewer 1. Prashanth N Suravajhala
Is there a clear statement of need explaining what problems the software is designed to solve and who the target audience is? Yes. The authors could describe Minimum Information about bioinformatics investigation (MIABI) guidelines. Is the source code available, and has an appropriate Open Source Initiative license been assigned to the code? github and Zenodo, yes!
I tested git, forked it and as I didn't test the graphical version, ensured all python libraries are working! Is the documentation provided clear and user friendly? Yes. Yes, a white paper could be more friendly! Is there a clearly-stated list of dependencies, and is the core functionality of the software documented to a satisfactory level? Yes. yes with README version! Have any claims of performance been sufficiently tested and compared to other commonly-used packages? Yes, as described by the authors Are there (ideally real world) examples demonstrating use of the software? Yes. The Molecular Property Dynamic Suite (MPDS) is a welcome initiative which would serve chemical space for research community. While the authors aimed to deploy it in Galaxy, there is no Galaxy reference cited in first few introductory lines. A strong rationale on Galaxy-MPDS connect could be a value addition The port 8085/8080 are ephemeral and it would be nice if the authors deploy it on a more permanent base An absolute strength for the suite is availability of source code so that end-users can fine tune and reinstantiate the codes. In the architecture, could the end user have a chance to deploy biopython modules for drug discovery/modeling
In Page 4, the authors can define what are the tools precisely used in MPDS 2.3 section: The PPI is not abbreviated as first use The results are exploited well for disease dependent/independent use. However, the major challenge for ligand use/preparation is the use of ncRNAs. Could MPDS provide such instances where ncRNAs could be used fpr targeted ligands? L28 in section 4.1: Pluralis for features ( as one of is used) Also a word or two on aadhar card for perhaps naive users may be mentioned and it may be italicized as it may be a domestic word. Does MPDS suite augur well for Anvaya that Government of India launched, or Tavexa or Taverna? A word to two on local setting up of cloud instance may be a nice addition
Scores on a scale of 0-5 with 5 being the best
Language: 4 Novelty: 4.5 Brevity: 4 Scope and relevance: 4 Language/Brevity checks: Page 9 L6: fulfill misspelt webserver are two words, IMHO
Page 10: CADD which IS available
Tabl S2/S4: from THE coronavirdiae space between anticoronavirusdrugs
Figure S3: remove OF (identifying OF existing) Supporting information may be corrected High resolution Figures esp GA, Figures 2-4 may be inserted
Reviewer 2. Abdul Majeed
Is the language of sufficient quality? Yes. Some changes are needed to make the writing more scientific. Is the code executable? Unable to test Is installation/deployment sufficiently outlined in the paper and documentation, and does it proceed as outlined? Unable to test
Additional comments: In this paper, the authors introduced a Molecular Property Diagnostic Suite (MPDS), which is a Galaxy-based web portal that was conceived and developed as an open-source disease-specific web portal. MPDS is a customized web portal developed for COVID-19, which is a one-stop solution for drug discovery research. I read the article; it is well-written and well-presented. The enclosed contents can be very useful for researchers working in this field (e.g., COVID-19 systems development). However, I propose some comments/concerns to the current version that need correction during the revision. 1- In the abstract, please provide the technical description of the method’s working. Also, please mention the entities which can benefit from the system. 2- The introduction section doesn’t present the challenges/problems of the existing tools. Please discuss the challenges of the previous such tools and how are they addressed through this new system. 3- I could not find the concrete details of data modalities supported in the system. The authors are advised to include such details. 4- The authors mentioned the use of ML, but I couldn’t find any potential usage of ML models. Please add such analysis during the revision. 5- Also, please add some performance results like time complexity, storage, I/O cost, etc. 6- One comprehensive diagram should be included to better illustrate the working of the proposed tool. 7- Please add limitations of the proposed tool in the revised work. 8- Please add the potential implications of this tool in the context of current/future pandemics.
Re-review: I have carefully checked the revised work and the author's responses. The authors have made the desired modifications. I have no major concerns on this paper. In the previous review round, Comment #: 3 has not been properly responded by the authors. By data modality, I meant tabular data, graph data, audio data, video data, etc. Authors should add this aspect clearly in the paper about each data modality processed in their system. In Figure 4, some contents (e.g., protein information, PPI interaction, etc.) are unreadable. The abbreviations are not consistently written in terms of small and capital letters. In the paper, the authors are advised to clearly describe the purpose of this tool, who will benefit and in what capacity, why these kinds of tools are needed, etc. I suggest adding such information in abstract to clearly convey the message to readers. In the title, please recheck one word, Open Access or Open Source. The journals are open access while the software are usually open source .
Reviewer 3. Agastya P Bhati
Is there a clear statement of need explaining what problems the software is designed to solve and who the target audience is? Yes. As noted in my comments, it would be beneficial to clarify what new capabilities are provided by this new portal over and above what is already available currently. Is the source code available, and has an appropriate Open Source Initiative license been assigned to the code? No. There is a github repository (https://github.com/gnsastry/MPDS-18Compound_Library), however, I am unable to access it currently. As Open Source Software are there guidelines on how to contribute, report issues or seek support on the code? Yes. A github repository provides such capabilities. However, it is inaccessible currently. Is the code executable? Unable to test Is installation/deployment sufficiently outlined in the paper and documentation, and does it proceed as outlined? Unable to test Have any claims of performance been sufficiently tested and compared to other commonly-used packages? Is automated testing used or are there manual steps described so that the functionality of the software can be verified? No Additional comments: Molecular Property Diagnostic Suite for COVID-19 (MPDSCOVID19) is an open-source disease specific web portal aiming to provide a collection of all tools and databases relevant for COVID-19 that are available online along with a few in-house scripts at a single portal. It is built upon another platform called "Galaxy" that provides similar services for data intensive biomedical research. MPDSCOVID19 is in continuation to two other similar disease-specific portals that this group has published earlier - for Tuberculosis and Diabetes mellitus. Overall, MPDSCOVID19 is an interesting and useful resource that could be helpful for biomedical community in conducting COVID-19 related research. It brings together all the databases and relevant tools that may make a researcher's life easier as exemplified through the various case studies included.
I recommend publishing this article after the following revisions noted. Please note that any mention of page numbers below is referring to the reviewer PDF version.
Major revisions:
(1) One main issue in this manuscript is the lack of a clear description of the "new" capabilities provided by MPDSCOVID19 over and above what Galaxy provides. I think a clear distinction between the capabilities/features of Galaxy and MPDSCOVID19 would help improve the manuscript substantially and help readers better understand the capabilities of this new COVID-19 portal.
Further, a description of the additions in the new portal over the earlier TB and Diabetes portals is mentioned on page 7. However, I think more details on such advancements/additions would be beneficial. It could be in the form of a table.
(2) It is mentioned that a major advancement in this new portal is the inclusion of ML/AI models/approaches, however no details have been provided. It would be beneficial to briefly describe what ML based capabilities are included in MPDS and how they can be used by any general user. An additional case study demonstrating the same would be helpful.
(3) MPDS portal provides a collection of tools and databases for COVID-19. However, such resources are ever-growing and hence constant updating of the portal's capabilities/resources would be a necessary requirement for its sustainability. There is no mention of any such plans. Do authors have a modus operandi for the same? Have there been further releases of the previous MPDS portals for TB and Diabetes that may be relevant here?
(4) Page 6 - lines 3-4: I suggest replacing "are going to witness" with "are witnessing". There are several recent advancements in applying ML/AI based approaches to improve different aspects of drug discovery. I recommend including a few references here to this effect. Below are some relevant examples:
(a) 10.1021/acs.jcim.0c00915 (b) 10.1021/acs.jcim.1c00851 (c) 10.1038/s41598-023-28785-9 (d) 10.1098/rsfs.2021.0018 (e) 10.1145/3472456.3473524 (f) 10.1145/3468267.3470573
(5) Page 7 - line 8: I am assuming that the terms like "updates", "additions", etc., used in this paragraph are comparing MPDS with its older versions. If so, it would be beneficial to clarify this explicitly. In addition, I suggest that the authors include a brief literature survey to describe what other tools and/or webservers are available already and how MPDS compares with them. This has not been done so far.
(6) The github repository is currently inaccessible publicly. This needs rectification.
Minor revisions:
(1) Page 4: Before introducing MPDSCOVID19 it makes sense to briefly describe Galaxy and its main features. For instance moving forward lines 19-20 (page 4) and lines 3-6 (page 5) to line 12 (page 4).
(2) Page 5 - line 22: I suggest that authors mention the total number of databases/servers that are covered by MPDSCOVID19 as of now. From Table S1, it appears that there are 15 currently (items 5 and 7 are repeated so the 13 seems the wrong total - needs rectification).
(3) Page 5 - line 30: It would make sense to specify details of the MPDS local server. For instance, how many cores/GPUs are available and what are their hardware architectures? Also, it would be beneficial for the users to know if it is possible to use MPDS tools on their own or public infrastructures for large scale implementations. I suggest authors comment on this aspect too.
(4) Page 6 - lines 16-19: The sentence "Galaxy platform.......extend the availability." needs some rephrasing. It is too long and the hard to comprehend.
(5) Page 7 - line 18: I don't understand the word "colloids". Please clarify.
(6) Page 8 - line 30: "section 2.3" is referred to but I don't see any section numbering the PDF provided. This needs rectification.
Re-review: I am satisfied with the changes made to the manuscript and recommend publishing it in its current form if all other reviewers are happy with that.
-
-
pressbooks.library.torontomu.ca pressbooks.library.torontomu.ca
-
Two-factor authentication, or two-step authentication, is a login process where the user is asked to provide two authentication points, such as a password and a code shared through a text message. Two-factor authentication enhances login security.
I think a Two-Factor Authentication is very important because hackers can easily get through usernames and passwords but having a specific code sent to your email or phone number is something only yo have access to. I like these because it personally makes me feel like my information is safe!
-
-
www.education.gouv.fr www.education.gouv.fr
-
www.fcpe.asso.fr www.fcpe.asso.fr
-
La composition du conseil de classeArt. R. 421-50 du code de l’éducation
-
-
prometheus.io prometheus.io
-
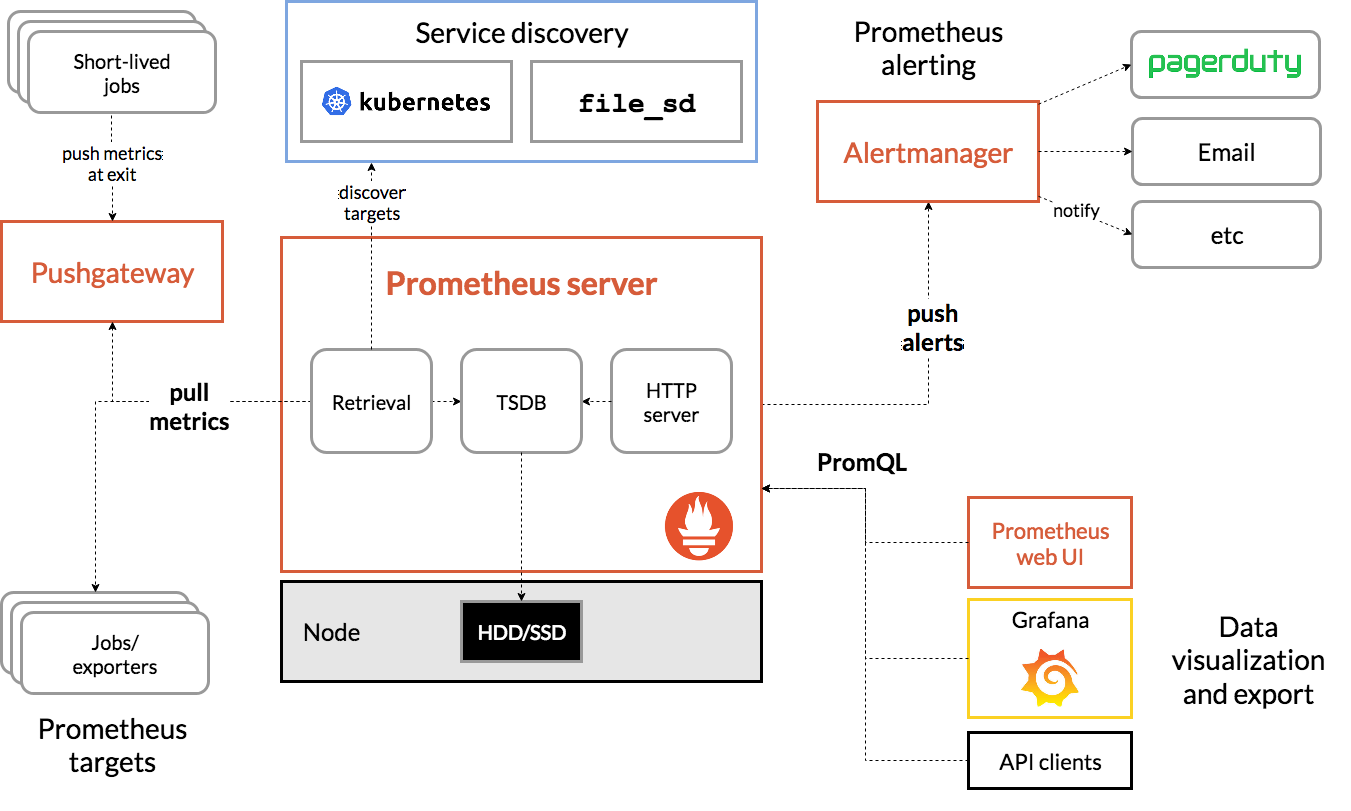
- the main Prometheus server which scrapes and stores time series data
- client libraries for instrumenting application code
- a push gateway for supporting short-lived jobs
- special-purpose exporters for services like HAProxy, StatsD, Graphite, etc.
- an alertmanager to handle alerts
- various support tools
Tags
Annotators
URL
-
-
storage.courtlistener.com storage.courtlistener.com
-
109. On information and belief, in addition to her extensive online presence, she has aGitHub (a software code hosting platform) account called, “anarchivist,” and she developed arepository for a python module for interacting with OCLC’s WorldCat® Affiliate web services.
Matienzo has a GitHub account with code that interacts with OCLC’s API
Is this really the extent of the connection between Matienzo and Anna’s Archive? I don’t know what is required at the Complaint stage of a civil lawsuit to prove someone is connected to an anonymous collective, but surely something more than this plus public statements and an internet handle (“anarchivist”) is required to convict. Does Ohio have SLAPP protections?
-
-
www.researchsquare.com www.researchsquare.com
-
Author Response
Reviewer #1 (Public Review):
Weaknesses:
The manuscript needs proper editing and is not complete. Some wordings lack precision and make it difficult to follow (e.g. line 98 "we assembled a chromosome-scale genome of ..." should read instead "we assembled a chromsome-scla genome sequence of ...". Also, panel Figure 2E is missing.
We will make the suggested change of adding “sequence”. Concerning additional changes, we have carefully edited our manuscript and looked for any incomplete sections. Unfortunately, it is difficult to see what other issues are being raised here without any further information. And the example given is not helpful to ascertain what other changes may be necessary, since we cannot see any problem with the sentence “we assembled a chromosome-scale genome of” as this phrase is widely used in many similar publications.
As for panel E of figure 2, it is not missing. The panel located to the right, just below “Target Cells”.
The shortcomings of the manuscripts are not limited to the writing style, and important technical and technological information is missing or not clear enough, thereby preventing a proper evaluation of the resolution of the genomic resources provided:
- Several RNASeq libraries from different tissues have been built to help annotate the genome and identify transcribed regions. This is fine. But all along the manuscript, gene expression changes are summarized into a single panel where it is not clear at all which tissue this comes from (whole embryo or a specific tissue ?), or whether it is a cumulative expression level computed across several tissues (and how it was computed) etc. This is essential information needed for data interpretation.
No fertilised eggs or embryos have been sequenced, individual tissues derived from juvenile fish were used for the genome annotation and whole larval fish for the developmental analysis. We will specify in the figures and text that the results shown are from whole larvae, and add more detail to the material and methods section about which type of sample was analysed in which way.
- The bioinformatic processing, especially of the assemble and annotation, is very poorly described. This is also a sensitive topic, as illustrated by the numerous "assemblathon" and "annotathon" initiatives to evaluate tools and workflows. Importantly, providing configuration files and in-depth description of workflows and parameter settings is highly recommended. This can be made available through data store services and documents even benefit from DOIs. This provides others with more information to evaluate the resolution of this work. No doubt that it is well done,but especially in the field of genome assembly and annotation, high resolution is VERY cost and time-intensive. Not surprisingly, most projects are conditioned by trade-offs between cost, time, and labor. The authors should provide others with the information needed to evaluate this.
We will upload the code used to assemble and annotate this genome to a public repository or add it to the supplementary material.
The genome assembly did not use a specific workflow (e.g., nextflow), but was done with a simple command and standard parameters in IPA. Scaffolding was carried out by Phase Genomics using their standardised proprietary workflow, of which a detailed description provided by Phase Genomics can be found in the supplementary material. The annotation workflow has been described in a previous publication already, but an in-depth description can also be found in the Material and methods section, including parameters used for specific steps. The RNA-seq mapping and analysis part has also been described in the Material and Methods section, including parameters and models for DESEq2.
- Quantifications of T3 and T4 levels look fairly low and not so convincing. The work would clearly benefit from a discussion about why the signal is so low and what are the current technological limitations of these quantifications. This would really help (general) readers.
We will add a comment on this in the manuscript as suggested. Basically, the T3/T4 levels are consistent with other published work in fish. In the present manuscript for grouper we have a peak level of 1.2 ng/g (1,200 pg/g) of T4 and 0.06 ng/g (60 pg/g) of T3. This is a higher level of T4 and comparable level of T3 to what was found in convict tang (Holzer et al. 2017; Figure 2) with 30 pg/g of T4 and 100 pg/g of T3. Of course, there are also examples with higher levels, such as clownfish (Roux et al. 2023; Figure 1), with 10 ng/g (10,000 pg/g) of T4 and 2 ng/g (2,000 pg/g) of T3.
The differences could be due to different structure of fish tissues and therefore different hormone extraction efficiency, different hormone measurement protocols, different fish physiology, different fish size (e.g., the weighting of tiny grouper larvae is difficult and less precise than in convict tang). What is important is not the absolute level but the relative level, which shows the change within different larval stages of a species with identical extraction and measurement protocols. Which means our data is internally consistent and coherent with what the grouper literature says.
Holzer, Guillaume, et al. "Fish larval recruitment to reefs is a thyroid hormone-mediated metamorphosis sensitive to the pesticide chlorpyrifos." Elife 6 (2017): e27595.
Roux, Natacha, et al. "The multi-level regulation of clownfish metamorphosis by thyroid hormones." Cell Reports 42.7 (2023).
- Differential analysis highlights up to ~ 15,000 differentially expressed genes (DEG), out of a predicted 26k genes. This corresponds to more than half of all genes. ANOVA-based differential analysis relies on the simple fact that only a minority of genes are DEG. Having >50% DEG is well beyond the validity of the method. This should be addressed, or at least discussed.
As the reviewer notes, there are a large number of differentially expressed genes due to the fact that this is coming from a larval developmental transcriptome going from one day old larva to fully metamorphosed juveniles at around day 60.
While DESeq2 indeed works on an assumption that most genes are not differentially expressed, this affects normalization but not hypothesis testing (Wald-test, LRT tests or ANOVA). Normalisation in DESeq2 is fairly robust to this assumption. According to the author of DESeq2, Micheal Love, DESeq2 is using the median ratio for normalisation, and as long as the number of up and down regulated genes is relatively even, DESeq2 will be able to handle the data. As part of our general quality control for this project we consulted the MA plots, which do not show any overrepresented up or down expression patterns. Additionally see Michael Love comment on comparing different tissues, which is also applicable here when comparing vastly different larval stages (https://support.bioconductor.org/p/63630/): “For experiments where all genes increase in expression across conditions, the median ratio method will not be able to capture this difference, but this is typically not the case for a tissue comparison, as there are many "housekeeping" genes with relatively similar expression pattern across tissues.”
Reviewer #3 (Public Review):
Weaknesses:
However, the authors make substantial considerations that are not proven by experimental or functional data. In fact, this is a descriptive study that does not provide any functional evidence to support the claims made.
We agree with the reviewer that our paper lacks functional experiments but despite that, the transcriptomic data clearly show the activation of TH and corticoid pathways during two distinct periods; an early activation between D1 and D10, and a second one between D32 and juvenile stage. These data are interesting as they call for further examination of 1) the possible interaction of corticoids and TH during metamorphosis, a question that is certainly not settled yet in teleost fishes, and 2) the existence of an early larval developmental step also involving TH and corticosteroids.
Especially 2) is of interest and importance, since this early activation (unique to our knowledge in any teleost fish studied so far) raises a lot of new questions and once again will certainly be scrutinised by other groups in the years to come, therefore ensuring a good citation impact of our study. We hope that the reviewer, while disagreeing with some our statements, will recognize that our study will be stimulating at that level and that this is what scientific studies should do.
The consideration that cortisol is involved in metamorphosis in teleosts has never been shown, and the only example cited by the authors (REF 20) clearly states that cortisol alone does not induce flatfish metamorphosis. In that work, the authors clearly state that in vivo cortisol treatment had no synergistic effect with TH in inducing metamorphosis. Moreover, in Senegalensis, the sole pre-otic CRH neuron number decreases during metamorphosis, further arguing that, at least in flatfish, cortisol is not involved in flatfish metamorphosis (PMID: 25575457).
We will do our best to improve the clarity of the revised manuscript to avoid any misunderstanding about our claims. However, we would like to point out the semantic shift in the reviewer first sentence: Indeed “being involved” is not the same as “cortisol alone does not induce”. In ref 20 the authors explicitly wrote that “Cortisol further enhanced the effects of both T4 and T3, but was ineffective in the absence of thyroid hormones” and in our view this indeed corresponds to ”being involved in metamorphosis”.
We are not claiming that cortisol alone is involved in metamorphosis as the reviewer suggests, but simply that there is a possible involvement of cortisol together with TH in metamorphosis. We stand on this claim as we indeed observed an activation of corticoid pathway genes around D32, which is sufficient to say it is involved. We do agree that functional experiments will be needed to properly demonstrate the involvement of corticoids in grouper metamorphosis, but this was not possible in the current study as it would imply to set up a full grouper life cycle in lab conditions which is impossible for the scope of this manuscript.
We also mentioned in the discussion that the role of corticoids in fish larval development is still debated, and we agree that this remain a contentious issue.
We wrote that “there is contrasting evidence of communication between these two pathways [TH and corticosteroids] in teleost fish with some data suggesting a synergic and other an antagonistic relationship. In terms of synergy, an increase in cortisol level concomitantly with an increase in TH levels has been observed in flatfish (ref 19), golden sea bream (ref 100) and silver sea bream (ref 101). Cortisol was also shown to enhance in vitro the action of TH on fin ray resorption (phenomenon occurring during flatfish metamorphosis) in flounder (ref 20). TH exposure increases MR and GR genes expression in zebrafish embryo (ref 55). It has also been shown that cortisol regulates local T3 bioavailability in the juvenile sole via regulation of deiodinase 2 in an organ-specific manner (ref 56) On the antagonistic side, it has been shown that experimentally induced hyperthyroidism in common carp, decreasing cortisol levels (ref 57), whereas cortisol exposure decreases TH levels in European eel (ref 58). Given this scattered evidence, the existence of a crosstalk active during teleost metamorphosis has never been formally demonstrated. The results we obtained in grouper are clearly indicating that HPI axis and cortisol synthesis are activated (i) during early development and (ii) during metamorphosis. This may suggest that in some aspect cortisol synthesis can work in concert with TH, as has been shown in several different contexts in amphibians (ref 17).” In the revised manuscript, we will also add the interesting case of the Senegal sole mentioned by the reviewer.
In the last revision, we had also added that our results “brought a first insight into the potential role of corticoids in the metamorphosis of E. malabaricus and call for functional experiments directly testing a possible synergy” meaning that we clearly acknowledge that we are only revealing a hypothesis that remains to be tested. We later follow up with a discussion about the most novel observation and focus of our study, the increase in THs and cortisol during early development, which was unexpected and very intriguing. Again, these results suggest that there might be a link between the two, as has been shown in amphibians. This is typically the kind of results that should encourage more investigations into other fish species. Indeed, this has been pointed out by other authors and in particular by Bob Denver (probably the foremost expert on this topic) in Crespi and Denver 2012: “Elevation in HPA/I axis activity has been described prior to Metamorphosis in amphibians and fish, birth in mammals (reviewed in Crespi & Denver 2005a; Wada 2008)”. B. Denver also adds that: “Experiments in which GCs were elevated prior to metamorphosis or prior to hatching or birth (e.g. Weiss, Johnston & Moore 2007) or inhibited by treatments with GC synthesis blockers (e.g. metyrapone) or receptor antagonists (e.g. RU486, Glennemeir & Denver 2002) demonstrate that GCs play a causal role in precipitating these life-history transitions (also reviewed in Crespi & Denver 2005a; Wada 2008).” We believe the reviewer will be convinced by these elements coming from a colleague unanimously respected in the field.
Furthermore, the authors need to recognise that the transcriptomic analysis is whole-body and that HPA axis genes are upregulated, which does not mean they are involved in regulating the HPT axis. The authors do not show that in thyrotrophs, any CRH receptor is expressed or in any other HPT axis-relevant cells and that changes in these genes correlate with changes in TSH expression. An in-situ hybridisation experiment showing co-expression on thyrotrophs of HPA genes and TSH could be a good start. However, the best scenario would be conducting cortisol treatment experiments to see if this hormone affects grouper metamorphosis.
We agree that functional experiments are needed to validate our hypothesis. As the early peaks of expression levels observed for many genes were very intriguing for us, we did carry out thyroid hormones and goitrogenic treatment on young grouper larvae to test their effect on the morphological changes. Unfortunately, such experiments, already tricky on metamorphosing larvae, are even more risky on such tiny individuals just after hatching and we encountered high mortality rates. We must add that because we cannot establish a full grouper life cycle under lab conditions, we have done these experiment in the context of a commercial husbandry system in Japan, which while excellent limits the scope of possible experiments. We were thus not able to provide functional validation of our hypothesis. Such experiments will be a full project in itself, requiring setting up a rearing system suitable for both larval survival and economical constraints related to drug treatments. We were further limited by the spawning times of the grouper in the operational aquaculture farm, which are limited to a short time during each year. So even if we strongly agree with the necessity of conducting such experiments, we think that this is not in the scope of the present paper, but something future research can explore.
High TSH and Tg levels usually parallel whole-body TH levels during teleost metamorphosis. However, in this study, high Tg expression levels are only achieved at the juvenile stage, whereas high TSH is achieved at D32, and at the juvenile stage, they are already at their lowest levels.
This is exactly our point. We observe two peaks in TSH expression, one at D3 and one at D32. The peak at D3 coincides with high thyroid hormone levels on the same day, and while we have not measured TH at D32, existing literature shows that there is a peak in TH during that time (e.g., de Jesus et al., 1998). Similarly, there is a small peak of Tg at D3. Our manuscript focused more on the upregulation of these genes at D3, which has not been reported before in the literature and raised the question of the role of TH so early in the larval development, outside of the metamorphosis period.
Regarding the respective levels of TSH and Tg, we first would like to add that their respective order of appearance before metamorphosis (TSH at D32, Tg after) is consistent with what we would expect. We agree however that the strong increase of Tg and TPO expression is later than expected. We will make this clear in the revised manuscript.
It is very difficult to conclude anything with the TH and cortisol levels measurements. The authors only measured up until D10, whereas they argue that metamorphosis occurs at D32. In this way, these measurements could be more helpful if they focus on the correct developmental time. The data is irrelevant to their hypothesis.
We respectfully disagree with the reviewer, considering that 1) TH levels have already been investigated in groupers coinciding with pigmentation changes and fin rays resorption, 2) that there is also evidence in numerous fish species that TH level increase is concomitant with increase of TH related genes, and 3) that we observed in our data an increase in the expression of TH related genes as well as pigmentation changes and fin rays resorption. Based on our experience in fish metamorphosis and the literature we can say confidently that those observations indicate that metamorphosis is occurring between D32 and the juvenile stage. To reinforce our point, we plan to add a figure to the revised manuscript, which puts our data in the context of earlier studies done in grouper. This will clearly show that our inference is correct. Additionally, we would like to point out that from our experience in several fish species transcriptomic data are more robust and precise than hormone measurements.
However, as we were surprised by the activation of TH and corticoid pathway genes very early in the larval development (at D3), which is clearly outside of the metamorphosis period, we decided to measure TH and cortisol levels during this period of time to determine if whether or not there this surprising early activation was indeed corresponding to an increase in both TH and cortisol. As such observation has never been made in other teleost species (to our knowledge), and as we were wondering if gene activation was accompanied by hormonal increase, the measurements we did for TH and cortisol between D1 and D10 are relevant. We will make sure to improve the clarity of the revised version of the manuscript to avoid any confusion between the two periods we are studying: early larval development (between D1 and D10) and metamorphosis (between D32 and juvenile stage).
Moreover, as stated in the previous review, a classical sign of teleost metamorphosis is the upregulation of TSHb and Tg, which does not occur at D32 therefore, it is very hard for me to accept that this is the metamorphic stage. With the lack of TH measurements, I cannot agree with the authors. I think this has to be toned down and made clear in the manuscript that D32 might be a putative metamorphic climax but that several aspects of biology work against it. Moreover, in D10, the authors show the highest cortisol level and lowest T4 and T3 levels. These observations are irreconcilable, with cortisol enhancing or participating in TH-driven metamorphosis.
We thank the reviewer for this comment, but we think that there might be a misunderstanding here.
(1) We clearly observed an increase of TSHb (that occurs between D18 and juvenile stage) and an increase of tg from D32 which coincide with the activation of other genes involved in TH pathway (dio2, dio3, and also a strong increase of TRb). All this and put in the context of what we know from previous grouper studies, clearly supports our conclusion that TH-regulated metamorphosis is starting at around D32 in grouper. We also observed morphological changes such as fin rays resorption and pigmentation changes between D32 and juvenile stage. Such morphological changes have already been associated as corresponding to metamorphosis in groupers (De Jesus et al 1998) as they occur during TH level increase, and they also happen to be under the control of TH in grouper (De Jesus et al 1998). Based on this study but also on studies (conducted on many other teleost species) showing that the increase of TH levels is always associated with an activation of TH pathway genes and morphological and pigmentation changes we concluded that metamorphosis of E. malabaricus occurs between D32 and juvenile stage. We will improve the clarity of the manuscript to make sure that our conclusion is based on our transcriptomic and morphological data plus the available literature.
(2) We clearly observed another activation of TH related gene earlier in the development (between D1 and D10, with a surge of trhrs, tg and tpo at D3. As this activation was very unexpected for us, we decided to focus the analysis of TH levels between D1 and D10 and very interestingly we observed high level of T4 at D3 indicating that THs are instrumental very precociously in the larval development of the malabar grouper which has never been shown before. We declared line 195 that our “data reinforce the existence of two distinct periods of TH signalling activity, one early on at D3 and one late corresponding to classic metamorphosis at D32”. However, we agree that we could have been clearer and clearly explained that this early activation was very intriguing for us and that we wanted to investigate hormonal levels around that period. However, we never claimed anywhere in the manuscript that this early developmental period corresponds to metamorphosis. Something else is occurring and both TH and cortisol seem to be involved but further experiments need to be conducted to understand their role and their possible interaction.
(3) Finally, regarding the comment about cortisol enhancing or participating in TH driven metamorphosis, our data clearly showed an activation of the corticoid pathway genes around metamorphosis (between D32 and juvenile stage) suggesting a potential implication of corticoids in metamorphosis, but we agree with the reviewer that further experiment are needed to test that. We never claimed that cortisol was enhancing or participating in metamorphosis, on the contrary we are “suggesting a possible interaction between TH and corticoid pathway during metamorphosis”. And we also say that our “results brought a first insight into the potential role of corticoids in the metamorphosis of E. malabaricus and call for functional experiments directly testing a possible synergy.” Nonetheless, we agree that some parts of our manuscript can be confusing in regards of cortisol synthesis during metamorphosis as we did not measure cortisol levels between D32 and juvenile stage. We will correct this in the revised version.
Given this, the authors should quantify whole-body TH levels throughout the entire developmental window considered to determine where the peak is observed and how it correlates with the other hormonal genes/systems in the analysis.
We did not measure TH levels at later stages as it has already been measured during Epinephelus coioides metamorphosis and the morphological changes observed in this species around the TH peak corresponds to what we observed in Epinephelus malabaricus around the peak of expression of TH pathway genes (see De Jesus et al., 1998 General and Comparative Endocrinology, 112:10-16). We are planning to add a figure reconciling all these data together. However, the main focus of this manuscript is the novel observation of the existence of an early activation period observed at D3, and for which we needed TH levels to determine if they were involved in another early developmental process (not related to metamorphosis). Our hypothesis is that this early activation might be related to the growth of fin rays necessary to enhance floatability during the oceanic larval dispersal. As we may have arrived at the explanation of this hypothesis too rapidly without setting up the context well enough, we will pay attention to improve that part too.
Even though this is a solid technical paper and the data obtained is excellent, the conclusions drawn by the authors are not supported by their data, and at least hormonal levels should be present in parallel to the transcriptomic data. Furthermore, toning down some affirmations or even considering the different hypotheses available that are different from the ones suggested would be very positive.
We thank the reviewer for acknowledging the solidity of the method of our paper and the quality of the results. We agree that there were several parts where our message is unclear, which we will address in the revised version of the manuscript to make sure there is no more confusion between the two distinct periods we studied in this paper (early larval development and metamorphosis). We will also make sure that our claims about TH/corticoids interaction during both periods remain hypothetical as we cannot yet, despite trials, sustain them with functional experiment.
-
-
-
Résumé de la vidéo [00:00:00][^1^][1] - [00:23:39][^2^][2]:
Cette vidéo présente une conférence sur l'avenir des transports, en se concentrant sur le véhicule autonome. Patrick Perez, directeur scientifique chez Valeo, discute des avantages et des défis de cette technologie, en soulignant son potentiel pour améliorer la sécurité, le confort et l'efficacité des transports. Il aborde l'histoire du véhicule autonome, les progrès techniques, les niveaux d'automatisation et les défis techniques tels que la perception, la prédiction et la prise de décision.
Points forts: + [00:00:22][^3^][3] Introduction de la conférence * Présentation de Patrick Perez * Discussion sur le véhicule autonome + [00:02:31][^4^][4] Histoire et développement * Projet Prometheus en Europe * Challenges DARPA aux États-Unis + [00:05:12][^5^][5] Évolution récente et acteurs clés * Transition de Google Car à Waymo * Concurrence internationale dans le domaine + [00:07:14][^6^][6] Diversité des véhicules autonomes * Véhicules de livraison, navettes autonomes * Camions et équipements agricoles + [00:11:08][^7^][7] Niveaux d'automatisation * Explication des différents niveaux * Importance de la sécurité et de la légalité + [00:13:00][^8^][8] Défis techniques et solutions * Perception, prédiction et décision * Utilisation de l'intelligence artificielle et des capteurs Résumé de la vidéo [00:23:43][^1^][1] - [00:35:24][^2^][2]:
La partie 2 de la vidéo aborde les avancées dans les systèmes de transport, en mettant l'accent sur les véhicules autonomes et l'intelligence artificielle. Elle explique le rôle des capteurs, l'importance des données pour l'entraînement des modèles d'IA, et les défis liés à la fiabilité et la certification des systèmes automatisés.
Points forts: + [00:23:43][^3^][3] Les capteurs et la loi * Importance de respecter le code de la route et les lois de la physique * Présentation des différents types de capteurs utilisés dans les véhicules + [00:25:00][^4^][4] Perception et adaptation * Les capteurs ont des résolutions et des modes de fonctionnement variés * Adaptation de domaine pour améliorer la perception des systèmes d'IA + [00:28:08][^5^][5] Prédiction de la confiance * Développement de réseaux de neurones pour prédire la confiance dans la détection * Importance de savoir quand le système ne sait pas pour une utilisation fiable + [00:30:23][^6^][6] Explicabilité des décisions * Problématique de l'interprétabilité des décisions des réseaux de neurones * Utilisation d'explications contrefactuelles pour comprendre les décisions d'IA
-
-
docdrop.org docdrop.org
-
Résumé de la vidéo [00:00:00][^1^][1] - [00:21:47][^2^][2]:
Cette vidéo, présentée par Stanislas Dehaene, explore la perception des objets mathématiques élémentaires et leurs mécanismes cérébraux. Elle examine comment les humains reconnaissent rapidement des motifs géométriques et sonores, en utilisant des exemples comme les dessins géométrisés des enfants et les motifs anciens trouvés sur des objets datant de 70 000 à 100 000 ans. Dehaene discute de la capacité extraordinaire de percevoir des motifs abstraits et comment cela est traité dans le cerveau, en s'appuyant sur des études comportementales et des modèles de langage de la pensée.
Points forts: + [00:00:26][^3^][3] Introduction au sujet * Focus sur les formes géométriques élémentaires * Importance des mécanismes cérébraux dans la perception + [00:01:41][^4^][4] Sensibilité aux motifs * Illustration avec une vidéo musicale * Reconnaissance immédiate des motifs spatiaux et sonores + [00:03:50][^5^][5] Étude des séquences géométriques * Utilisation d'un test avec des enfants pour étudier la perception * Capacité à anticiper des motifs géométriques complexes + [00:07:04][^6^][6] Langage de la géométrie * Nécessité d'un langage pour expliquer la mémoire des séquences * Présentation d'un modèle de langage de la pensée + [00:10:58][^7^][7] Performances et anticipation * Corrélation entre la complexité des séquences et la performance * Étude des mouvements oculaires et de l'IRM fonctionnelle + [00:16:09][^8^][8] Inférence de programme * Exploration de la capacité à inférer des motifs géométriques * Discussion sur la nature des primitives du langage de la pensée Résumé de la vidéo [00:21:50][^1^][1] - [00:42:52][^2^][2]:
La partie 2 de la vidéo se concentre sur la perception des objets mathématiques élémentaires et leur représentation dans le cerveau. Stanislas Dehaene explore les régions cérébrales activées par les tâches géométriques et mathématiques, la magnéto-encéphalographie pour suivre l'activité cérébrale, et comment les séquences spatiales sont codées et anticipées par le cerveau. Il discute également des limites de la compréhension des séquences spatiales chez les animaux par rapport aux humains.
Points saillants: + [00:22:00][^3^][3] Activation cérébrale et langage * Les régions activées par le langage ne sont pas les mêmes que celles utilisées pour les tâches géométriques * Les tâches géométriques activent des régions liées au calcul mental * L'anticipation cérébrale joue un rôle dans la complexité des séquences + [00:23:25][^4^][4] Magnéto-encéphalographie * Permet de suivre l'activité cérébrale milliseconde par milliseconde * Révèle comment le cerveau code et anticipe les séquences spatiales * Montre que le cerveau fonctionne sur un mode prédictif + [00:27:01][^5^][5] Anticipations cérébrales * Le cerveau anticipe les positions dans les séquences avant qu'elles ne surviennent * La capacité d'anticipation est corrélée avec la complexité de la séquence * Les représentations mentales abstraites comme la rotation et la symétrie sont décodables + [00:33:00][^6^][6] Comparaison avec les animaux * Les animaux ont des difficultés à comprendre les structures géométriques complexes * Les humains ont une capacité unique à former des structures récursives et à comprimer l'information * Cette capacité pourrait être propre à l'espèce humaine Résumé de la vidéo [00:42:54]¹[1] - [01:03:55]²[2]:
Cette partie de la vidéo explore la perception des objets mathématiques élémentaires dans la musique et la géométrie, en se concentrant sur la théorie de la compression de l'information et son application dans la compréhension des séquences musicales et géométriques. Stanislas Dehaene discute de l'ancienneté du phénomène musical chez Homo sapiens, comparable à celle du langage géométrique, et présente des expériences de laboratoire simplifiant ces phénomènes pour tester la mémoire de travail et la détection de régularités.
Points saillants: + [00:42:54]³[3] Ancienneté de la musique et de la géométrie * Présentation de flûtes anciennes comme preuve de la culture musicale préhistorique * Comparaison de l'ancienneté du langage musical avec le langage géométrique + [00:43:36]⁴[4] Expériences de laboratoire sur la musique * Utilisation de séquences musicales binaires pour tester la mémoire et la perception des régularités * Proposition d'un système de compression de l'information pour les séquences musicales + [00:45:01]⁵[5] Complexité des séquences musicales * Discussion sur la longueur de description minimale et la complexité subjective perçue * Exclusion de modèles alternatifs et confirmation de la théorie de la compression + [00:49:55]⁶[6] Systèmes de règles et statistiques dans la perception * Distinction entre le traitement des règles et des statistiques dans la perception des séquences * Présentation d'un modèle bayésien pour inférer les règles et les probabilités de transition
Source : conversation avec Bing, 14/03/2024 (1) undefined. https://www.education.gouv.fr/education-la-sexualite-en-milieu-scolaire-341103. (2) undefined. https://soseducation.org/docs/notes-etudes-entretiens-tribunes/education-a-la-sexualite-danger-ou-prevention-final.pdf. (3) undefined. https://www.planning-familial.org/sites/default/files/2023-11/LIVRE_BLANC_WEB.pdf. (4) undefined. https://www. Résumé de la vidéo [01:03:56][^1^][1] - [01:21:55][^2^][2]:
Cette partie de la vidéo aborde la perception des objets mathématiques élémentaires à travers une étude sur la structure des séquences musicales et leur traitement cérébral. Stanislas Dehaene discute des résultats d'IRM montrant comment le cerveau humain traite la complexité des séquences musicales, révélant un réseau de régions cérébrales impliquées dans la compréhension de la structure et de la syntaxe des séquences.
Points saillants: + [01:03:56][^3^][3] Traitement des séquences musicales * Étude des parenthèses placées par les sujets dans des séquences * Révélation de la structure perçue par le cerveau * Différences entre séquences simples et complexes + [01:05:11][^4^][4] Résultats d'IRM et prédiction de la complexité * Activation cérébrale croissante avec la complexité des séquences * Réseau de régions cérébrales associées à la perception de la structure * Inversion de l'activation pour les séquences complexes + [01:07:55][^5^][5] Séparation entre musique et langage * Études indiquant une distinction claire dans le cortex temporal supérieur * Régions cérébrales spécialisées pour le traitement du langage et de la musique * Peu de recouvrement entre les aires du langage et celles traitant la musique + [01:11:56][^6^][6] Implications en éducation et mathématiques * Corrélation entre la capacité de traiter les motifs et les performances mathématiques * Potentiel impact de l'entraînement musical sur le développement des compétences abstraites * Importance de l'enseignement précoce des motifs géométriques et musicaux
-
-
arxiv.org arxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
Reviewer #1
More details about the classification and how it is trained
We included a sentence in the introduction to clarify which data we are using: "In order to demonstrate this improvement, we apply our methods to two classification datasets: a synthetic dataset and a public clinical dataset where the predicted outcome is the survival of the patient"
And about how the classifier is trained in the "Results" section: "we used the default parameters of the classifier, since our focus is comparing the different imputation methods"
Availability of the code
Now the code is publicly available in a github repository https://github.com/AstraZeneca/dpp_imp/ (see Availability of Data and Code section)
Reviewer #2
Clarifying that Determinantal Point Processes and their deterministic version have been introduced before but are applied for the first time for data imputation in this work:
We added explanation in the 6th paragraph of the introduction that we use pre-existing DPP and deterministic-DPP algorithms for our imputation methods and include the references to avoid confusion
We also added a paragraph at the end of the introduction to summarize this work's contribution
Explaining the claim about the computational advantage of using quantum determinantal point processes for the imputation methods:
In the fourth paragraph of the "Discussion" section (page 8), we give an imputation example by numerically comparing the classical and quantum algorithms running time for DPP sampling, which shows the advantage of using the quantum algorithm.
Regarding running time for classical DPP and quantum DPP sampling algorithms:
We included Table VIII (page 13) that compares the preprocessing and sampling complexities for both classical and quantum DPP algorithms, we consider the case where we sample d rows from an (n,d) matrix and n=O(d) which is usually the case for our DPP-Random Forest algorithm
We added some details regarding the quantum advantage in the first paragraph of page 12
Regarding the comment about the modest improvement of the DPP methods and questions about their practical benefit:
As mentioned in the third paragraph of the "Discussion" section, we point out that the consistency of the improvement and the removal of variance as a result of using the DPP and deterministic DPP methods make our methods very beneficial to use on clinical data. Further exploration with different data sets can provide a more result in a more complete understanding of the practical advantages of the methods
Algorithmic complexity of the deterministic DPP algorithm:
Detailed in the last sentence of the "Determinantal Point Processes" subsection of the "Methods" section: O(N^2 d) for the preprocessing step and O(Nd^3) for the sampling step
Running time for the quantum deterministic DPP sampling and how it is done in practice:
While it is difficult to assess the real running time for the quantum detDPP algorithm for large circuits (100 or more qubits), due to the unavailability of such devices, we give more details about our practical implementation in the last paragraph of the "Methods" section. In our case (up to 10 qubits) we used 1000 shots to sample the highest probability elements.
On which quantum simulator was used
We point out in the first paragraph of page 5 that we employ the qiskit noiseless simulator
Tags
Annotators
URL
-
-
m-earnest.github.io m-earnest.github.io
-
Data code might not adhere to the same standards or structure
what do you mean by that?
-
-
www.blackpast.org www.blackpast.org
-
(1804) Ohio Black Codes
They Ohio government wrote the Ohio Black Code. It was written in 1804. Making restriction on blacks and mulatto who are living in the state.
-
-
www.biorxiv.org www.biorxiv.org
-
eLife assessment
In this important study, the authors manually assessed randomly selected images published in eLife between 2012 and 2020 to determine whether they were accessible for readers with deuteranopia, the most common form of color vision deficiency. They then developed an automated tool designed to classify figures and images as either "friendly" or "unfriendly" for people with deuteranopia. While such a tool could be used by publishers, editors or researchers to monitor accessibility in the research literature, the evidence supporting the tools' utility was incomplete. The tool would benefit from training on an expanded dataset that includes different image and figure types from many journals, and using more rigorous approaches when training the tool and assessing performance. The authors also provide code that readers can download and run to test their own images. This may be of most use for testing the tool, as there are already several free, user-friendly recoloring programs that allow users to see how images would look to a person with different forms of color vision deficiency. Automated classifications are of most use for assessing many images, when the user does not have the time or resources to assess each image individually.
-
-
zhuanlan.zhihu.com zhuanlan.zhihu.com
-
MAC码
在密码学中,消息认证码(英语:Message authentication code,缩写为MAC),又译为消息鉴别码、文件消息认证码、讯息鉴别码、信息认证码,是经过特定算法后产生的一小段信息,检查某段消息的完整性,以及作身份验证。它可以用来检查在消息传递过程中,其内容是否被更改过,不管更改的原因是来自意外或是蓄意攻击。同时可以作为消息来源的身份验证,确认消息的来源。
-
-
ipese-web.epfl.ch ipese-web.epfl.ch
-
You can add posts to this Quarto blog in any of the menus on the navbar. Blog articles are written in Quarto, which is similar to Markdown. You can also run R or Python code chunks in your posts, import .bib files, and use many other features.
this is my comments
-
-
pharo.org pharo.org
-
The immersive programming experience Pharo is a pure object-oriented programming language and a powerful environment, focused on simplicity and immediate feedback (think IDE and OS rolled into one).
from - Writing Code Without Plain Tex…

-
-
itnext.io itnext.io
-
no code for you to forget to change or update to reflect the new change
yes
-
Unison doesn’t store source code; it stores ASTs
ASTs
-
Writing Code Without Plain Text FilesThe Unison programming language doesn’t store code in files, but in a database. What is that like?
source - "Writing Code Without Plain Tex…" (itnext.io)

-
Writing Code Without Plain Text Files
The Unison programming language doesn’t store code in files, but in a database. What is that like?
personal.archived - https://ipfs.indy0.net/ipfs/QmQz4CcH6Buwb48hi4skRuXbwf5wVvMPpDqJDbKcBbb4uB?p=QmaZHodquC4dhjn8Ct6GG9zFvJjfYD6j4AoAmAakeTygdG - in: https://hypothes.is/a/UY8YsuBMEe6Jnkt8qYLGRA - from: https://hypothes.is/a/UY8YsuBMEe6Jnkt8qYLGRA - by: gyuri
-
-
erikexplores.substack.com erikexplores.substack.com
-
What Makes Smalltalk Unique?Exploring the implication of software development without files

-
-
www.scaledrone.com www.scaledrone.com
-
Introducing Chatacular — a no-code solution for adding stunning group chats to your website.
-
-
myd2l.lcc.edu myd2l.lcc.edu
-
The audience of this passage is the entire French Caribbean colonies. It's evident from the document's title, "The Black Code of Louisiana," which suggests it's intended for widespread dissemination and application across the region. This code isn't meant for private conversation; rather, it serves as a set of laws directed at both slaves and colonists in the area. These regulations were designed to govern the behavior and interactions of individuals within the French Caribbean colonies. It's significant to note that these laws remained in effect until 1803 when the United States acquired possession of Louisiana, indicating their long-standing impact and influence on the region's social and legal structures.
-
This statement seems hypocritical because it claims to forbid masters from forcing slaves into marriage against their will, yet the entire Black Code imposes strict control on slaves and colonists. It's contradictory that they care about not forcing someone to do something against their will, yet they enforce laws that dictate exactly what slaves and colonists must do. This raises doubts about the true intentions behind such laws and the ethics of those who created them. It shows how systems of oppression often have double standards and prioritize maintaining power over individual freedoms. Overall, it highlights the need to question and critically analyze historical narratives of power and control.
-
This puzzling situation really shows how messed up things were for women and slaves back in the 1700s. It's strange that if a woman was free but her husband was a slave, their kids would be free, but if it was the other way around, the kids would be slaves. It just doesn't make sense. The Black Code of Louisiana had a tight grip on African-American folks during that time, controlling their lives in big ways. Looking back, it's clear how unfair and messed up those laws were, and it makes us realize how far we still have to go for fairness and equality.
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
eLife assessment
This manuscript presents a valuable approach to exploring CD4+ T-cell response in mice across stimuli and tissues through the analysis of their T-cell receptor repertoires. The authors use a transgenic mouse model, in which the possible diversity of the T-cell receptor repertoire is reduced, such that each of a diverse set of immune exposures elicits more detectably consistent T-cell responses across different individuals. However, whereas the proposed experimental system could be utilized to study convergent T-cell responses, the analyses done in this manuscript are incomplete and do not support the claims due to limitations in the statistical analyses and lack of data/code access.
We worked to address the reviewers' concerns below, point-by-point.
All data on immune repertoires are deposited here: https://figshare.com/articles/dataset/Convergence_plasticity_and_tissue_residence_of_regulatory_and_effector_T_cell_response/22226155
We added the Data availability statement to the manuscript.
Public Reviews:
Reviewer #1 (Public Review):
The authors investigate the alpha chain TCR landscape in conventional vs regulatory CD4 T cells. Overall I think it is a very well thought out and executed study with interesting conclusions. The authors have investigated CDR3 alpha repertoires coupled with a transgenic fixed CDR3beta in a mouse system.
Strengths:
-
One of a kind evidence and dataset.
-
State-of-the-art analyses using tools that are well-accepted in the literature.
-
Interesting conclusions on the breadth of immune response to challenges across different types of challenges (tumor, viral and parasitic).
Thank you for the positive view.
Weaknesses:
- Some conclusions regarding the eCD4->eTreg transition are not so strong using only the data.
The overlaps between the top-nucleotide clones in both LLC and PYMT challenges are prominently above the average, and this result is reproducible in lungs and skin, so we have no doubts based on these data. Further experiments with different methods, including tracking the clonal fates, should clarify and confirm/correct/disprove our findings.
- Some formatting issues.
We are working on the manuscript to correct minor errors and formatting.
Reviewer #2 (Public Review):
This study investigates T-cell repertoire responses in a mouse model with a transgenic beta chain, such that all T-cells in all mice share a fixed beta chain, and repertoire diversity is determined solely by alpha chain rearrangements. Each mouse is exposed to one of a few distinct immune challenges, sacrificed, and T-cells are sampled from multiple tissues. FACS is used to sort CD4 and Treg cell populations from each sample, and TCR repertoire sequencing from UMI-tagged cDNA is done.
Various analyses using repertoire diversity, overlap, and clustering are presented to support several principal findings: 1) TCR repertoires in this fixed beta system have highly distinct clonal compositions for each immune challenge and each cell type, 2) these are highly consistent across mice, so that mice with shared challenges have shared clones, and 3) induction of CD4-to-Treg cell type transitions is challenge-specific.
The beta chain used for this mouse model was previously isolated based on specificity for Ovalbumin. Because the beta chain is essential for determining TCR antigen specificity, and is highly diverse in wildtype mice, I found it surprising that these mice are reported to have robust and consistently focused clonal responses to very diverse immune challenges, for which a fixed OVA-specific beta chain is unlikely to be useful. The authors don't comment on this aspect of their findings, but I would think it is not expected a priori that this would work. If this does work as reported, it is a valuable model system: due to massively reduced diversity, the TCR repertoire response is much more stereotyped across individual samples, and it is much easier to detect challenge-specific TCRs via the statistics of convergent responses.
This was to some extent expected, since these mice live almost normally and have productive adaptive immune responses and protection. In real life, there are frequent TCR-pMHC interactions where the TCR-alpha chain dominates (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5701794/; https://pubmed.ncbi.nlm.nih.gov/37047500/). On the fixed TCR-beta background this mechanics starts working full-fledged, essentially substituting TCR-beta diversity, at the extent of relatively simplified TCRab repertoire and probably higher cross-reactivity.
We agree that this is a valuable model, for sure, and indicated this in the last sentence of our Discussion. Now we are also adding this point to the abstract.
While the data and analyses present interesting signals, they are flawed in several ways that undermine the reported findings. I summarize below what I think are the most substantive data and analysis issues.
(1) There may be systematic inconsistencies in repertoire sampling depth that are not described in the manuscript. Looking at the supplementary tables (and making some plots), I found that the control samples (mice with mock challenge) have consistently much shallower sampling-in terms of both read count and UMI count-compared with the other challenge samples. There is also a strong pattern of lower counts for Treg vs CD4 cell samples within each challenge.
The immune response of control mice is less extensive, as it should be. Just like the fact that the number of Tregs in tissues is lower than CD4, this is normal. So this all follows the expectations. But please note that we were very accurate everywhere with appropriate data normalisation, using all our previous extensive experience (https://pubmed.ncbi.nlm.nih.gov/29080364/).
In particular (now adding more relevant details to Methods):
For diversity metrics calculations, we randomly sampled an equal number of 1000 UMI from each cloneset. Samples with UMI < 700 were excluded from analysis.
For amino acid overlap metrics calculations, we selected top-1000 largest clonotypes from each cloneset. Samples with clonotype counts < 700 were excluded from analysis.
For nucleotide overlaps metrics calculations (eCD4-eTreg), we selected top-100 clonotypes from each cloneset. Samples with clonotypes < 100 were excluded from analysis.
The top N clonotypes were selected as the top N clonotypes after randomly shuffling the sequences and aligning them in descending order. This was done in order to get rid of the alphabetical order for clonotypes with equal counts (e.g. count = 1 or 2).
Downsampling was carried out using software vdjtools v.1.2.1.
(2) FACS data are not reported. Although the graphical abstract shows a schematic FACS plot, there are no such plots in the manuscript. Related to the issue above, it would be important to know the FACS cell counts for each sample.
Yes, we agree that this is valuable information that should be provided. Unfortunately, this data has not been preserved.
(3) For diversity estimation, UMI-wise downsampling was performed to normalize samples to 1000 random UMIs, but this procedure is not validated (the optimal normalization would require downsampling cells). What is the influence of possible sampling depth discrepancies mentioned above on diversity estimation? All of the Treg control samples have fewer than 1000 total UMIs-doesn't that pose a problem for sampling 1000 random UMIs?
Indeed, I simulated this procedure and found systematic effects on diversity estimates when taking samples of different numbers of cells (each with a simulated UMI count) from the same underlying repertoire, even after normalizing to 1000 random UMIs. I don't think UMI downsampling corrects for cell sampling depth differences in diversity estimation, so it's not clear that the trends in Fig 1A are not artifactual-they would seem to show higher diversity for control samples, but these are the very same samples with an apparent systematic sampling depth bias.
We evaluated this approach through all our work, and summarised in the ref: https://pubmed.ncbi.nlm.nih.gov/29080364/. Altogether, normalising to the same count of randomly sampled UMI seems to be the best approach (although, preferably, the initial sequencing depth should be essentially higher for all samples than the sampling threshold used). Initial sorting of identical numbers of cells and ideally uniform library preparation and sequencing is generally not realistic and does not work in the real world, while UMI downsampling does the same work much better.
(4) The Figures may be inconsistent with the data. I downloaded the Supplementary Table corresponding to Fig 1 and made my own version of panels A-C. This looked quite different from the diversity estimations depicted in the manuscript. The data does not match the scale or trends shown in the manuscript figure.
There was a wrong column for Chao1, now correcting. Also, please note that we only used samples with > 700 UMI. Supplementary Table now corrected accordingly. Also, please note that Figure 1 shows the results for lung samples only.
(5) For the overlap analysis, a different kind of normalization was performed, but also not validated. Instead of sampling 1000 UMIs, the repertoires were reduced to their top 1000 most frequent clones. It is not made clear why a different normalization would be needed here. There are several samples (including all Treg control samples) with only a couple hundred clones. It's also likely that the noted systematic sampling depth differences may drive the separation seen in MDS1 between Treg and CD4 cell types. I also simulated this alternative downsampling procedure and found strong effects on MDS clustering due to sampling effects alone.
That’s right, for the overlap analysis (which values are mathematically proportional to the clonotype counts in both compared repertoires, so the difference in the counts causes major biases) the right way to do it is to choose the same number of clonotypes. See Ref. https://pubmed.ncbi.nlm.nih.gov/29080364/.
We kept only samples with > 700 for the overlap analyses. Some relatively poor samples are present in all challenges, while MDS1 localization has clear reproducible logic, so we are confident in these results.
It is not made clear how the overlap scores were converted to distances for MDS. It's hard to interpret this without seeing the overlap matrix.
This is a built-in feature in VDJtools software (https://pubmed.ncbi.nlm.nih.gov/26606115/). See also here: https://vdjtools-doc.readthedocs.io/en/master/overlap.html.
(6) The cluster analysis is superficial, and appears to have been cherry-picked. The clusters reported in the main text have illegibly small logo plots, and no information about V/J gene enrichments. More importantly, as the caption states they were chosen from the columns of a large (and messier-looking) cluster matrix in the supplementary figure based on association with each specific challenge. There's no detail about how this association was calculated, or how it controlled for multiple tests. I don't think it is legitimate to simply display a set of clusters that visually correlate; in a sufficiently wide random matrix you will find columns that seem to correlate with any given pattern across rows.
Particular CDR3 sequences and VJ segments do not mean much for the results of this manuscript. Logos are given just for visual explanation of how the consensus motifs of the clusters look like.
We now add two more Supplementary Tables and a Supplementary Figure with full information about clusters.
We disagree that the Supplementary Figure 1 (representing all the clusters) looks “messy”. Vice versa, it is surprisingly “digital”, showing the clear patterns of responses and homings. This becomes clear if you visually study it for a while. But yes, it is too big to let the reader focus on this or that aspect. That is why we need to select TCR clusters to illustrate this or that aspect discussed in the work, but they were selected from the overall already structured picture.
(7) The findings on differential plasticity and CD4 to Treg conversion are not supported. If CD4 cells are converting to Tregs, we expect more nucleotide-level overlap of clones. This intuition makes sense. But it seems that this section affirms the consequent: variation in nucleotide-level clone overlap is a readout of variation in CD4 to Treg conversion. It is claimed, based on elevated nucleotide-level overlap, that the LLC and PYMT challenges induce conversion more readily than the other challenges. It is not noted in the textual interpretations, but Fig 4 also shows that the control samples had a substantially elevated nucleotide-level overlap. There is no mention of a null hypothesis for what we'd expect if there was no induced conversion going on at all. This is a reduced-diversity mouse model, so convergent recombination is more likely than usual, and the challenges could be expected to differ in the parts of TCR sequence space they induce focus on. They use the top 100 clones for normalization in this case, but don't say why (this is the 3rd distinct normalization procedure).
Your point is absolutely correct: “This is a reduced-diversity mouse model, so convergent recombination is more likely than usual”. Distinct normalisation procedure was required to focus on the most expanded clonotypes to avoid the tail of (presumably cross-reactive) and identical TCRs present in all repertoires in these limited-repertoire mice. So we downsampled as strictly as possible to minimise this background signal of nucleotide overlap, and only this strict downsampling to the top-100 clonotypes allowed us to visualise the difference between the challenges. This is a sort of too complicated explanation that would overload the manuscript. But your comments and our answers will be available to the reader who wants to go into all the details.
The observed (at this strict downsampling) overlaps between the top-nucleotide clones in both LLC and PYMT challenges are prominently above the average, and this result is reproducible in lungs and skin, so we have no doubts in interpretations based on these data. Further experiments with different methods, including tracking the clonal fates, should clarify and confirm/correct/disprove our findings.
Although interpretations of the reported findings are limited due to the issues above, this is an interesting model system in which to explore convergent responses. Follow-up experimental work could validate some of the reported signals, and the data set may also be useful for other specific questions.
Yes, thank you for your really thorough analysis. We fully agree with your conclusion.
Reviewer #3 (Public Review):
Nakonechnaya et al present a valuable and comprehensive exploration of CD4+ T cell response in mice across stimuli and tissues through the analysis of their TCR-alpha repertoires.
The authors compare repertoires by looking at the relative overlap of shared clonotypes and observe that they sometimes cluster by tissue and sometimes by stimulus. They also compare different CD4+ subsets (conventional and Tregs) and find distinct yet convergent responses with occasional plasticity across subsets for some stimuli.
The observed lack of a general behaviour highlights the need for careful comparison of immune repertoires across cell subsets and tissues in order to better understand their role in the adaptive immune response.
In conclusion, this is an important paper to the community as it suggests several future directions of exploration.
Unfortunately, the lack of code and data availability does not allow the reproducibility of the results.
Thank you for your positive view.
All data on immune repertoires are deposited here: https://figshare.com/articles/dataset/Convergence_plasticity_and_tissue_residence_of_regulatory_and_effector_T_cell_response/22226155
We added the Data availability statement to the manuscript.
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
- In the manuscript at "yielding 13,369 {plus minus} 1,255 UMI-labeled TCRα cDNA molecules and 3233 {plus minus} 310 TCRα CDR3 clonotypes per sample" I'm not sure how can there be fewer unique DNA molecules than clonotypes in each sample.
That was our mistake for sure, now corrected.
- In the manuscript at "This indicates that the amplitude and focused nature of the effector and regulatory T cell response in lungs is generally comparable."
I'm not sure it's possible to conclude that a drop in diversity in all conditions necessarily signals a focused nature. Since at this stage, the nature of the colotypes was not compared between conditions, it is not possible to claim a focused nature of the response.
We have softened the wording:
"This could indicate that the amplitude and focused nature of the effector and regulatory T cell response in lungs is generally comparable."
- What are your thoughts on why there is such a large overlap between Treg and Teff in the Lung in control? For some replicates it is almost as much as a post-LLC challenge!
There is some natural dispersion in the data, which is generally expectable. The overlaps between the top-nucleotide clones in both LLC and PYMT challenges are prominently above the average, and this result is reproducible in lungs and skin, so we have no doubts based on these data. Further experiments with different methods, including tracking the clonal fates, should clarify and confirm/correct/disprove our findings.
- In the manuscript at "These results indicate that distinct antigenic specificities are generally characteristic for eTreg cells that preferentially reside in particular lymphatic niches" I'm not sure we can conclude this from the Figure. Wouldn't you expect the samples to be grouped by color (the different challenges)? Maybe I'm not understanding the sentence!
This is a different story, about resident Tregs, irrespective of the challenge.
The whole explanation is here in the text:
“Global CDR3α cluster analysis revealed that characteristic eTreg TCR motifs were present in distinct lymphatic tissues, including spleen and thymus, irrespective of the applied challenge (Supplementary Fig. 1). To better illustrate this phenomenon, we performed MDS analysis of CDR3α repertoires for distinct lymphatic tissues, excluding the lungs due to their otherwise dominant response to the current challenge. This analysis demonstrated close proximity of eTreg repertoires obtained from the same lymphatic tissues upon all lung challenges and across all animals (Fig. 5a, b). These results indicate that distinct antigenic specificities are generally characteristic for eTreg cells that preferentially reside in particular lymphatic niches. Notably, the convergence of lymphatic tissue-resident TCR repertoires was less prominent for the eCD4 T cells (Fig. 5c, d).”
And in the abstract:
“Additionally, our TCRα repertoire analysis demonstrated that distinct antigenic specificities are characteristic for eTreg cells residing in particular lymphatic tissues, regardless of the challenge, revealing the homing-specific, antigen-specific resident Treg populations. ”
- In the manuscript at " Notably, the convergence of lymphatic tissue-resident TCR repertoires was less prominent for the eCD4 T cells ":
5b and 5d seem to have the same pattern: Spleen and MLN group together, AxLN and IgLN together and thymus is separate. Do you mean to say that the groups are more diffuse? I feel like the pattern really is the same and it's likely due to some noise in the data…
Yes, we just mean here that eTreg groups are less diffuse - means more convergent.
- I'm not sold on the eCD4 to eTreg conversion evidence. Why only limit to the top 100 clones? The top 1000 clones were used in previous analyses! Moreover, the authors claim that calculating relative overlap (via F2) of matching CDR3+V+J genes is evidence of a conversion between eCD4 and eTreg. I think to convince myself of a real conversion, I would track the cells between groups, unfortunately, I'm not sure how to track this.. Maybe looking at the thymus population? For example, what is the overlap in the thymus vs. after the challenge? I don't have an answer on how to verify but I feel that this conclusion is a bit on the weaker end.
Distinct normalisation procedure was required to focus on the most expanded clonotypes to avoid the tail of (presumably cross-reactive) and identical TCRs present in all repertoires in these limited-repertoire mice. So we downsampled as strictly as possible to minimise this background signal of nucleotide overlap, and only this strict downsampling to the top-100 clonotypes allowed us to visualise the difference between the challenges. This is a sort of too complicated explanation that would overload the manuscript. But your comments and our answers will be available to the reader who wants to go into all the details.
The observed (at this strict downsampling) overlaps between the top-nucleotide clones in both LLC and PYMT challenges are prominently above the average, and this result is reproducible in lungs and skin, so we have no doubts in interpretations based on these data. Further experiments with different methods, including tracking the clonal fates, should clarify and confirm/correct/disprove our findings.
- There is a nuance in the analysis between Figure 3 and Figure 5 which I think I am not grasping. Both Figures use the same method and the same data but what is different? I think the manuscript would benefit from making this crystal clear. The conclusions will likely be more evident as well!
As explained in the text and above, on Figure 5 “we performed MDS analysis of CDR3α repertoires for distinct lymphatic tissues, excluding the lungs due to their otherwise dominant response to the current challenge.”
The idea of this mini-chapter of the manuscript is to reveal tissue-resident Tregs, distinct for distinct tissues, resident there in all these mice, irrespectively of the challenge we applied. And they are really there (!).
- Do the authors plan to share their R scripts?
All calculations were performed in VDJtools. R was only used to build figures. Corrected this in Methods.
Minor typos and formatting issues to address:
- Typo in Figure 2a the category should read "worm" instead of "warm"
Corrected.
- Figure 2a heatmap is missing a color bar indicating the value ranges
The detailed information can be found in additional Supplementary materials.
- Figure 2f is never mentioned in the manuscript!
Corrected.
- "eTreg repertoire upon lung challenge is reflected in the draining lymph node" - the word upon is of a lower size
Corrected.
- The authors should make the spelling of eTreg uniform across the manuscript (reg in subscript vs just lower case letters. Same goes for CDR3a vs CDR3\alpha
Corrected.
- Figure 4a-d p-values annotations are not shown. Is it because they are not significant?
Corrected.
- The spelling of FACS buffer should be uniform (FACs vs FACS, see methods)
Corrected.
- In the gating strategy, I would make a uniform annotation for the cluster of differentiation, for example, "CD44 high" vs "CD44^{hi}", pos vs + etc.
Corrected.
- Citation for MIGEC software (if available) is missing from methods
Present in the text so probably sufficient.
Reviewer #2 (Recommendations For The Authors):
I noticed the data was made available via Figshare in the preprint, but there is no data availability statement in the current ms.
We provided Data availability statement.
The methods state that custom scripts were written to perform the various analyses. Those should be made available in a code repository, and linked in the ms.
All calculations were performed in VDJtools. R was only used to build figures. Corrected this in Methods.
The title mentioned "TCR repertoire prism", so I thought "prism" was the name of a new method or software. But then the word "prism" didn't appear anywhere in the ms.
We just mean viewing or understanding something from a different perspective or through a lens that reveals different aspects or nuances.
Figure 1D lacks an x-axis label.
Worked on the figures in general.
Reviewer #3 (Recommendations For The Authors):
- The paper is very concise, possibly a bit too much. It could use additional explanations to properly affirm its relevance, for example:
why the choice of fixing the CDR3beta background?
To make repertoire more similar across the mice, and to track all the features of repertoire using only one chain.
to what it is fixed?
As explained in Methods:
“C57BL/6J DO11.10 TCRβ transgenic mice (kindly provided by Philippa Marrack) and crossed to C57BL/6J Foxp3eGFP TCRa-/- mice.”
What do you expect to see and not to see in this specific system and why it is important?
As stated above: we expected repertoire to be more similar across the mice, and it is important to find antigen-specific TCR clusters across mice, and to be able to track all the features of the TCR repertoire using only one chain.
Does this system induce more convergent responses? If so, can we extrapolate the results from this system to the full alpha-beta response?
Such a model, compared to conventional mice, is much more powerful in terms of the ability of monitoring convergent TCR responses. At the same time, it behaves natural, mice live almost normally, so we believe it reflects natural behaviour of the full fledged alpha-beta T cell repertoire.
- Is the lack of similarity of other tissues to Lung/MLN due to a lack of a response?
As indicated in the title of the corresponding mini-chapter: “eTreg repertoire upon lung challenge is reflected in the draining lymph node”. And conclusion of this mini-chapter is that “these results demonstrate the selective tissue localization of the antigen-focused Treg response. ”
Can you do a dendrogram like 2a for the other tissues to better clarify what is going on there? There is space in the supplementary material.
We built lots of those, but in such single dimension mostly they are less informative compared to 2D MDS plots.
- Figure 5 seems a bit out of place as it looks more related to Figure 2. It could maybe be integrated there, sent to supplementary or become Figure 3?
This is a different story, about resident Tregs, irrespective of the challenge.
The whole explanation is here in the text:
“Global CDR3α cluster analysis revealed that characteristic eTreg TCR motifs were present in distinct lymphatic tissues, including spleen and thymus, irrespective of the applied challenge (Supplementary Fig. 1). To better illustrate this phenomenon, we performed MDS analysis of CDR3α repertoires for distinct lymphatic tissues, excluding the lungs due to their otherwise dominant response to the current challenge. This analysis demonstrated close proximity of eTreg repertoires obtained from the same lymphatic tissues upon all lung challenges and across all animals (Fig. 5a, b). These results indicate that distinct antigenic specificities are generally characteristic for eTreg cells that preferentially reside in particular lymphatic niches. Notably, the convergence of lymphatic tissue-resident TCR repertoires was less prominent for the eCD4 T cells (Fig. 5c, d).”
And in the abstract:
“Additionally, our TCRα repertoire analysis demonstrated that distinct antigenic specificities are characteristic for eTreg cells residing in particular lymphatic tissues, regardless of the challenge, revealing the homing-specific, antigen-specific resident Treg populations. ”
- Have you explored more systematically the role of individual variability? If you stratify by individual, do you observe any trend? If not this is also an interesting observation to highlight and discuss.
This is inside the calculations and figures/ one dot = 1 mice, so this natural variation is there inside.
- Regarding the MDS plots: why are 2 dimensions the right amount? Maybe with 3, you can see both tissue specificity and stimuli contributions. Can you do a stress vs # dimensions plot to check what should be the right amount of dimensions to more accurately reproduce the distance matrix?
Tissue specificity and stimuli contribution is hard to distinguish without focussing on appropriate samples, as we did on Fig. 3 and 5. The work is already not that simple as is, and attempting to analyse this in multidimensional space is far beyond our current abilities. But this is an interesting point for future work, thank you.
- Figure 2: A better resolution is needed in order to properly resolve the logo plots at the bottom.
Yes, we worked on Figures, and also provide new Supplementary Figure with all the logos.
- No code or data are made available. There is also a lack of supplementary figures that complement and expand the results presented in the main text.
We believe that the main text, although succinct, contains lots of information to analyse and conclusions (preliminary) to make. So we do not see it rational to overload it further.
-
-
-
wiki.linuxfoundation.org wiki.linuxfoundation.org
-
anyone can modify the code anytime
??? Just try to modify the Linux Kernel source code and you may be banned https://www.theverge.com/2021/4/30/22410164/linux-kernel-university-of-minnesota-banned-open-source
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
We would like to express our sincere appreciation for the invaluable comments provided by the reviewers and their constructive suggestions to enhance the quality of our manuscript. In response to their feedback, we have diligently revised and resubmitted our paper as an article, introducing five primary figures, seven supplementary figures, and two supplementary data files. Importantly, this work represents a noteworthy contribution to the field, presenting novel findings for the first time without any prior publication.
Within the enclosed document, we have provided a comprehensive response to the reviewer comments, addressing each point in a meticulous and specific manner. We extend our sincere gratitude to the reviewers for their diligent examination of our manuscript and for offering insightful recommendations.
In our latest revision, we have taken great care to respond to every reviewer's comment, ensuring that we clarify the manuscript and provide robust evidence where required. The primary focus of these revisions was to provide additional context regarding the cooperative role between PR-Set-7 and PARP-1 in the repression of metabolic genes, accompanied by a thorough description of the current state of the field. Substantial modifications and new analyses, presented in the supplemental figures, have been included to comprehensively address this concern.
Another concern raised was regarding the interaction between PARP-1 and mono-methylated active histone marks, which was not adequately described in the previous version of our manuscript. In this revised version, we have updated our Fig. 1 and Supplemental Fig. S1 and introduced Supplemental Fig. S2 to properly demonstrate that PARP-1 binds to all mono-methylated active histone marks tested. Furthermore, we extensively revised the Discussion section of our manuscript to discuss the implications of this discovery and how it fits into the broader context of PARP-1 research.
Addressing another reviewer's concern about the potential indirect regulation of transcription by PARP1 and PR-SET7, we revised the discussion section and incorporated findings from our recent study. These findings clearly demonstrate PARP1's binding to the loci of misregulated genes, suggesting a direct involvement in their regulation.
Furthermore, we have improved the description of the reagents and Drosophila lines used in this study to provide a more comprehensive understanding for readers. Finally, we conducted a comprehensive revision of the entire manuscript to rectify the identified typos and grammatical errors.
Enclosed, you will find a detailed, point-by-point response to each of the reviewer's comments, showcasing our commitment to addressing their concerns with precision.
We firmly believe that our revisions successfully resolve all the concerns raised by the reviewers, and we are confident that this improved version of our manuscript contributes significantly to the scientific discourse.
Reviewer #1:
The study investigates the role of PARP-1 in transcriptional regulation. Biochemical and ChIP-seq analyses demonstrate specific binding of PARP-1 to active histone marks, particularly H4K20me, in polytene chromosomes of Drosophila third instar larvae. Under heat stress conditions, PARP-1's dynamic repositioning from the Hsp70 promoter to its gene body is observed, facilitating gene activation. PARP-1, in conjunction with PR-Set7, plays a crucial role in the activation of Hsp70 and a subset of heat shock genes, coinciding with an increase in H4K20me1 levels at these gene loci. This study proposes that H4K20me1 is a key facilitator of PARP-1 binding and gene regulation. However, there are several critical concerns that are yet to be addressed. The experimental validation and demonstration of results in the main manuscript are scant. Recent developments in the area are omitted, as an important publication hasn't been discussed anywhere in the work (PMID: 36434141). The proposed mechanism operates quite selectively, and any extrapolations require intensive scientific evidence.
Major Comments:
(1) PARP1 hypomorphic mutant validation data must be provided at RNA levels as the authors have mentioned about its global reduction in RNA levels.
We sincerely appreciate Reviewer 1 for their meticulous review of our manuscript and for providing valuable insights. In response to the raised concern, we would like to highlight that the validation data for the PARP1 hypomorphic mutant at the RNA level has been previously documented in our study (PMID: 20371698), where we found that PARP1 RNA level was deeply impacted in parp1C03256. To enhance clarity, we have made corresponding modifications to the Materials and Methods section to explicitly articulate this aspect: parp-1C03256 significantly lowers the level of PARP-1 RNA and protein level (14) but also significantly diminishes the level of pADPr (11).
We hope these revisions effectively address the reviewer's suggestion and contribute to a more comprehensive understanding of our findings.
(2) The authors should provide immunoblot data for global Poly (ADP) ribosylation levels in PARP1 hypomorphic mutant condition as compared to the control. They must also provide the complete details of the mouse anti-pADPr antibody used in their immunoblot in Figure 5B.
We extend our gratitude to Reviewer 1 for drawing attention to aspects requiring further clarification. In response to the inquiry about global Poly (ADP) ribosylation levels in the PARP1 hypomorphic mutant condition, we want to emphasize that our study extensively reported on the diminished levels of pADPr in comparison to the wildtype, as documented in our previous work (PMID: 21444826). To address this, we have incorporated pertinent details in the Materials and Methods section, providing a comprehensive account of our findings. parp-1C03256 significantly lowers the level of PARP-1 RNA and protein level (14) but also significantly diminishes the level of pADPr (11).
Furthermore, in addressing the request for complete details of the mouse anti-pADPr antibody (10H) used in Figure 5B, we have taken steps to enhance transparency. The Materials and Methods section has been revised to incorporate more comprehensive information about the antibody, ensuring a clearer understanding of our experimental procedures. anti-pADPr (Mouse monoclonal, 1:500, 10H - sc-56198, Santa Cruz).
We appreciate the reviewer's diligence in ensuring the robustness of our methodology, and we believe these modifications strengthen the overall quality and transparency of our study.
(3) PR-Set7 mutant validation results should be provided in the main manuscript, as done by the authors using qRT-PCR. Also, immunoblot data for the PR-set7 null condition should be supplemented in the main manuscript as the authors have already mentioned their anti-PR-Set7 (Rabbit, 1:1000, Novus Biologicals, 44710002) antibody in the materials and methods section.
We appreciate Reviewer 1's thorough examination of our manuscript and their constructive feedback. The pr-set7 null mutant has been rigorously characterized in a study conducted by Dr. Ruth Steward's laboratory (PMID: 15681608). Additionally, we employed our PR-SET7 antibody to validate the mutant, and the corresponding data can be found in Supplemental Figure 3. To enhance clarity, we have made necessary modifications to both the results and Materials and Methods sections, providing explicit details on the validation process. Result section: To validate our hypothesis, we initially confirmed that the pr-set720 mutant not only eliminated PR-SET7 RNA and protein but also abrogated H4K20me1 modification (Supplemental Fig.S3).
Material and methods section: The pr-set720 null mutant was validated in (15) and we confirmed that this mutant abolishes PR-SET7 RNA and protein level but also leads to the absence of H4K20me1 (Supplemental Fig. S3).
We believe these revisions address the reviewer's concerns and contribute to a more comprehensive presentation of our study.
(4) The authors have probably missed out on a very important recent report (PMID: 36434141), suggesting the antagonistic nature of the PARP1 and PR-SET7 association. In light of these important observations, the authors must check for the levels of PR-SET7 in PARP1 hypomorphic conditions.
We appreciate the insightful comment from Reviewer 1, drawing our attention to the recent study by Estève et al. (PMID: 36434141) highlighting the potential antagonistic relationship between PARP1 and PR-SET7. To address this important point, we have carefully examined the levels of PR-SET7 in PARP1 hypomorphic conditions.
In response to this concern, we have added two new supplemental figures, Supplemental Fig. S4 and S5, which specifically address the impact of PARP1 deficiency on PR-Set7 expression. These figures clearly demonstrate that there were no significant changes observed in PR-SET7 RNA (Fig. S4) or protein levels (Fig. S5) in the absence of Parp1. This finding supports the conclusion that Parp1 is not directly involved in the regulation of PR-SET7 in Drosophila.
Furthermore, we have updated the Results section to explicitly mention this observation:
Interestingly, in the absence of PARP-1, neither PR-SET7 RNA nor protein levels were affected (Supplemental Fig. S4-5), indicating that PARP-1 is not directly implicated in the regulation of PR-SET7.
Additionally, we have included information about the anti-H3 antibody used in Supplemental Fig. S4 in the Materials and Methods section: anti-H3 (Rabbit polyclonal, 1/1000, FL-136 sc-10809 Santa Cruz).
We believe that these modifications effectively address the raised concern and provide a more comprehensive understanding of the relationship between PARP1 and PR-SET7 in our study. We hope these clarifications enhance the overall robustness and clarity of our findings.
(5) Also, the results of the aforementioned study should be adequately discussed in the present study along with its implications in the same.
We appreciate Reviewer 1's valuable suggestion to discuss the implications of the study by Estève et al. (PMID: 36434141) within the context of our own findings. Estève et al. reported a potential antagonistic relationship between PARP1 and PR-SET7, showing that a decrease in PARP1 proteins leads to an increase in PR-SET7 protein levels. In our investigation, however, we did not observe significant changes in PR-SET7 RNA and protein levels in the parp1C03256 mutant, as demonstrated in the newly added Supplemental Fig. S3 and S4.
We acknowledge the discrepancy between our results and those of Estève et al., and we propose that this difference may be due to distinct experimental approach: Estève et al.'s study focused on mammalian cell populations and in vitro experiments, whereas our investigation employed Drosophila third-instar larvae as the whole organism model. It is plausible that regulatory mechanisms governing PR-SET7 differ between mammals and Drosophila. Another possibility is that PARP-1 may cooperate with PR-SET7 in the context of Drosophila development but could exhibit antagonistic roles against PR-SET7 in specific cell lines and under certain biological or developmental conditions.
In the Discussion section, we have incorporated this information, stating: A recent study demonstrated that in human cells overexpressing PARP-1, PR-SET7/SET8 is degraded (33). This implies that the absence of PARP-1 might lead to increased levels of PR-SET7. However, in our study involving parp-1 mutant in Drosophila third-instar larvae, we observed a slightly different scenario: we detected a minor but not significant reduction in both PR-SET7 RNA and protein levels (Supplemental Fig.S4 and S5). This outcome stands in stark contrast to the previous study's findings. The discrepancy could be due to the distinct experimental approaches used: the previous research focused on mammalian cells and in vitro experiments, whereas our study examined the functions of PARP-1 in whole Drosophila third-instar larvae during development. Consequently, while PARP-1 may cooperate with PR-SET7 in the context of Drosophila development, it could exhibit antagonistic roles against PR-SET7 in specific cell lines and under certain biological or developmental conditions.
We believe these modifications provide a comprehensive discussion of the observed discrepancies and enhance the overall interpretation of our findings. We hope that these clarifications satisfactorily address the concerns raised by Reviewer 1.
(6) Gene transcriptional activation requires open chromatin and RNA polymerase II binding to the promoter. Since, differentially expressed genes in both PR-Set7 null and PARP1 hypomorph mutants, co-enriched with PARP-1 and H4K20me1 were mainly upregulated, the authors should provide RNA polymerase II occupancy data of these genes via RNA-Pol II ChIP-seq to further attest their claims.
We appreciate the insightful comment from Reviewer 1 regarding the necessity for RNA-polymerase II (PolII) occupancy data to further support our claims on gene transcriptional activation. To address this concern, we conducted an analysis of PolII occupancy around genes co-enriched with PARP-1 and H4K20me1 that are upregulated in both pr-set720 and parp-1C03256 mutants during the third instar larvae stage. The results of this analysis have been included in the newly added supplemental Fig. S5.
Our findings reveal that these upregulated genes exhibit higher PolII occupancy compared to other genes, both at their promoter regions and gene bodies, suggesting heightened activity during third instar larval stage in wild type animals (Supplemental Fig. S6). To further validate these results, we cross-referenced publicly available RNA-seq data at the same developmental stage, confirming that, on average, these upregulated genes display a 40% higher expression compared to other genes (supplemental Fig. S6B).
Moreover, we would like to highlight the consistency of our current findings with our previous study (PMID: 38012002), where we reported the critical involvement of PARP-1 in tempering the expression of active metabolic genes at the end of the third instar larvae. The current data, suggesting a role for PR-SET7 in this regulatory process, adds another layer to our understanding of the nuanced control exerted by PARP-1 on the expression of active metabolic genes during this critical developmental transition.
In light of these results, we have modified the Results section to emphasize these findings: Intriguingly, under wild-type conditions, these genes displayed expression levels approximately 40% higher than the average and demonstrated increased RNA-Polymerase II occupancy both at their promoter regions and gene bodies compared to other genes (supplemental Fig.S6), indicating their high activity in wild type context.
Additionally, we have incorporated this information into the Discussion section to underscore the cooperative role of PARP-1 and PR-SET7 in repressing the expression of active metabolic genes: Notably, genes co-enriched with PARP-1 and H4K20me1, and are upregulated in both parp-1C03256 and pr-set720 mutants, are predominantly metabolic genes exhibiting high expression levels under wild-type conditions and a high occupancy of polymerase II both at their promoter region and gene body (Supplemental Fig. S6). In our previous study, we discovered that PARP-1 plays a crucial role in repressing highly active metabolic genes during the development of Drosophila by binding directly to their loci (34). Also, PARP-1 is required for maintaining optimum glucose and ATP levels at the third-instar larval stage (34). During Drosophila development, repression of metabolic genes is crucial for larval to pupal transition (35, 36). This repression is linked to the reduced energy requirements as the organism prepares for its sedentary pupal stage (35, 37). Notably, we observed that PARP-1 shows a high affinity for binding to the gene bodies of these metabolic genes (34).
Our data indicates that in both parp-1 and pr-set7 mutant animals, there was a preferential repression of metabolic genes at sites where PARP-1 and H4K20me1 are co-bound (Fig.3E), while these metabolic genes are highly active during third-instar larval stage (Supplemental Fig.S6). Thus, we propose that the presence of H4K20me1 may be essential for the binding of PARP-1 at these gene bodies, contributing to their repression. Importantly, this mechanism of gene repression has broader developmental implications. As earlier stated, mutant animals lacking functional PARP-1 and PR-SET7 undergo developmental arrest during larval to pupal transition. This arrest could be directly linked to the disruption of the normal metabolic gene repression during development. Without the repressive action of PARP-1 and PR-SET7, key metabolic processes might remain unchecked, leading to metabolic imbalances that are incompatible with the normal progression to the pupal stage.
Finaly, we have updated the Materials and Methods section to include information about the RNA-seq and PolII ChIP-seq datasets used: GSE15292 (RNA-polymerase II). In addition, we used the Developmental time-course RNA-seq dataset (54), SRP001065.
We believe that these modifications comprehensively address Reviewer 1's concern and provide a more robust foundation for our claims regarding the role of PARP-1 and PR-SET7 in the transcriptional regulation of co-enriched genes during the critical developmental transition.
(7) As discussed in Figure 4, the authors found transcriptional activation of group B genes even after a significant reduction of H3K20me1 in their gene body after heat shock. Given the dynamic equilibrium shift in epigenetic marks that regulate gene expression and their locus-specific transcriptional regulation, the authors should further look for the enrichment of other epigenetic marks and even H4K20me1 specific demethylases such as PHF8 (PMID: 20622854), and their cross-talk with PARP1 to further bridge the missing links of this tale. This will add more depth to this work.
We appreciate the thoughtful input provided by Reviewer 1 and acknowledge the importance of exploring additional epigenetic marks and potential cross-talk association with PARP1 to enhance the depth of our study. Our investigation has primarily focused on the interplay between PR-SET7/H4K20me1 and PARP-1, as evidenced by the colocalization and robust binding affinity observed between PARP-1 and H4K20me1 (Fig 1C, 2B, and 3A). This interaction is particularly noteworthy in the context of regulating specific heat shock genes, as highlighted in Figure 4A. While we recognize the potential significance of examining a broader spectrum of epigenetic marks and considering the involvement of specific demethylases, such as PHF8 (PMID: 20622854), in this regulatory network, our research strategy is intentionally tailored to leverage the unique characteristics of the PR-SET7/H4K20me1 and PARP-1 interplay in Drosophila. A key consideration is the technical advantage afforded by the fact that PR-SET7 is the exclusive methylase responsible for H4K20 in Drosophila (PMID: 15681608), allowing for specific depletion of H4K20me1 without the confounding influence of other methyltransferases.
This specificity is pivotal, especially given the similar developmental arrest patterns observed in both PR-SET7 and PARP-1 mutants. Such parallel phenotypes provide a distinct opportunity to delve deeply into the intricacies of their interaction during organismal development and in response to heat stress. Additionally, the identity of the demethylase for H4K20me1 in Drosophila remains unknown, further underscoring the rationale for our focused approach.
While we acknowledge the broader implications of exploring additional epigenetic marks, we believe that our deliberate focus on the PR-SET7/H4K20me1 and PARP-1 pathway provides a unique and valuable perspective on the regulation of gene expression in Drosophila. We hope that this clarification addresses the concerns raised by Reviewer 1 and conveys the rationale behind our chosen research strategy.
Reviewer #2:
Summary:
This study from Bamgbose et al. identifies a new and important interaction between H4K20me and Parp1 that regulates inducible genes during development and heat stress. The authors present convincing experiments that form a mostly complete manuscript that significantly contributes to our understanding of how Parp1 associates with target genes to regulate their expression.
Strengths:
The authors present 3 compelling experiments to support the interaction between Parp1 and H4K20me, including:
(1) PR-Set7 mutants remove all K4K20me and phenocopy Parp mutant developmental arrest and defective heat shock protein induction.
(2) PR-Set7 mutants have dramatically reduced Parp1 association with chromatin and reduced poly-ADP ribosylation.
(3) Parp1 directly binds H4K20me in vitro.
Weaknesses:
(1) The histone array experiment in Fig1 strongly suggests that PARP binds to all mono-methylated histone residues (including H3K27, which is not discussed). Phosphorylation of nearby residues sometimes blocks this binding (S10 and T11 modifications block binding to K9me1, and S28P blocks binding to K27me1). However, H3S3P did not block H3K4me1, which may be worth highlighting. The H3K9me2/3 "blocking effect" is not nearly as strong as some of these other modifications, yet the authors chose to focus on it. Rather than focusing on subtle effects and the possibility that PARP "reads" a "histone code," the authors should consider focusing on the simple but dramatic observation that PARP binds pretty much all mono-methylated histone residues. This result is interesting because nucleosome mono-methylation is normally found on nucleosomes with high turnover rates (Chory et al. Mol Cell 2019)- which mostly occurs at promoters and highly transcribed genes. The author's binding experiments could help to partially explain this correlation because PARP could both bind mono-methylated nucleosomes and then further promote their turnover and lower methylation state.
We appreciate the comprehensive review and valuable insights provided. In response to the comments, we have made substantial revisions to address the concerns and enhance the clarity of our findings. In Figure 1B, C, D, F, and G, we have expanded our data presentation to demonstrate PARP-1's binding affinity for H3K27me1. This addition is now incorporated into the revised results section. Additionally, we have updated Supplemental Fig.S1 and introduced new supplemental data (Supplemental Fig.S2) to illustrate the inhibition of PARP-1 binding by H3S10P, H3S28P, and H3T11P. The comprehensive exploration of PARP-1's interaction with mono-methylated histones, as suggested by the reviewer, is now more robustly documented in our revised figures and supplementary materials.
Our Discussion section has been refined to articulate more clearly how PARP-1 may be selectively recruited to active chromatin domains through its interaction with mono-methylated histone marks. We have proposed a model where PARP-1 actively participates in the turnover process, contributing to the maintenance of an active chromatin environment. This proposed mechanism involves PARP-1 selectively binding to mono-methylated active histone marks associated with highly transcribed genes. Upon activation, PARP-1 undergoes automodification, leading to its release from chromatin and facilitating the reassembly of nucleosomes carrying the mono-methylated marks. The enzymatic action of Poly(ADP)-ribose glycohydrolase (PARG) subsequently cleaves pADPr, allowing for the restoration of PARP-1's binding affinity to mono-methylated active histone marks. This proposed hypothesis is consistent with existing research across various model organisms and aligns with the known association of PARP-1 with highly expressed genes, as well as its role in mediating nucleosome dynamics and assembly.
Our Discussion section is modified a followed: Finaly, highly transcribed genes have been reported to present a high turnover of mono-methylated modifications, maintaining a state of low methylation (50). Then, our findings suggest that PARP-1 might actively participate in the turnover process to uphold an active chromatin environment. The proposed mechanism unfolds as follows: 1) PARP-1 selectively binds to mono-methylated active histone marks associated with highly transcribed genes. 2) Upon activation, PARP-1 undergoes automodification and is subsequently released from chromatin, facilitating the reassembly of nucleosomes carrying the mono-methylated marks. 3) The enzymatic action of Poly(ADP)-ribose glycohydrolase (PARG) cleaves pADPr, allowing for the restoration of PARP-1's binding affinity to mono-methylated active histone marks. This proposed hypothesis aligns cohesively with existing research conducted across various model organisms, including mice, Drosophila, and Humans (7, 23, 29, 51-53). Notably, previous studies have consistently demonstrated that PARP-1 predominantly associates with highly expressed genes and plays a crucial role in mediating nucleosome dynamics and assembly. Thus, our proposed model provides a molecular framework that may contribute to understanding the relationship between PARP-1 and the epigenetic regulation of gene expression. Further experimental validation is warranted to elucidate the precise details of this proposed mechanism and its implications in the broader context of chromatin dynamics and transcriptional control.
We hope that these revisions address the reviewer's concerns and contribute to the overall strength and clarity of our manuscript.
(2) The RNAseq analysis of Parp1/PR-Set7 mutants is reasonable, but there is a caveat to the author's conclusion (Line 251): "our results indicate H4K20me1 may be required for PARP-1 binding to preferentially repress metabolic genes and activate genes involved in neuron development at co-enriched genes." An alternative possibility is that many of the gene expression changes are indirect consequences of altered development induced by Parp1 or PR-Set7 mutants. For example, Parp1 could activate a transcription factor that represses the metabolic genes that they mention. The authors should consider discussing this possibility.
We hope that these revisions address the reviewer's concerns and contribute to the overall strength and clarity of our manuscript.
We extend our gratitude to Reviewer 2 for their thoughtful consideration of our manuscript and the insightful suggestion. In response to the raised concern regarding the conclusion on Line 251, where we proposed that "our results indicate H4K20me1 may be required for PARP-1 binding to preferentially repress metabolic genes and activate genes involved in neuron development at co-enriched genes," we acknowledge the alternative possibility suggested by the reviewer. It is plausible that many of the observed gene expression changes are indirect consequences of altered development induced in parp-1 or pr-set7 mutants. For example, PARP-1 could activates a transcription factor that represses the mentioned metabolic genes.
To address this concern, we have revisited our data and incorporated relevant findings from one of our recent studies that utilized a ChIP-seq approach. The results from this study suggest a direct binding of PARP-1 to the loci of metabolic genes, providing support for the notion that PARP-1 may indeed directly regulate their expression (PMID: 37347109). We have updated the Discussion section to reflect this information, aiming to provide a more comprehensive perspective on the potential mechanisms underlying the observed gene expression changes: In our previous study, we discovered that PARP-1 plays a crucial role in repressing highly active metabolic genes during the development of Drosophila by binding directly to their loci (34). Also, PARP-1 is required for maintaining optimum glucose and ATP levels at the third-instar larval stage (34). During Drosophila development, repression of metabolic genes is crucial for larval to pupal transition (35, 36). This repression is linked to the reduced energy requirements as the organism prepares for its sedentary pupal stage (35, 37). Notably, we observed that PARP-1 shows a high affinity for binding to the gene bodies of these metabolic genes (34).
We believe these modifications contribute to a more informed interpretation of our findings.
(3) The section on the inducibility of heat shock genes is interesting but missing an important control that might significantly alter the author's conclusions. Hsp23 and Hsp83 (group B genes) are transcribed without heat shock, which likely explains why they have H4K20me without heat shock. The authors made the reasonable hypothesis that this H4K20me would recruit Parp-1 upon heat shock (line 270). However, they observed a decrease of H4K20me upon heat shock, which led them to conclude that "H4K20me may not be necessary for Parp1 binding/activation" (line 275). However, their RNA expression data (Fig4A) argues that both Parp1 and H40K20me are important for activation. An alternative possibility is that group B genes indeed recruit Parp1 (through H4K20me) upon heat shock, but then Parp1 promotes H3/H4 dissociation from group B genes. If Parp1 depletes H4, it will also deplete H4K20me1. To address this possibility, the authors should also do a ChIP for total H4 and plot both the raw signal of H4K20me1 and total H4 as well as the ratio of these signals. The authors could also note that Group A genes may similarly recruit Parp1 and deplete H3/H4 but with different kinetics than Group B genes because their basal state lacks H4K20me/Parp1. To test this possibility, the authors could measure Parp association, H4K20methylation, and H4 depletion at more time points after heat shock at both classes of genes.
We thank Reviewer 2 for their valuable comment on our manuscript. We acknowledge your hypothesis suggesting that PARP-1 may induce H3/H4 dissociation from group B genes, potentially leading to a reduction in H4K20me1. However, our findings support a different interpretation.
Our data indicate that while H4K20me1 is present under normal conditions at group B genes, its reduction following heat shock does not appear to hinder PARP-1's role in transcriptional activation (Fig 4A, C and E). We propose that the observed decrease in H4K20me1 might reflect a regulatory shift in chromatin structure that is conducive to transcriptional activation during heat shock, facilitated by PARP-1 independently of sustained H4K20me1 levels at group B genes. Additionally, the literature suggests a dual role for H4K20me1 in gene regulation, from facilitating transcriptional elongation in certain contexts to acting as a repressor in others.
Unlike in group A genes which had low enrichment of H4K20me1 before heat shock (Fig 4B and D), the high enrichment of H4K20me1 in group B genes (Fig 4C and E) could imply a repressive role for this mark prior to heat stress. Thus, in the context of group B genes, it's conceivable that the removal of H4K20me1 might be necessary for their activation during heat stress. Thus, PR-SET7 may possess functions beyond its role as a histone methylase, which are crucial for activating group B genes under heat stress conditions. These functions could include methylation of non-histone substrates and non-catalytic activities.
Furthermore, our analysis of gene expression in pr-set720 and parp-1C03256 mutants indicates that while PARP-1 and H4K20me1 interaction may have overlapping roles in gene regulation, they also possess distinct functions in the modulation of gene expression (Fig 3E). Thus, we propose that the relationship between PR-SET7 and PARP-1 in transcriptional regulation involves a complex regulatory mechanism that extends beyond the presence of H4K20me1.
We modified the discussion section to address this point: Another plausible explanation could be that the recruitment of PARP-1 to group B genes loci promotes H4 dissociation and then leads to a reduction of H4K20me1. However, our findings suggest an alternative interpretation: the decrease in H4K20me1 at group B genes during heat shock does not seem to impede PARP-1's role in transcriptional activation, (Fig.4A, C and E). Rather than disrupting PARP-1 function, we propose that this reduction in H4K20me1 may signify a regulatory shift in chromatin structure, priming these genes for transcriptional activation during heat shock, with PARP-1 playing an independent facilitating role. Moreover, existing studies have highlighted the dual role of H4K20me1, acting as a promoter of transcription elongation in certain contexts and as a repressor in others (13, 25, 38, 39, 41-45). The elevated enrichment of H4K20me1 in group B genes under normal conditions may indicate a repressive state that requires alleviation for transcriptional activation. Additionally, we cannot discount the possibility of unique regulatory functions associated with PR-SET7, extending beyond its recognized role as a histone methylase. Non-catalytic activities and potential interactions with non-histone substrates might contribute to the nuanced control exerted by PR-SET7 on group B genes during heat stress (46, 47). Furthermore, our exploration of pr-set720 and ParpC03256 mutants reveals distinct roles for PARP-1 and H4K20me1 in modulating gene expression (Fig 3E). This reinforces the notion that the interplay between PR-SET7 and PARP-1 involves a multifaceted regulatory mechanism. Understanding the intricate relationship between these molecular players is crucial for elucidating the complexities of gene expression modulation under heat stress conditions.
We hope that this modification will adequately address Reviewer 2 concerns and enhance the clarity of our conclusions.
Reviewer #1 (Recommendations For The Authors):
(1) Please check the entire manuscript for grammatical errors and typos. PR-set7 has been wrongly written as PR-ste7 in quite a few places in the manuscript. Poly (ADP)-ribosylation has been written as poly(ADP-ribosyl)ation in the last result heading. There are more such errors. Please rectify them.
We express our sincere appreciation to Reviewer 1 for their meticulous review of our manuscript, and we acknowledge the importance of ensuring grammatical accuracy and clarity. We have taken your feedback seriously and conducted a comprehensive revision of the entire manuscript to rectify the identified typos and grammatical errors. We hope that these revisions contribute to an improved overall presentation of our research, and we appreciate the reviewer's diligence in ensuring the accuracy of the manuscript.
(2) The authors can also look up publicly available mammalian ChIP-seq data for H4K20me1 and PARP1, in order to further ossify their findings and increase the breadth of their work.
We appreciate the suggestion from Reviewer 1 and have taken steps to further validate and broaden the scope of our findings. Specifically, we compared the distribution of PARP1 and H4K20me1 in Human K562 cells. The results of this analysis revealed a correlation in their distribution, supporting the idea that the observed correlation between PARP-1 and H4K20me1 is not limited to fruit flies. We have incorporated these findings into the Results section and added a new Supplemental Fig. S6 to visually highlight this correlation: Finally, to extend the generalizability of our observations beyond Drosophila, we compared the distribution of PARP1 and H4K20me1 in Human K562 cells. Strikingly, we observed a correlation in their distribution, suggesting that the interplay between PARP-1 and H4K20me1 is not limited to fruit flies (Supplemental Fig. S6).
We believe that this modification addresses Reviewer 1's suggestion by providing additional evidence that supports the broader relevance of our findings beyond the Drosophila model system.
(3) Please discuss in greater detail how the PARP1-H4K20me1 axis orchestrates the repression program (metabolic pathways in this case) with proper references.
We appreciate Reviewer 1's continued engagement with our manuscript and have adjusted the discussion section to provide a more detailed insight into how the PARP1-H4K20me1 axis orchestrates the repression program, particularly focusing on metabolic pathways. The modified discussion section now reads: In our previous study, we discovered that PARP-1 plays a crucial role in repressing highly active metabolic genes during the development of Drosophila by binding directly to their loci (34). Also, PARP-1 is required for maintaining optimum glucose and ATP levels at the third-instar larval stage (34). During Drosophila development, repression of metabolic genes is crucial for larval to pupal transition (35, 36). This repression is linked to the reduced energy requirements as the organism prepares for its sedentary pupal stage (35, 37). Notably, we observed that PARP-1 shows a high affinity for binding to the gene bodies of these metabolic genes (34). Our data indicates that in both parp-1 and pr-set7 mutant animals, there was a preferential repression of metabolic genes at sites where PARP-1 and H4K20me1 are co-bound (Fig.3E), while these metabolic genes are highly active during third-instar larval stage (Supplemental Fig.S6). Thus, we propose that the presence of H4K20me1 may be essential for the binding of PARP-1 at these gene bodies, contributing to their repression. Importantly, this mechanism of gene repression has broader developmental implications. As earlier stated, mutant animals lacking functional PARP-1 and PR-SET7 undergo developmental arrest during larval to pupal transition. This arrest could be directly linked to the disruption of the normal metabolic gene repression during development. Without the repressive action of PARP-1 and PR-SET7, key metabolic processes might remain unchecked, leading to metabolic imbalances that are incompatible with the normal progression to the pupal stage.
We hope these modifications provide a more comprehensive discussion on how the PARP1-H4K20me1 axis influences the repression program, particularly within metabolic pathways, and how this mechanism contributes to the broader context of Drosophila development.
-
-
www.biorxiv.org www.biorxiv.org
-
Reviewer #2 (Public Review):
In this manuscript, the authors propose a computational method based on deep convolutional neural networks (CNNs) to automatically detect cell divisions in two-dimensional fluorescence microscopy timelapse images. Three deep learning models are proposed to detect the timing of division, predict the division axis, and enhance cell boundary images to segment cells before and after division. Using this computational pipeline, the authors analyze the dynamics of cell divisions in the epithelium of the Drosophila pupal wing and find that a wound first induces a reduction in the frequency of division followed by a synchronised burst of cell divisions about 100 minutes after its induction.
Comments on revised version:
Regarding the Reviewer's 1 comment on the architecture details, I have now understood that the precise architecture (number/type of layers, activation functions, pooling operations, skip connections, upsampling choice...) might have remained relatively hidden to the authors themselves, as the U-net is built automatically by the fast.ai library from a given classical choice of encoder architecture (ResNet34 and ResNet101 here) to generate the decoder part and skip connections.
Regarding the Major point 1, I raised the question of the generalisation potential of the method. I do not think, for instance, that the optimal number of frames to use, nor the optimal choice of their time-shift with respect to the division time (t-n, t+m) (not systematically studied here) may be generic hyperparameters that can be directly transferred to another setting. This implies that the method proposed will necessarily require re-labeling, re-training and re-optimizing the hyperparameters which directly influence the network architecture for each new dataset imaged differently. This limits the generalisation of the method to other datasets, and this may be seen as in contrast to other tools developed in the field for other tasks such as cellpose for segmentation, which has proven a true potential for generalisation on various data modalities. I was hoping that the authors would try themselves testing the robustness of their method by re-imaging the same tissue with slightly different acquisition rate for instance, to give more weight to their work.
In this regard, and because the authors claimed to provide clear instructions on how to reuse their method or adapt it to a different context, I delved deeper into the code and, to my surprise, felt that we are far from the coding practice of what a well-documented and accessible tool should be.
To start with, one has to be relatively accustomed with Napari to understand how the plugin must be installed, as the only thing given is a pip install command (that could be typed in any terminal without installing the plugin for Napari, but has to be typed inside the Napari terminal, which is mentioned nowhere). Surprisingly, the plugin was not uploaded on Napari hub, nor on PyPI by the authors, so it is not searchable/findable directly, one has to go to the Github repository and install it manually. In that regard, no description was provided in the copy-pasted templated files associated to the napari hub, so exporting it to the hub would actually leave it undocumented.
Regarding now the python notebooks, one can fairly say that the "clear instructions" that were supposed to enlighten the code are really minimal. Only one notebook "trainingUNetCellDivision10.ipynb" has actually some comments, the other have (almost) none nor title to help the unskilled programmer delving into the script to guess what it should do. I doubt that a biologist who does not have a strong computational background will manage adapting the method to its own dataset (which seems to me unavoidable for the reasons mentioned above).
Finally regarding the data, none is shared publicly along with this manuscript/code, such that if one doesn't have a similar type of dataset - that must be first annotated in a similar manner - one cannot even test the networks/plugin for its own information. A common and necessary practice in the field - and possibly a longer lasting contribution of this work - could have been to provide the complete and annotated dataset that was used to train and test the artificial neural network. The basic reason is that a more performant, or more generalisable deep-learning model may be developed very soon after this one and for its performance to be fairly compared, it requires to be compared on the same dataset. Benchmarking and comparison of methods performance is at the core of computer vision and deep-learning.
-
-
-
Writing Code Without Plain Text Files
The Unison programming language doesn’t store code in files, but in a database. What is that like?
personal.archived - from: https://itnext.io/writing-code-without-plain-text-files-cb8f1ed2c0ad - by: gyuri
-
-
www.frontiersin.org www.frontiersin.org
-
I also did the other analysis regarding the quality of sleep, i'll link the notebook: [(https://www.kaggle.com/code/manuiaccarino/eda-and-predicting-sleep-quality) I was surprised by the predictive ability of a "simple model" without any preprocessing. The data seams to be real (and not syntetic, at least that's what the source of the data say) and nonetheless the data size being small I figure it out that the variables were highly correlated with each other, so either the sleep quality variable was just obtained through a similar algorithms or predicting the quality of sleep can be quite easy with a few relevant variables. Waiting for ur feedback on this one as well. I'm sorry I can't work also on the other dataset but I don't have a lot of time this week and, by a first look, there seams like a lot of work on preprocessing to do there
-
I just finished to analyze one of the dataset I suggested last week, i'll link the Kaggle notebook: [(https://www.kaggle.com/code/manuiaccarino/interesting-finding-predicting-activity-calories?scriptVersionId=166551904) I made some interesting find while trying to predict the Activity detected by the sportwatch depending by the measured data. I don't want to spoiler anything, I hope you can read the work and provide me a feedback so we can discuss about it . Thank you
-
-
eiko-fried.com eiko-fried.com
-
Without exaggeration, I believe that the majority of published works in my field (broadly defined as psychology) do not add value. Many papers draw conclusions that are not supported by evidence, which cascades through the literature, because these papers are cited for the conclusions, not the evidence. The majority of published works are not reproducible, in the sense that authors conduct science behind closed doors without sharing data or code. Many published works are not replicable, i.e., will not hold up to scrutiny over time. Theories are verbal and vague, which means they can never get properly rejected. Instead, as Paul Meehl famously wrote, they sort of just slowly fade away as people lose interest. Let me try to convince you that it is an entirely reasonable position, based on the evidence we have.
Tags
Annotators
URL
-
-
runestone.academy runestone.academy
-
8-2-6: Consider the following code segment. What is the last row of numbers printed when this code segment is executed?
?
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
eLife assessment
This computational study is a valuable empirical investigation into the common trait of neurons in brains and artificial neural networks: responding effectively to both objects and their mirror im- ages and it focuses on uncovering conditions that lead to mirror symmetry in visual networks and the evidence convincingly demonstrates that learning contributes to expanding mirror symmetry tuning, given its presence in the data. Additionally, the paper delves into the transformation of face patches in primate visual hierarchy, shifting from view specificity to mirror symmetry to view invariance. It empirically analyzes factors behind similar effects in two network architec- tures, and key claims highlight the emergence of invariances in architectures with spatial pooling, driven by learning bilateral symmetry discrimination and importantly, these effects extend be- yond faces, suggesting broader relevance. Despite strong experiments, some interpretations lack explicit support, and the paper overlooks pre-training emergence of mirror symmetry.
As detailed above, we have now analyzed several convolutional architectures and made a direct link between the artificial neural networks and neuronal data to further support our claims (refer to Figure 6, S10- 13).
To address the concern about pre-training emergence of mirror symmetry, we conducted a new analysis inspecting unit-level response profile, following Baek and colleagues (2021). This analysis is described in detail below (response to R3). In brief, we found that the first fully connected layer in trained networks exhibits twice the number of mirror symmetric units found before training. In addition to our population-level observations (Fig. S2) and explicit training- dataset manipulations (Fig. 4), this finding supports the interpretation of training to discriminate among mirror- symmetric object categories as a major factor behind the emergence of mirror symmetric viewpoint tuning.
Reviewer 1 (Public Review):
By using deep convolutional neural networks (CNNs) as model for the visual system, this study aims at understanding and explaining the emergence of mirror-symmetric viewpoint tuning in the brain.
Major strengths of the methods and results:
1) The paper presents comprehensive, insightful and detailed analyses investigating how mirror- symmetric viewpoint tuning emergence in artificial neural networks, providing significant and novel insights into this complex process.
2) The authors analyze reflection equivariance and invariance in both trained and untrained CNNs’ convolutional layers. This elucidates how object categorization training gives rise to mirror-symmetric invariance in the fully-connected layers.
3) By training CNNs on small datasets of numbers and a small object set excluding faces, the authors demonstrate mirror-symmetric tuning’s potential to generalize to untrained categories and the necessity of view-invariant category training for its emergence.
4) A further analysis probes the contribution of local versus global features to mirror-symmetric units in the first fully-connected layer of a network. This innovative analysis convincingly shows that local features alone suffice for the emergence of mirror-symmetric tuning in networks.
5) The results make a clear prediction that mirror-symmetric tuning should also emerge for other bilaterally symmetric categories, opening avenues for future neural studies.
We are grateful for your insightful feedback and the positive evaluation of our study on mirror-symmetric viewpoint tuning in neural networks. Your constructive comments considerably improved the manuscript. We eagerly look forward to exploring the future research avenues you have highlighted.
Major weaknesses of the methods and results:
Point 1.1) The authors propose a mirror-symmetric viewpoint tuning index, which, although innovative, complicates comparison with previous work and this choice is not well motivated. This index is based on correlating representational dissimilarity matrices (RDMs) with their flipped versions, a method differing from previous approaches.
We have revised the Methods section to clarify the motivation for the mirror-symmetric viewpoint tuning index we introduced.
Manuscript changes:
Previous work quantified mirror-symmetry in RDMs by comparing neural RDMs to an idealized mirror- symmetric RDM (see Fig. 3c-iii in [14]). Although highly interpretable, such an idealized RDM encompasses implicit assumptions about representational geometry that are unrelated to mirror-symmetry. For example, consider a neural RDM reflecting perfect mirror-symmetric viewpoint tuning and wherein for each view, the distances among all of the exemplars are equal. Such a neural RDM would fit an idealized mirror- symmetric RDM better than a neural RDM reflecting perfect mirror-symmetric viewpoint tuning but with non-equidistant exemplars. In contrast, the measure proposed in Eq. 2 equals 1.0 in both cases.
Point 1.2> Faces exhibit unique behavior in terms of the progression of mirror-symmetric viewpoint tuning and their training task and dataset dependency. Given that mirror-symmetric tuning has been identified in the brain for faces, it would be beneficial to discuss this observation and provide potential explanations.
We revised the caption of Figure S1 to explicitly address this point:
Manuscript changes:
For face stimuli, there is a unique progression in mirror-symmetric viewpoint tuning: the index is negative for the convolutional layers and it abruptly becomes highly positive when transitioning to the first fully connected layer. The negative indices in the convolutional layers can be attributed to the image-space asymmetry of non-frontal faces; compared to other categories, faces demonstrate pronounced front-back asymmetry, which translates to asymmetric images for all but frontal views (Fig. S8). The features that drive the highly positive mirror-symmetric viewpoint tuning for faces in the fully connected layers are training-dependent (Fig. S2), and hence, may reflect asymmetric image features that do not elicit equivariant maps in low-level representations; for example, consider a profile view of a nose. Note that cars and boats elicit high mirror- symmetric viewpoint tuning indices already in early processing layers. This early mirror-symmetric tuning is independent of training (Fig. S2), and hence, may be driven by low-level features. Both of these object categories show pronounced quadrilateral symmetry, which translates to symmetric images for both frontal and side views (Fig. S8).
Point 1.3: 3. Previous work reported critical differences between CNNs and neural represen- tations in area AL indicating that mirror-symmetric viewpoint tuning is less present than view invariance in CNNs compared to area AL. While such findings could potentially limit the use- fulness of CNNs as models for mirror-symmetric viewpoint tuning in the brain, they are not addressed in the study.
This point is now addressed explicitly in the caption of Figure S9:
Manuscript changes:
Yildirim and colleagues [14] reported that CNNs trained on faces, notably VGGFace, exhibited lower mirror- symmetric viewpoint tuning compared to neural representations in area AL. Consistent with their findings, our results demonstrate that VGGFace, trained on face identification, has a low mirror-symmetric viewpoint tuning index. This is especially notable in comparison to ImageNet-trained models such as VGG16. This difference between VGG16 and VGGFace can be attributed to the distinct characteristics of their training datasets and objective functions. The VGGFace training task consists of mapping frontal face images to identities; this task may exclusively emphasize higher-level physiognomic information. In contrast, training on recognizing objects in natural images may result in a more detailed, view-dependent representation. To test this potential explanation, we measured the average correlation-distance between the fc6 representations of different views of the same face exemplar in VGGFace and VGG16 trained on ImageNet. The average correlation-distance between views is 0.70±0.04 in VGGFace and 0.93±0.04 in VGG16 trained on ImageNet. The converse correlation distance between different exemplars depicted from the same view is 0.84±0.14 in VGGFace and 0.58±0.06 in VGG16 trained on ImageNet. Therefore, as suggested by Yildirim and colleagues, training on face identification alone may result in representations that cannot explain intermediate levels of face processing.
Point 1.4) The study’s results, while informative, are qualitative rather than quantitative, and lack direct comparison with neural data. This obscures the implications for neural mechanisms and their relevance to the broader field.
We addressed this point by conducting a quantitative comparison between the architectures of various networks and neural response patterns in monkey face patches (see Figures 6, S10-S13, appearing above).
Point 1.5) The study provides compelling evidence that learning to discriminate bilaterally symmetric objects (beyond faces) induces mirror-symmetric viewpoint tuning in the networks, qualitatively similar to the brain. Moreover, the results suggest that this tuning can, in principle, generalize beyond previously trained object categories. Overall, the study provides important conclusions regarding the emergence of mirror-symmetric viewpoint tuning in networks, and potentially the brain. However, the conducted analyses and results do not entirely address the question why mirror-symmetric viewpoint tuning emerges in networks or the brain. Specifically, the results leave open whether mirror-symmetric viewpoint tuning is indeed necessary to achieve view invariance for bilaterally symmetric objects.
We believe that mirror-symmetric viewpoint tuning is not strictly necessary for achieving view-invariance. However, it is a plausible path from view-dependence to view invariance. We addressed this point in the updated limitations subsection of the discussion.
Manuscript changes:
A second consequence of the simulation-based nature of this study is that our findings only establish that mirror-symmetric viewpoint tuning is a viable computational means for achieving view invariance; they do not prove it to be a necessary condition. In fact, previous modeling studies [10, 19, 61] have demonstrated that a direct transition from view-specific processing to view invariance is possible. However, in practice, we observe that both CNNs and the face-patch network adopt solutions that include intermediate representations with mirror-symmetric viewpoint tuning.
Taken together, this study moves us a step closer to uncovering the origins of mirror-symmetric tuning in networks, and has implications for more comprehensive investigations into this neural phenomenon in the brain. The methods of probing CNNs are innovative and could be applied to other questions in the field. This work will be of broad interest to cognitive neuroscientists, psychologists, and computer scientists.
We appreciate your acknowledgment of our study’s contribution to understanding mirror-symmetric tuning in networks and its wider implications in the field.
Reviewer 2 (Public Review);
Strengths
1) The statements made in the paper are precise, separating observations from inferences, with claims that are well supported by empirical evidence. Releasing the underlying code repository further bolsters the credibility and reproducibility. I especially appreciate the detailed discussion of limitations and future work.
2) The main claims with respect to the two convolutional architectures are well supported by thorough analyses. The analyses are well-chosen and overall include good controls, such as changes in the training diet. Going beyond ”passive” empirical tests, the paper makes use of the fully accessible nature of computational models and includes more ”causal” insertion and deletion tests that support the necessity and sufficiency of local object features.
3) Based on modeling results, the paper makes a testable prediction: that mirror-symmetric viewpoint tuning is not specific to faces and can also be observed in other bilaterally symmetric objects such as cars and chairs. To test this experimentally in primates (and potentially other model architectures), the stimulus set is available online.
We express our gratitude for your constructive feedback. Your acknowledgment of the clarity of our statements and the robustness of our empirical evidence is greatly appreciated. We are also thankful for your recognition of our comprehensive analyses and the testable predictions arising from our work.
Point 2.1: Weaknesses
My main concern with this paper is in its choice of the two model architectures AlexNet and VGG. In an earlier study, Yildirim et al. (2020) found an inverse graphics network ”EIG” to better correspond to neural and behavioral data for face processing than VGG. All claims in the paper thus relate to a weaker model of the biological effects since this work does not analyze the EIG model. Since EIG follows an analysis-by-synthesis approach rather than standard classification training, it is unclear whether the claims in this paper generalize to this other model architecture. It is also unclear if the claims will hold for: 1) transformer architectures, 2) the HMAX architecture by Leibo et al. (2017) which has also been proposed as a computational explanation for mirror-symmetric tuning, and, as the authors note in the Discussion, 3) deeper architectures such as ResNet-50 which tend to better align to neural and behavioral data in general. These architectures include different computational motifs such as skip connections and a much smaller proportion of fully-connected layers which are a major focus of this work.
Overall, I thus view the paper’s claims as limited to AlexNet- and VGG-like architectures, both of which fall behind state-of-the-art in their alignment to primates in general and also specifically for mirror-symmetric viewpoint tuning.
We understand your concern regarding the choice of AlexNet and VGG architectures. The decision to focus on these models was driven by the need for a straightforward macroscopic correspondence between the layer structure of the artificial networks and the ventral visual stream. However, acknowledging this potential limitation of generality, we have expanded our analysis to include the EIG model, a transformer architecture, the HMAX model, and deeper convolutional architectures like ResNet-50 and ConvNeXt. Our revised analysis, detailed in Figures S1, S9, and S10-S13, incorporates these additional models and offers a comprehensive evaluation of their brain alignment and mirror-symmetric viewpoint tuning. We found that while the architectures indeed vary in their computational motifs, the emergence of mirror-symmetric viewpoint tuning is not exclusive to AlexNet and VGG. It occurs for every CNN we tested, exactly at the stage where equivariant feature maps are pooled globally. We believe that the new analyses extend the generality of our findings and remove the concern that our claims apply only to older, shallower networks.
For details, please refer to Point 1 in the ’Essential Revisions’ section.
Point 2.2: Minor weaknesses
1) Figure 1A: since the relevance to primate brains is a major motivator of this work, the results from actual neural recordings should be shown and not just schematics. For instance, the mirror symmetry in AL is not as clean as the illustration (compare with Fig. 3 in Yildirim et al. 2020), and in the paper’s current form, this is not easily accessible to the reader.
Thank you for your feedback regarding the presentation of neural recordings in Figure 1A. We have updated Figure 1A to include actual neural RDMs instead of the previous schematic representations.
Point 2.3: 2. Figure 4 L832-845: The claims for the effect of training on mirror-symmetric viewpoint tuning are with respect to the training data only, but there are other differences between the models such as the number of epochs (250 for CIFAR-10 training, 200 for all other datasets), the learning rate (2.5 ∗ 10−4 for CIFAR-10, 10−4 for all others), the batch size (128 vs 64), etc. I do not expect these choices to make a major difference for your claims, but it would be much cleaner to keep everything but the training dataset consistent. Especially the different test accuracies worry me a bit (from 81% to 92%, and they appear different from the accuracy numbers in figure S4 e.g. for CIFAR-10 and asymSVHN), at the very least those should be comparable.
We addressed this point by retraining the models while holding most of the hyperparameters constant. Specifically, we standardized the number of epochs, batch size, and weight decay. The remaining differences are necessitated by the characteristics of the specific training image sets used (natural images versus digits). Please note that we do not directly contrast models trained on CIFAR-10 and SVHN; the controlled comparisons are conducted while holding the SVHN training images constant, and are not confounded by hyperparameter choice.
Manuscript changes:
The networks’ weights and biases were initialized randomly using the uniform He initialization [70]. We trained the models using 250 epochs and a batch size of 256 images. The CIFAR-10 network was trained using stochastic gradient descent (SGD) optimizer starting with a learning rate of 10−3 and momentum of 0.9. The learning rate was halved every 20 epochs. The SVHN/symSVHN/asymSVHN networks were trained using the Adam optimizer. The initial learning rate was set to 10−5 and reduced by half every 50 epochs. The hyper-parameters were determined using the validation data. The models reached around 83% test accuracy (CIFAR-10: 81%, SVHN: 89%, symSVHN: 83%, asymSVHN: 80%). Fig. S4 shows the models’ learning curves.
Point 2.4: 3. L681-685: The general statement made in the paper that ”deeper models lose their advantage as models of cortical representations” is not supported by the cited limited comparison on a single dataset. There are many potential confounds here with respect to prior work, e.g. the recording modality (fMRI vs electrodes), the stimulus set (62 images vs thousands), the models that were tested (9 vs hundreds), etc.
We agree that the recording modality and stimulus set may play a critical role in determining model ranking. Since we generalized the analyses to deeper models, we removed this statement from the paper. While we still believe that shallower networks may prove to be better models of the visual cortex, this empirical question is out of the scope of the current manuscript.
Reviewer 3
This study aimed to explore the computational mechanisms of view invariance, driven by the observation that in some regions of monkey visual cortex, neurons show comparable responses to (1) a given face and (2) to the same face but horizontally flipped. Here they study this known phenomenon using AlexNet and other shallow neural networks, using an index for mirror symmetric viewpoint tuning based on representational similarity analyses. They find that this tuning is enhanced at fully connected- or global pooling layers (layers which combine spatial information), and that the invariance is prominent for horizontal- but not vertical- or rotational transformations. The study shows that mirror tuning can be learned when a given set of images are flipped horizontally and given the same label, but not if they are flipped and given different labels. They also show that networks learn this tuning by focusing on local features, not global configurations.
We are grateful for your thorough reading, reflected by the comprehensive summary of our study and its main findings.
Point 3.1) I found the study to be a mixed read. Some analyses were fascinating: for example, it was satisfying to see the use of well-controlled datasets to increase or decrease the rate of mirror-symmetry tuning. The insertion- and deletion¬ experiments were elegant tests to probe the mechanisms of mirror symmetry, asking if symmetry could arise from (1) global feature configurations (in a holistic sense) vs. (2) local features, with stronger evidence for the latter. These two sets of results were successful and interpretable. They stand in contrast with the first analysis, which relies on observations that do not seem justified. Specifically, Figure 2D shows mirror-symmetry tuning across 11 stages of image processing, from pixels space to fully connected layers. It shows that images from different object categories evoke considerably different tuning index values. The explanation for this result is that some categories, such as ”tools,” have ”bilaterally symmetric structure,” but this is not explicitly measured anywhere. ”Boats” are described as having ”front-back symmetry,” more so than flowers. One imagines flowers being extremely symmetric, but perhaps that depends on the metric. What is the metric? At first I thought it was the mirror-symmetric viewpoint tuning index in the image (pixel) space, but this cannot be, as the index for faces and flowers is negative, cars have no symmetry, and boats are positive. To support these descriptions, one must have an independent variable (for object class symmetry) that can be related to the dependent variable (the mirror-symmetric viewpoint tuning index). If it exists, it is not a part of the Results section. This omission undermines other parts of the Results section: ”some car models have an approximate front-back symmetry...however, a flower typically does not...” ”Some,” ”typically:” how many in the dataset exactly, and how often?
We thank you for your insightful observation. You are correct that we did not refer to pixel-space symmetry; our descriptions relate to the 3D structure of the objects used in the study.
Following this comment, we objectively quantified the symmetry planes of the 3D objects. Unfortunately, we do not have direct access to the proprietary 3D meshes of these objects, only to their renders. Therefore, we devised measures that assess the symmetry of the 3D objects through the symmetry they elicit in the different 2D renders.
This analysis is described in the new supplemental figure S8. We believe that these measurements support the qualitative claims we made in the previous draft.
Point 3.2) The description of CIFAR-10 as having bilaterally symmetric categories - are all these categories equally symmetric? If not, would such variability matter in terms of these results?
When considering their 3D structure, all ten CIFAR10 categories exhibit pronounced left-right symmetry. These categories encompass vertebrate animals (birds, cats, deer, dogs, frogs, horses); They also include man-made vehicles (airplanes, cars, ships, and trucks), which, at least externally, are nearly perfectly symmetric by design. It is important to note that this symmetry pertains to the photographed 3D objects, rather than the images themselves, which could be highly asymmetric. Other axes of symmetry (e.g., back-front) in CIFAR10 cannot be measured without 3D representations of the objects.
Point 3.3) These assessments of object category symmetry values are made before experiments are presented, so they are not interpretations of the results, and it would be circular to write it otherwise.
We have changed the order so that the explanations follow the experimental results. This includes the relevant main text paragraph, as well as the relevant figure—both the order of panels and the phrasing of the figure caption.
Point 3.4) Overall, my bigger concern is that the framing is misleading or at best incomplete. The manuscript successfully showed that if one introduces left-right symmetry to a dataset, the network will develop population-level representations that are also bilaterally symmetric. But the study does not explain that the model’s architecture and random weight distribution are sufficient for symmetry tuning to emerge, without training, just to a much more limited degree. Baek et al. showed in 2021 that viewpoint-invariant face-selective units and mirror-symmetric units emerge in untrained networks (”Face detection in untrained deep neural networks”; this current manuscript cites this paper but does not mention that mirror symmetry is a feature of the 2021 study). This current study also used untrained networks as controls (Fig. 3), and while they were useful in showing that learning boosts symmetry tuning, the results also clearly show that horizontal-reflection invariance is far from zero. So, the simple learning-driven explanation for the mirror-symmetric viewpoint tuning for faces is wrong: while (1) network training and (2) pooling are mechanisms that charge the development of mirror-symmetric tuning, the lottery ticket hypothesis is enough for its emergence. Faces and numbers are simple patterns, so the overparameterization of networks is enough to randomly create units that are tuned to these shapes and to wire many of them together. How learning shapes this process is an interesting direction, especially now that this current study has outlined its importance.
We agree with the reviewer that random initialization may result in units that show mirror-symmetric viewpoint tuning for faces in the absence of training. In the revised manuscript, we quantify the occurrence of such units, first reported by Baek et al, in detail, and discuss the relation between Baek et al., 2021 and our work. In brief, our analysis affirms that units with mirror-symmetric viewpoint tuning for faces appear even in untrained CNNs, although we believe their rate is lower than previously reported. Regardless of the question of the exact proportion of such units, we believe it is unequivocal that at the population level, mirror-symmetric viewpoint tuning to faces (and other objects with a single plane of symmetry) is strongly training-dependent.
First, we refer the reviewer to Figure S2, which directly demonstrates the effect of training on the population-level mirror symmetric viewpoint tuning:
Note the non-mirror-symmetric reflection invariant tuning profile for faces in the untrained network.
Second, the above-zero horizontal reflection-invariance referred by the reviewer (Figure 3) is distinct from mirror- symmetric viewpoint tuning; the latter requires both reflection-invariance and viewpoint tuning. More importantly, it was measured with respect to all of the object categories grouped together; this includes objects with quadrilateral symmetry, which elicit mirror-symmetric viewpoint tuning even in shallow layers and without training. To clarify the confusion that this grouping might have caused, we repeated the measurement of invariance in fc6, separately for each 3D object category:
Disentangling the contributions of different categories to the reflection-invariance measurements, this analysis under-scores the necessity of training for the emergence of mirror-symmetric viewpoint symmetry.
Last, we refer the reviewer to Figure S5, which shows that the symmetry of untrained convolutional filters has a narrow, zero-centered distribution. Indeed, the upper limit of this distribution includes filters with a certain degree of symmetry. This level of symmetry, however, becomes the lower limit of the filters’ symmetry distribution following training.
Therefore, we believe that training induces a shift in the tuning of the unit population that is qualitatively distinct from, and not explained by, random-lottery-related mirror-symmetric viewpoint tuned units. In the revised manuscript, we clarify the distinction between mirror-symmetric viewpoint tuning at the population level and the existence of individual units showing pre-training mirror symmetric viewpoint tuning, as shown by Baek et al.
Manuscript changes: (Discussion section)
Our claim that mirror-symmetric viewpoint tuning is learning-dependent may seem to be in conflict with findings by Baek and colleagues [17]. Their work demonstrated that units with mirror-symmetric viewpoint tuning profile can emerge in randomly initialized networks. Reproducing Baek and colleagues’ analysis, we confirmed that such units occur in untrained networks (Fig. S15). However, we also identified that the original criterion for mirror-symmetric viewpoint tuning employed in [17] was satisfied by many units with asymmetric tuning profiles (Figs. S14 and S15). Once we applied a stricter criterion, we observed a more than twofold increase in mirror-symmetric units in the first fully connected layer of a trained network compared to untrained networks of the same architecture (Fig. S16). This finding highlights the critical role of training in the emergence of mirror-symmetric viewpoint tuning in neural networks also at the level of individual units.
Point 3.5) Finally, it would help to cite other previous demonstrations of equivariance and mirror symmetry in neural networks. Chris Olah, Nick Cammarata, Chelsea Voss, Ludwig Schubert, and Gabriel Goh of OpenAI wrote of this phenomenon in 2020 (Distill journal).
We added a reference to the study by Olah and colleagues (2020).
Manuscript changes: (Discussion section)
(see Olah and colleagues (2020) [60] for an exploration of emergent equivariance using activation maximiza- tion).
Point 3.6) Some other observations that might help:
I am enthusiastic about the experiments using different datasets to increase or decrease the rate of mirror-symmetry tuning (sets including CIFAR10, SVHN, symSVHN, asymSVHN); it is worth noting, however, that the lack of a ground truth metric for category symmetry is a problem here too. In the asymSVHN dataset, images are flipped and given different labels. If some categories are naturally symmetric after horizontal flips, such as images containing ”0” or ”8”, then changing the label is likely to disturb training. This would explain why the training loss is larger for this condition (Figure S4D).
We now acknowledge that the inclusion of digits 0 and 8 reduces the accuracy of asymSVHN:
Manuscript changes: (Figure S4 caption)
Note that the accuracy of asymSVHN might be negatively affected by the inclusion of relatively symmetric categories such as 0 and 8.
Our rationale for retaining these digits in the dataset was to manipulate the symmetry of the learned categories (compared to symSVHN) while keeping the images themselves constant.
Regarding ground-truth symmetry of these dataset: For CIFAR-10, the relevant measure of symmetry pertains to the 3D structure of the photographed objects, which we believe is unequivocally symmetric (see Point 3.2). Note that 2D, pixel-space image symmetry is not directly indicative of symmetry in 3D.
For SVHN, which consists of two-dimensional characters, the pixel-space symmetry of the images indeed reflects the objects’ symmetry. However, since we are worried that some readers might confuse our claims that relate to the symmetry of objects with claims (we did not make) about symmetry of 2D images, we prefer to avoid reporting measurements of image-space symmetry. We believe that our interpretation of the experiments with SVHN/symSVHN/asymSVHN holds even in the absence of such measurements.
For your reference, we include here a quantification of image-space horizontal symmetry for each category of CIFAR-10 and SVHN:
Point 3.7) It is puzzling why greyscale 3D rendered images are used. By using greyscale 3D render (at least as shown in the figures) the study proceeds as if the units are invariant under color transformations. Unfortunately, this is not true and using greyscale images impact the activations of different layers of Alexnet in a way that is not fully defined. Moreover, many units in shallow networks focus on color and exactly these units could be invariant to other transformation like the mirror symmetry, but grey scaling the images makes them inactive.
We use grayscale 3D rendered images to align with the setting in other studies investigating mirror- symmetric viewpoint tuning, including Freiwald et al. (2010), Leibo et al. (2017), and Yildirim et al. (2020). The choice of using grayscale images in these studies is motivated by the need to dissociate face-processing from lower-level, hue-specific responses.
-
Reviewer #2 (Public Review):
Strengths
(1) The statements made in the paper are precise, separating observations from inferences, with claims that are well supported by empirical evidence. Releasing the underlying code repository further bolsters the credibility and reproducibility. I especially appreciate the detailed discussion of limitations and future work.
(2) The main claims with respect to the two convolutional architectures are well supported by thorough analyses. The analyses are well-chosen and overall include good controls, such as changes in the training diet. Going beyond "passive" empirical tests, the paper makes use of the fully accessible nature of computational models and includes more "causal" insertion and deletion tests that support the necessity and sufficiency of local object features.
(3) Based on modeling results, the paper makes a testable prediction: that mirror-symmetric viewpoint tuning is not specific to faces and can also be observed in other bilaterally symmetric objects such as cars and chairs. To test this experimentally in primates (and potentially other model architectures), the stimulus set is available online.
Weaknesses
My main concern with this paper is in its choice of the two model architectures AlexNet and VGG. In an earlier study, Yildirim et al. (2020) found an inverse graphics network "EIG" to better correspond to neural and behavioral data for face processing than VGG. All claims in the paper thus relate to a weaker model of the biological effects since this work does not analyze the EIG model. Since EIG follows an analysis-by-synthesis approach rather than standard classification training, it is unclear whether the claims in this paper generalize to this other model architecture. It is also unclear if the claims will hold for: 1) transformer architectures, 2) the HMAX architecture by Leibo et al. (2017) which has also been proposed as a computational explanation for mirror-symmetric tuning, and, as the authors note in the Discussion, 3) deeper architectures such as ResNet-50 which tend to better align to neural and behavioral data in general. These architectures include different computational motifs such as skip connections and a much smaller proportion of fully-connected layers which are a major focus of this work.
Overall, I thus view the paper's claims as limited to AlexNet- and VGG-like architectures, both of which fall behind state-of-the-art in their alignment to primates in general and also specifically for mirror-symmetric viewpoint tuning.
Minor weaknesses
(1) Figure 1A: since the relevance to primate brains is a major motivator of this work, the results from actual neural recordings should be shown and not just schematics. For instance, the mirror symmetry in AL is not as clean as the illustration (compare with Fig. 3 in Yildirim et al. 2020), and in the paper's current form, this is not easily accessible to the reader.
(2) Figure 4 / L832-845: The claims for the effect of training on mirror-symmetric viewpoint tuning are with respect to the training data only, but there are other differences between the models such as the number of epochs (250 for CIFAR-10 training, 200 for all other datasets), the learning rate (2.5 * 10^-4 for CIFAR-10, 10^-4 for all others), the batch size (128 vs 64), etc. I do not expect these choices to make a major difference for your claims, but it would be much cleaner to keep everything but the training dataset consistent. Especially the different test accuracies worry me a bit (from 81% to 92%, and they appear different from the accuracy numbers in figure S4 e.g. for CIFAR-10 and asymSVHN), at the very least those should be comparable.
(3) L681-685: The general statement made in the paper that "deeper models lose their advantage as models of cortical representations" is not supported by the cited limited comparison on a single dataset. There are many potential confounds here with respect to prior work, e.g. the recording modality (fMRI vs electrodes), the stimulus set (62 images vs thousands), the models that were tested (9 vs hundreds), etc.
-
-
docdrop.org docdrop.org
-
Résumé de la vidéo [00:00:01][^1^][1] - [00:22:59][^2^][2]:
Cette vidéo présente les objectifs et les activités de la Fabrique des Mobilités (FabMob), une association qui vise à promouvoir une mobilité durable et moins émettrice de carbone. Elle explique le concept de commun numérique et son application pratique dans le secteur de la mobilité, en mettant l'accent sur la coopération entre acteurs hétérogènes et la gouvernance partagée des ressources numériques.
Points forts: + [00:00:01][^3^][3] Introduction de FabMob * Présentation des objectifs * Définition d'un commun numérique + [00:04:00][^4^][4] Rôle de la DGITM * Collaboration avec FabMob * Importance des communs dans la mobilité + [00:08:03][^5^][5] Modalités de participation * Encouragement des questions * Cycle de travail sur les outils numériques + [00:09:01][^6^][6] Définition académique d'un commun * Trois piliers : ressource, communauté, gouvernance * Exemples de communs numériques + [00:13:20][^7^][7] Panorama institutionnel * Diverses institutions impliquées dans les communs numériques * Exemples européens et français + [00:20:36][^8^][8] Distinction entre Open Data, Open Source et commun numérique * Explication des termes * Importance de la gouvernance des données Résumé de la vidéo [00:23:01][^1^][1] - [00:45:09][^2^][2]:
La partie 2 de la vidéo aborde la logique d'Open Data, d'OP source, et de commun numérique dans le contexte français, en mettant l'accent sur l'importance de l'ouverture, des licences variées, et de la gouvernance collective pour le partage des ressources numériques.
Points forts: + [00:23:01][^3^][3] Open Data et OP source * Accès libre aux logiciels * Licences variées + [00:23:37][^4^][4] Commun numérique * Service de sa communauté * Pas nécessairement ouvert + [00:25:02][^5^][5] Avantages du numérique * Effets de réseau * Coûts de réplication faibles + [00:27:00][^6^][6] Gouvernance collective * Importance de la fédération * Gestion de la ressource + [00:31:11][^7^][7] Exemples concrets * Affluence TC à Grenoble * Intelligence artificielle dans les transports + [00:43:42][^8^][8] Politiques publiques par les communs * Réduction des coûts * Transparence et pérennité Résumé de la vidéo [00:45:11][^1^][1] - [01:05:36][^2^][2]:
La vidéo discute de l'importance de rendre les données de réglementation routière accessibles et utilisables pour les collectivités, en particulier pour l'intégration dans les systèmes GPS. Elle souligne la nécessité d'une collaboration communautaire pour créer une base de données exhaustive et utile.
Points forts: + [00:45:11][^3^][3] Accessibilité des données * Simplifier l'utilisation des données pour les collectivités * Créer des outils de navigation intuitifs + [00:46:01][^4^][4] Intégration GPS * Intégrer les règles de circulation dans les GPS * Adapter la navigation aux spécificités des véhicules + [00:47:03][^5^][5] Avantages logistiques * Faciliter la traduction des règlements pour les chauffeurs étrangers * Améliorer la coordination entre les services de gestion du réseau + [00:48:00][^6^][6] Applications futures * Imaginer des usages réglementaires dynamiques * Permettre une créativité réglementaire avec les données numériques Résumé de la vidéo [01:05:38][^1^][1] - [01:25:41][^2^][2]:
Cette vidéo discute des incitations financières pour le covoiturage en France, des défis de fraude et de la création d'un registre de preuve de covoiturage pour sécuriser les trajets et encourager l'adoption du covoiturage.
Points forts: + [01:05:38][^3^][3] Incitations pour le covoiturage * Gratuité pour les passagers * Rémunération pour les conducteurs + [01:06:14][^4^][4] Forfait mobilité durable * Jusqu'à 800 € par an pour les salariés + [01:06:28][^5^][5] Primes de l'État * 100 € pour les nouveaux covoitureurs + [01:07:10][^6^][6] Défis de fraude * Risques liés aux incitations financières + [01:07:58][^7^][7] Registre de preuve de covoiturage * Infrastructure numérique contre la fraude + [01:11:02][^8^][8] Communauté et gouvernance * Plus de 700 collectivités impliquées Résumé de la vidéo [01:15:00][^1^][1] - [01:22:59][^2^][2]:
La vidéo aborde le concept des communs numériques, leur importance dans la transition écologique et la mobilité, et comment ils favorisent la coopération entre divers acteurs. Elle souligne l'importance de la gouvernance collective et présente des exemples concrets de communs numériques dans le secteur des transports.
Points clés: + [01:15:00][^3^][3] Définition des communs numériques * Trois piliers : ressource partagée, communauté hétérogène, règles de gouvernance + [01:17:00][^4^][4] Exemples de communs numériques * Open Street Map, logiciels, données, serveurs + [01:19:00][^5^][5] Institutions et communs numériques * Directions ministérielles, agences nationales, collectivités + [01:21:00][^6^][6] Différence entre Open Data, Open Source et communs numériques * Open Data : données en accès libre; Open Source : code source ouvert; Communs numériques : gestion collective de ressources numériques
-
-
www.biorxiv.org www.biorxiv.org
-
Reviewer #3 (Public Review):
Induced pluripotent stem cells, or iPSCs, are cells that scientists can push to become new, more mature cell types like neurons. iPSCs have a high potential to transform how scientists study disease by combining precision medicine gene editing with processes known as high-content imaging and drug screening. However, there are many challenges that must be overcome to realize this overall goal. The authors of this paper solve one of these challenges: predicting cell types that might result from potentially inefficient and unpredictable differentiation protocols. These predictions can then help optimize protocols.
The authors train advanced computational algorithms to predict single-cell types directly from microscopy images. The authors also test their approach in a variety of scenarios that one may encounter in the lab, including when cells divide quickly and crowd each other in a plate. Importantly, the authors suggest that providing their algorithms with just the right amount of information beyond the cells' nuclei is the best approach to overcome issues with cell crowding.
The work provides many well-controlled experiments to support the authors' conclusions. However, there are two primary concerns: (1) The model may be relying too heavily on the background and thus technical artifacts (instead of the cells) for making CNN-based predictions, and (2) the conclusion that their nucleocentric approach (including a small area beyond the nucleus) is not well supported, and may just be better by random chance. If the authors were to address these two concerns (through additional experimentation), then the work may influence how the field performs cell profiling in the future.
Additionally, the impact of this work will be limited, given the authors do not provide a specific link to the public source code that they used to process and analyze their data.
-
-
opendatastructures.org opendatastructures.org
-
packIntoArray
把packIntoArray配合他的recursive code是如何增加i值的过程给我看?
ChatGPT
在这个修改后的packIntoArray方法中,我们可以看到递归过程如何增加i值:
开始访问根节点4,当前索引为0。 递归访问左子节点2,索引仍然是0。 继续递归访问更深层的左子节点1,索引仍是0。 添加值1到位置0,然后索引增加到1。 返回到节点2,添加值2到位置1,索引增加到2。 访问节点2的右子节点3,当前索引是2。 添加值3到位置2,索引增加到3。 返回到根节点4,添加值4到位置3,索引增加到4。 访问根节点的右子节点6,当前索引是4。 递归访问节点6的左子节点5,当前索引仍然是4。 添加值5到位置4,然后索引增加到5。 返回到节点6,添加值6到位置5,索引增加到6。 访问节点6的右子节点7,当前索引是6。 添加值7到位置6,索引增加到7(完成数组填充)。通过这个详细的步骤,我们可以看到i是如何随着每个节点的访问而递增的。这确保了所有节点都按照二叉树的中序遍历顺序被正确添加到数组中。每个节点在添加到数组后,索引i都会递增,以便为下一个节点留出空间。
-
-
matthewbjane.quarto.pub matthewbjane.quarto.pub
-
Φt−1[q,ν],
put R code here (ex. qt(p=p, df=nu)
-
-
-
“Code point” is a bit of a mouthful(拗口), so Go introduces a shorter term for the concept: rune.
rune
-
-
pressbooks.online.ucf.edu pressbooks.online.ucf.edu
-
The knight on his steed deemed it fair enough, if he might come to be sheltered within it to lodge there while that the Holy-day lasted. He called aloud, and soon there came a porter of kindly countenance, who stood on the wall and greeted this knight and asked his errand.
Sir Gawain and the Green Knight is a story of adventure and romance that exalts the chivalric values of honor, mercy and mercy. In this hero's journey, Gawain aspires to earn the honors of knighthood, but encounters difficulties and challenges along the way that test his mettle. Sir Gawain and the Green Knight becomes a story about the respect due to the chivalric code of honor and about the necessary submission to superiors (in this case, Gawain must passively lend himself to be beheaded by the green knight). It reflects the chivalric values espoused by King Edward III, which matured during the Hundred Years' War, in which the British aspired to occupy the throne of France. Sir Gawain and the Green Knight: An Appraisal Published online by Cambridge University Press: 02 December 2020 https://www.cambridge.org/core/journals/pmla/article/abs/sir-gawain-and-the-green-knight-an-appraisal/4D4501B59BE83BA09C48E7DD7FDE3545
-
“That is sooth,” quoth the other, “I grant you that same; and I have fairly won this within walls, and with as good will do I yield it to ye.” With that he clasped his hands round the lord’s neck and kissed him as courteously as he might. “Take ye here my spoils, no more have I won; ye should have it freely, though it were greater than this.”
In this section, the Green Knight reveals his true identity as Lord Bertilak, who had tested Sir Gawain's honor and integrity through a series of challenges. Sir Gawain, realizing that he had failed to uphold his end of the bargain by accepting the lady's green girdle as a token of her affection, confesses his mistake and offers to return it to Lord Bertilak. This moment is significant because of Sir Gawain's humility and honesty in admitting his fault, as well as his willingness to face the consequences of his actions. By returning the green girdle and accepting his mistake, Sir Gawain demonstrates his commitment to the code of chivalry and the importance of truth and honor in his knightly duties.
-
What makes Gawain so chivalrous
Gawain is known as being chivalrous because he was the proper hero, who morals matched his fight. Sir Gawain’s shield represented "the five virtues of chivalry, which were friendship, generosity, chastity, courtesy, and piety. These where the virtues people strove to live by and those they demanded of kings and knights."
-
But Sir Gawain said nay, he would in no wise do so; so they embraced and kissed, and commended each other to the Prince of Paradise
This is an important part of the poem as this is Gawain's return to being the model and chivalrous knight he is known to be. During his encounter with the Green Knight he believes himself to be a coward, and is uncouth when speaking to his adversary. When Gawain is told that the lord of the hall he was staying at was the Green Knight he confesses his wrong-doings. The Green Knight invites him to his hall again but Gawain refuses which can interpreted as him either fearing that he would be unable to control his "chivalry" or that he does not want to wrong the Green Knight anymore than he already has. So the both of them make up in the end and commend each other to Jesus to make themselves even.
Rouse, Robert. "Historical Context: The Middle Ages and the Code of Chivalry." Handbook of Arthurian Romance: King Arthur’s Court in Medieval European Literature (2017): 13-24.
-
gallant knights
I believe gallant knights refers to brave warriors who are at the highest rank of knights. These knights uphold a code of honor and loyalty. They are seen as role models to everyone. The "gallant" means how brave and well mannered they are known as great warriors and gentlemen.
"Pace University Project." Grendel's Den, csis.pace.edu/grendel/projf982b/passage.html.
-
chivalry.
"Chivalry" is often loosely defined and interpreted in different ways. According to Darcy Egan, this term refers to the code of honor which acts as a guideline for knights' endeavors such as battles. Having a clear understanding of this term will benefit readers throughout this Arthurian literature. It also helps to understand the message of the story as "chivalry' can be linked to how humanity treats others.
Egan, Darcy. Carroll Collected Senior Honors Projects Theses, Essays, and Senior Honors Projects Is Chivalry Really Dead? -an Exploration of Chivalry and Masculinity in Medieval and American Literature. 2012.
-
A lace was twined about it, that looped at the head, and all adown the handle it was clasped with tassels on buttons of bright green richly broidered.
This description continues on in representing the attire of the green knight, starting off by stating how a lace was twinned about it and looped at the end. This is describing a ribbon or string of some sorts, giving the sword a very medieval feel when imagining it like that. As stated by the poem, the handle was clasped with tassels and buttons of bright green richly broidered on, giving a sense of identity to the sword, something very important to knights. According to Knights Templar, "for a knight, his sword was not just a weapon—it was a symbol of honor, valor, and duty. The medieval knight’s sword was more than just a piece of metal; it was an embodiment of a warrior’s soul and a testament to his commitment to the chivalric code" (Oksana).
Reference: https://knightstemplar.co/medieval-knights-swords-blades-that-shaped-battles/
-
the harts they let pass them, and the stags with their spreading antlers, for the lord had forbidden that they should be slain
The lord only sought after the female deer since it adheres to environmental sustainability, "it is unlawful to hunt stags during fermysoun tyme, close-season—a period, in this case September fourteen through June twenty-four, when a particular animal cannot be hunted in order to protect the species" (Martinez 120-121). The Green Knight is portrayed as a nature figure in appearance as well as by actions, a landowner who leads by both control and care, upholding the use of environmental stewardship. It is important to recognize the Green Knights harmonious existence with the land given his stark contrast with Camelot. Camelot is upholding of the knightly, chivalric code and is therefore a representation of a human civilization that lives off order. The Green Knight is a representation of nature, the unpredictable and uncontrollable elements. This shows however, that he does not live a lawless life, instead seeking conditions that suit the environment as well as himself.
MARTINEZ, ANN M. “Bertilak’s Green Vision: Land Stewardship in ‘Sir Gawain and the Green Knight.’” Arthuriana (Dallas, Tex.), vol. 26, no. 4, 2016, pp. 114–29, https://doi.org/10.1353/art.2016.0052.
-
All looked on him as he stood, and drew near unto him wondering greatly what he might be; for many marvels had they seen, but none such as this
This is the foreshadowed event given to us by Arthur as he mentions in the beginning how he would not eat until he witnessed something marvelous in his court. In stating this line however, it is implied that Arthur assumes that there is nothing his court could not handle, “The Green Knight's appearance is at once the provision of this marvel and a challenge to the confidence, certainty, and light-heartedness with which Arthur assumes that his court will always be able to dominate and accommodate such a marvel” (Lander 48). The Green Knight appears as a marvelous, intimidating force that challenges the knights honor but also the very code they live by, he pushes the boundaries of the Round Table's chivalry and exposes its weaknesses. Arthur’s court is so closely knit to the chivalric code of conduct that any interruption that breaks the rules of this code poses a threat. It is this very code that Gawain struggles with throughout the story as his chivalry is tested by Morgain le Fay’s plan and the limitations are revealed.
Lander, Bonnie. “The Convention of Innocence and Sir Gawain and the Green Knight ’s Literary Sophisticates.” Parergon, vol. 24, no. 1, 2007, pp. 41–66, https://muse-jhu-edu.eu1.proxy.openathens.net/article/219967
-
For I am pledged by solemn compact sworn between us to meet that knight at the New Year if so I were on life; and of that same New Year it wants but little–I’faith, I would look on that hero more joyfully than on any other fair sight! Therefore, by your will, it behoves me to leave you, for I have but barely three days, and I would as fain fall dead as fail of mine errand.”
In this passage Gawain expresses his commitment to honor the agreement he made with the Green Knight, emphasizing the importance of fulfilling his pledge even at the risk of his own life. The concept of honor and fulfilling one's word, or oath, is central to the chivalric code of conduct that Gawain adheres to as a knight. Gawain's unwavering determination to meet the Green Knight at the agreed-upon time underscores the significance of honor and integrity in medieval society. In Benson's analysis of "Sir Gawain and the Green Knight," he explores the theme of chivalry and the importance of fulfilling one's word in medieval literature. He argues that Gawain's adherence to his pledge reflects the values of honor and integrity that were highly esteemed during the medieval period. This interpretation provides insight into the significance of Gawain's commitment to meeting the Green Knight, shedding light on the moral complexities of the text.
Benson, Larry D. "The Green Knight's Game and Gawain's God." PMLA, vol. 94, no. 3, 1979, pp. 209–222. JSTOR, www.jstor.org/stable/461908.
Tags
Annotators
- matthewwolf
- violetmae06
- ma696509
- Carmenescalante
- valesalas
- Luis_Buot
- georgereppas
- AshlynShephardshypothesis
- Ncamera04
URL
-
-
global.factiva.com global.factiva.comFactiva2
-
The software would scan a piece of text for two factors: “perplexity,” the randomness of word choice; and “burstiness,” the complexity or variation of sentences. Human writing tends to rate higher than AI writing on both metrics, which allowed Tian to guess how a piece of text had been created. Tian called the tool GPTZero—the “zero” signaled truth, a return to basics
breaking down code into a simple idea
-
n open source journalism project, where he'd written code to detect Twitter bots. As a junior, he'd taken classes on machine learning and natural language processing. And in the fall of 2022, he began to work on his senior thesis about detecting the differences between AI-generated and human-written text.
connects to in class discussion about general education and the purposes of it
-
-
help.zoho.com help.zoho.com
-
Create and embed custom capabilities across CRM with Kiosk Studio, our latest no-code tool
Kiosk studio is the Zoho Creator of widgets – for Zoho CRM.
Zoho Creator allows you to focus on custom backend functionality while having an out of the box front end.
Zoho CRM Kiosk allows you to create a small application interface with an out of the box front end, while allowing robust functionality on the backend.
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
Public Reviews:
We thank all the reviewers for taking the time to assess and provide valuable feedback on the manuscript. We believe these comments helped clarify the manuscript’s prose, and the suggestions on the functionality and aim of the toolbox were globally incorporated into the following updates of the toolbox. Particularly, we would like to point out some changes that will help all reviewers, independently of their individual comments, to understand the current state of the toolbox and some systematic changes that were made to the manuscript.
We have received a significant amount of feedback asking for a PyTorch implementation of the toolbox. Consequently, we decided to enact this, and the next version of the toolbox will be exclusively in PyTorch. We will maintain the Application Programming Interface (API) and tutorial documentation for the TensorFlow version of the toolbox on the online website. However, going forward we will focus exclusively on bug-fixing and expanding from the latest version of MotorNet, which will be in PyTorch. We now believe that the greater popularity of PyTorch in the academic community makes that choice more sustainable while helping a greater proportion of research projects.
These changes led to a significant alteration of the MotorNet structure, which is reflected by changes made throughout the manuscript, most particularly in Figure 3 and Table 1. A beneficial side-effect of this is a much simpler structure for MotorNet which ought to contribute positively toward its usability by researchers in the neuroscience community.
We also refactored some terminology to be more in line with current computational neuroscience vocabulary:
• The term “plant”, which comes from industrial engineering and is more niche in neuroscience, has been replaced by “effector”.
• The term “task” has been replaced by “environment” to match the gymnasium toolbox terminology, which MotorNet is now compatible with. Task objects essentially performed the same function as environment objects from the gymnasium toolbox.
• The term “controller” has been replaced by “policy” throughout, as this term is more general.
• The term “motor command” is very specific to the motor control subfield of neuroscience, and therefore is replaced by “action”, which is more commonplace for this modelling component in computational neuroscience and machine learning.
Reviewer #1 (Public Review):
Summary:
Codol et al. present a toolbox that allows simulating biomechanically realistic effectors and training Artificial Neural Networks (ANNs) to control them. The paper provides a detailed explanation of how the toolbox is structured and several examples that demonstrate its usefulness.
Main comments:
(1) The paper is well written and easy to follow. The schematics help in understanding how the toolbox works and the examples provide an idea of the results that the user can obtain.
We thank the reviewer for this comment.
(2) As I understand it, the main purpose of the paper should be to facilitate the usage of the toolbox. For this reason, I have missed a more explicit link to the actual code. As I see it, researchers will read this paper to figure out whether they can use MotorNet to simulate their experiments, and how they should proceed if they decide to use it. I'd say the paper provides an answer to the first question and assures that the toolbox is very easy to install and use. Maybe the authors could support this claim by adding "snippets" of code that show the key steps in building an actual example.
This is an important point, which we also considered when writing this paper. We instead decided to focus on the first approach, because it is easier to illustrate the scientific use of the toolbox using code or interactive (Jupyter) notebooks than a publication format. We find the “how to proceed” aspect of the toolbox can more easily and comprehensively be covered using online, interactive tutorials. Additionally, this allows us to update these tutorials as the toolbox evolves over different versions, while it is more difficult to update a scientific article. Consequently, we explicitly avoided code snippets on the article itself. However, we appreciate that the paper would gain in clarity if this was more explicitly stated early. We have modified the paper to include a pointer to where to find tutorials online. We added this at the last paragraph of the introduction section:
“The interested reader may consult the full API documentation, including interactive tutorials on the toolbox website at https://motornet.org.”
(3) The results provided in Figures 1, 4, 5 and 6 are useful, because they provide examples of the type of things one can do with the toolbox. I have a few comments that might help improving them:
(a) The examples in Figures 1 and 5 seem a bit redundant (same effector, similar task). Maybe the authors could show an example with a different effector or task? (see point 4).
The effectors from figures 1 and 5 are indeed very similar. However, the tasks in figure 1 and 5 present some important differences. The training procedure in figure 1 never includes any perturbations, while the one from figure 5 includes a wide range of perturbations of different magnitudes, timing and directions. The evaluation procedure of figure 1 includes center-out reaches with permanent viscous (proportional to velocity) external dynamics, while that of figure 5 are fixed, transient, square-shaped perturbation orthogonal to the reach direction. Finally, the networks in figure 1 undergo a second training procedure after evaluation while the network of figure 5 do not. While we agree that some variation of effectors would be beneficial, we do show examples of a point-mass effector in figure 6. Overall, figure 5 shows a task that is quite different from that of figure 1 with a similar effector, while the opposite is true for figure 6. We have modified the text to clarify this for the reader, by adding the following.
End of 1st paragraph, section 2.4.
“Therefore, the training protocol used for this task largely differed from section 2.1 in that the networks are exposed to a wide range of mechanical perturbations with varying characteristics.”
1st paragraph of section 2.5
[…] this asymmetrical representation of PMDs during reaching movements did not occur when RNNs were trained to control an effector that lacked the geometrical properties of an arm such as illustrated in Figure 4c-e and section 2.1.
(b) I missed a discussion on the relevance of the results shown in Figure 4. The moment arms are barely mentioned outside section 2.3. Are these results new? How can they help with motor control research?
We thank the reviewer for this comment. This relates to a point from reviewer 2 indicating that the purpose of each section was sometimes difficult to grasp as one reads. Section 2.3 explains the biomechanical properties that the toolbox implements to improve realism of the effector. They are not new results in the sense that other toolboxes implement these features (though not in differentiable formats) and these properties of biological muscles are empirically well-established. However, they are important to understand what the toolbox provides, and consequently what constraints networks must accommodate to learn efficient control policies. An example of this is the results in figure 6, where a simple effector versus a more biomechanically complex effector will yield different neural representations.
Regarding the manuscript itself, we agree that more clarity on the goal of every paragraph may improve the reader’s experience. Consequently, we ensured to specify such goals at the start of each section. Particularly, we clarify the purpose of section 2.3 by adding several sentences on this at the end of the first paragraph in that section. We also now clearly state the purpose of section 2.3 with the results of figure 6 and reference figure 4 in that section.
(c) The results in Figure 6 are important, since one key asset of ANNs is that they provide access to the activity of the whole population of units that produces a given behavior. For this reason, I think it would be interesting to show the actual "empirical observations" that the results shown in Fig. 6 are replicating, hence allowing a direct comparison between the results obtained for biological and simulated neurons.
These empirical observations are available from previous electrophysiological and modelling work. Particularly, polar histograms across reaching directions like panel C are displayed in figures 2 and 3 of Scott, Gribble, Graham, Cabel (2001, Nature). Colormaps of modelled unit activity across time and reaching directions like panel F are also displayed in figure 2 of Lillicrap, Scott (2013, Neuron). Electrophysiological recordings of M1 neurons during a similar task in non-human primates can also be seen on “Preserved neural population dynamics across animals performing similar behaviour” figure 2 B (https://doi.org/10.1101/2022.09.26.509498) and “Nonlinear manifolds underlie neural population activity during behaviour” figure 2 B as well (https://doi.org/10.1101/2023.07.18.549575). Note that these two pre-prints use the same dataset.
We have added these citations to the text and made it explicit that they contain visualizations of similar modelling and empirical data for comparison:
“This heterogeneous set of responses matches empirical observations in non-human primate primary motor cortex recordings (Churchland & Shenoy, 2007; Michaels et al., 2016) and replicate similar visualizations from previously published work (Fortunato et al., 2023; Lillicrap & Scott, 2013; Safaie et al., 2023).”
(4) All examples in the paper use the arm26 plant as effector. Although the authors say that "users can easily declare their own custom-made effector and task objects if desired by subclassing the base Plant and Task class, respectively", this does not sound straightforward. Table 1 does not really clarify how to do it. Maybe an example that shows the actual code (see point 2) that creates a new plant (e.g. the 3-joint arm in Figure 7) would be useful.
Subclassing is a Python process more than a MotorNet process, as python is an object-oriented language. Therefore, there are many Python tutorials on subclassing in the general sense that would be beneficial for that purpose. We have amended the main text to ensure that this is clearer to the reader.
Subclassing a MotorNet object, in a more specific sense, requires overwriting some methods from the base MotorNet classes (e.g., Effector or Environment classes, which correspond to the original Plant and Task object, respectively). Since we made the decision (mentioned above) to not include code in the main text, we added tutorials to the online documentation, which include dedicated tutorials for MotorNet class subclassing. For instance, this tutorial showcases how to subclass Environment classes:
(5) One potential limitation of the toolbox is that it is based on Tensorflow, when the field of Computational Neuroscience seems to be, or at least that's my impression, transitioning to pyTorch. How easy would it be to translate MotorNet to pyTorch? Maybe the authors could comment on this in the discussion.
We have received a significant amount of feedback asking for a PyTorch implementation of the toolbox. Consequently, we decided to enact this, and the next version of the toolbox will be exclusively in PyTorch. We will maintain the Application Programming Interface (API) and tutorial documentation for the TensorFlow version of the toolbox on the online website. However, going forward we will focus exclusively on bug-fixing and expanding from the latest version of MotorNet, which will be in PyTorch. We now believe that the greater popularity of PyTorch in the academic community makes that choice more sustainable while helping a greater proportion of research projects.
These changes led to a significant alteration of the MotorNet structure, which are reflected by changes made throughout the manuscript, notably in Figure 3 and Table 1.
(6) Supervised learning (SL) is widely used in Systems Neuroscience, especially because it is faster than reinforcement learning (RL). Thus providing the possibility of training the ANNs with SL is an important asset of the toolbox. However, SL is not always ideal, especially when the optimal strategy is not known or when there are different alternative strategies and we want to know which is the one preferred by the subject. For instance, would it be possible to implement a setup in which the ANN has to choose between 2 different paths to reach a target? (e.g. Kaufman et al. 2015 eLife). In such a scenario, RL seems to be a more natural option Would it be easy to extend MotorNet so it allows training with RL? Maybe the authors could comment on this in the discussion.
The new implementation of MotorNet that relies on PyTorch is already standardized to use an API that is compatible with Gymnasium. Gymnasium is a standard and popular interfacing toolbox used to link RL agents to environments. It is very well-documented and widely used, which will ensure that users who wish to employ RL to control MotorNet environments will be able to do so relatively effortlessly. We have added this point to accurately reflect the updated implementation, so users are aware that it is now a feature of the toolbox (new section 3.2.4.).
Impact:
MotorNet aims at simplifying the process of simulating complex experimental setups to rapidly test hypotheses about how the brain produces a specific movement. By providing an end-to-end pipeline to train ANNs on the simulated setup, it can greatly help guide experimenters to decide where to focus their experimental efforts.
Additional context:
Being the main result a toolbox, the paper is complemented by a GitHub repository and a documentation webpage. Both the repository and the webpage are well organized and easy to navigate. The webpage walks the user through the installation of the toolbox and the building of the effectors and the ANNs.
Reviewer #2 (Public Review):
MotorNet aims to provide a unified interface where the trained RNN controller exists within the same TensorFlow environment as the end effectors being controlled. This architecture provides a much simpler interface for the researcher to develop and iterate through computational hypotheses. In addition, the authors have built a set of biomechanically realistic end effectors (e.g., an 2 joint arm model with realistic muscles) within TensorFlow that are fully differentiable.
MotorNet will prove a highly useful starting point for researchers interested in exploring the challenges of controlling movement with realistic muscle and joint dynamics. The architecture features a conveniently modular design and the inclusion of simpler arm models provides an approachable learning curve. Other state-of-the-art simulation engines offer realistic models of muscles and multi-joint arms and afford more complex object manipulation and contact dynamics than MotorNet. However, MotorNet's approach allows for direct optimization of the controller network via gradient descent rather than reinforcement learning, which is a compromise currently required when other simulation engines (as these engines' code cannot be differentiated through).
The paper could be reorganized to provide clearer signposts as to what role each section plays (e.g., that the explanation of the moment arms of different joint models serves to illustrate the complexity of realistic biomechanics, rather than a novel discovery/exposition of this manuscript). Also, if possible, it would be valuable if the authors could provide more insight into whether gradient descent finds qualitatively different solutions to RL or other non gradient-based methods. This would strengthen the argument that a fully differentiable plant is useful beyond improving training time / computational power required (although this is a sufficiently important rationale per se).
We thank the reviewer for these comments. We agree that more clarity on the section goals may improve the reader’s experience and ensured this is the case throughout the manuscript. Particularly, we added the following on the first paragraph of section 2.3, for which an explicit goal was most missing:
“In this section we illustrate some of these biomechanical properties displayed by MotorNet effectors using specific examples. These properties are well-characterised in the biology and are often implemented in realistic biomechanical simulation software.”
Regarding the potential difference in solutions obtained from reinforcement or supervised learning, this would represent a non-trivial amount of work to do so conclusively and so may not be within the scope of the current article. We do appreciate however that in some situations RL may be a more fitting approach to a given task design. In relation to this point we now specify in the discussion that the new API can accommodate interfacing with reinforcement learning toolboxes for those who may want to pursue this type of policy training approach when appropriate (new section 3.2.4.).
Reviewer #3 (Public Review):
Artificial neural networks have developed into a new research tool across various disciplines of neuroscience. However, specifically for studying neural control of movement it was extremely difficult to train those models, as they require not only simulating the neural network, but also the body parts one is interested in studying. The authors provide a solution to this problem which is built upon one of the main software packages used for deep learning (Tensorflow). This allows them to make use of state-of-the-art tools for training neural networks.
They show that their toolbox is able to (re-)produce several commonly studied experiments e.g., planar reaching with and without loads. The toolbox is described in sufficient detail to get an overview of the functionality and the current state of what can be done with it. Although the authors state that only a few lines of code can reproduce such an experiment, they unfortunately don't provide any source code to reproduce their results (nor is it given in the respective repository).
The possibility of adding code snippets to the article is something we originally considered, and which aligns with comment two from reviewer one (see above). Hopefully this provides a good overview of the motivation behind our choice not to add code to the article.
The modularity of the presented toolbox makes it easy to exchange or modify single parts of an experiment e.g., the task or the neural network used as a controller. Together with the open-source nature of the toolbox, this will facilitate sharing and reproducibility across research labs.
I can see how this paper can enable a whole set of new studies on neural control of movement and accelerate the turnover time for new ideas or hypotheses, as stated in the first paragraph of the Discussion section. Having such a low effort to run computational experiments will be definitely beneficial for the field of neural control of movement.
We thank the reviewer for these comment.
-
Reviewer #2 (Public Review):
MotorNet aims to provide a unified interface where the trained RNN controller exists within the same TensorFlow environment as the end effectors being controlled. This architecture provides a much simpler interface for the researcher to develop and iterate through computational hypotheses. In addition, the authors have built a set of biomechanically realistic end effectors (e.g., a 2 joint arm model with realistic muscles) within TensorFlow that are fully differentiable.
MotorNet will prove a highly useful starting point for researchers interested in exploring the challenges of controlling movement with realistic muscle and joint dynamics. The architecture features a conveniently modular design and the inclusion of simpler arm models provides an approachable learning curve. Other state-of-the-art simulation engines offer realistic models of muscles and multi-joint arms and afford more complex object manipulation and contact dynamics than MotorNet. However, MotorNet's approach allows for direct optimization of the controller network via gradient descent rather than reinforcement learning, which is a compromise currently required when other simulation engines (as these engines' code cannot be differentiated through).
The paper has been reorganized to provide clearer signposts to guide the reader. Importantly, the software has been rewritten atop PyTorch which is increasingly popular in ML and computational neuroscience research.
One paragraph in the discussion regarding a "spinal cord" module is a bit perplexing. Quite sensibly, the software architecture partitions motor control into the plant or effector (the physical body being moved) and the controller (a model of the brain and spinal cord). Of course, the authors certainly appreciate this, though a reader from outside of neuro might not realize that control of movement is distributed throughout the central nervous system, spanning a network of spinal, subcortical (cerebellum, basal ganglia, thalamus, brainstem), and cortical brain regions. Casting the spinal cord as a pre-filter within the effector module would seem to belie its complex and dynamic role in these distributed neural circuits. This is particularly noticeable when contrasted with the subsequent paragraph on "Modular polices" (which is excellent). In my view, the spinal cord would be better treated as a module of this policy section rather than as part of the effector. I understand the nuance here, and suspect I'd see eye to eye with the authors for the most part. The choice of controller vs. plant depends on perspective (one could call the arm itself part of the controller, and treat the environment / manipulated object as the plant; similarly, one could treat the brain as controlling the cord rather than the body). However, I fear that someone lacking the appropriate neurophysiological/anatomical context might read the "Spinal Compartment" paragraph, think that it would be fine to introduce a simple filter module as the spinal cord, and then start referring to the MotorNet policy network as a model of motor cortex per se.
-
-
ipisresearch.be ipisresearch.be
-
Both artisanal cobalt and artisanal 3T mining in the DRC are regulated by the DRC Mining Code. The firstversion introduced in 2002 clearly aimed at attracting LSM to invest in the country, while the currentversion, enforced in 2018, aims to rebalance the benefits in favour of the Congolese state and the re-sponsibility of the LSM towards environmental issues.
2002 and 2018 mining code, differences
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
This important work presents a new methodology for the statistical analysis of fiber photometry data, improving statistical power while avoiding the bias inherent in the choices that are necessarily made when summarizing photometry data. The reanalysis of two recent photometry data sets, the simulations, and the mathematical detail provide convincing evidence for the utility of the method and the main conclusions, however, the discussion of the re-analyzed data is incomplete and would be improved by a deeper consideration of the limitations of the original data. In addition, consideration of other data sets and photometry methodologies including non-linear analysis tools, as well as a discussion of the importance of the data normalization are needed.
Thank you for the thorough and positive review of our work! We will incorporate this feedback to strengthen the manuscript. Specifically, we plan to revise the Discussion section to include a deeper consideration of the limitations of the original data, a description of the capacities of our method for conducting non-linear analyses, and the role data normalization plays in applicability of our tool.
Reviewer 1:
Strengths:
The framework the authors present is solid and well-explained. By reanalyzing formerly published data, the authors also further increase the significance of the proposed tool opening new avenues for reinterpreting already collected data.
Weaknesses:
However, this also leads to several questions. The normalization method employed for raw fiber photometry data is different from lab to lab. This imposes a significant challenge to applying a single tool of analysis.
Thank you for the positive feedback, we will address your comments in our revision. We agree that any data pre-processing steps will have down-stream consequences on the statistical inference from our method. Note, though, that this would also be the case with standard analysis approaches (e.g., t-tests, correlations) applied to summary measures like AUCs. For that reason, we do not believe that variability in pre-processing is an impediment to widespread adoption of a standard analysis procedure. Rather, we argue that the sensitivity of analysis results to pre-processing choices underscores the need for establishing statistical techniques that reduce the need for pre-processing, and properly account for structure in the data arising from experimental designs. The reviewer brings up an excellent point that we can further elaborate on how our methods actually reduce the need for such pre-processing steps. Indeed, our method provides smooth estimation results along the functional domain (i.e., across trial timepoints), has the ability to adjust for between-trial and -animal heterogeneity, and provides a valid statistical inference framework that quantifies the resulting uncertainty. For example, adjustment for session-to-session variability in signal magnitudes or dynamics could be accounted for, at least in part, through the inclusion of session-level random effects. This heterogeneity would then influence the width of the confidence intervals. This stands in contrast to “sweeping it under the rug” with a pre-processing step that may have an unknown impact on the final statistical inferences. Similarly, the level of smoothing is at least in part selected as a function of the data, and again is accounted for directly in the equations used to construct confidence intervals. In sum, our method provides both a tool to account for challenges in the data, and a systematic framework to quantify the additional uncertainty that accompanies accounting for those data characteristics.
Does the method that the authors propose work similarly efficiently whether the data are normalized in a running average dF/F as it is described in the cited papers? For example, trace smoothing using running averages (Jeong et al. 2022) in itself may lead to pattern dilution. The same question applies if the z-score is calculated based on various responses or even baselines.
This is an important question given how common this practice is in the field. Briefly, application of pre-processing steps will change the interpretation of the results from our analysis method. For example, if one subtracts off a pre-trial baseline average from each trial timepoint, then the “definition of 0”, and the interpretation of coefficients and their statistical significance, changes. Similarly, if one scales the signal (e.g., divides the signal magnitude by a trial- or animal-specific baseline), then this changes the interpretation of the FLMM regression coefficients to be in terms of an animal-specific signal unit as opposed to a raw dF/F. This is, however, not specific to our technique, and pre-processing would have a similar influence on, for example, linear regression (and thus t-tests, ANOVAs and Pearson correlations) applied to summary measures. We agree with the reviewer that explicitly discussing this point will strengthen the paper.
While it is difficult to make general claims about the anticipated performance of the method under all the potential pre-processing steps taken in the field, we believe that most common pre-processing strategies will not negatively influence the method’s performance or validity; they would, instead, change the interpretation of the results. We are releasing a series of vignettes to guide analysts through using our method and, to address your comment, we will add a section on interpretation after pre-processing.
How reliable the method is if the data are non-stationary and the baselines undergo major changes between separate trials?
This is an excellent question. We believe the statistical inferences will be valid and will properly quantify the uncertainty from non-stationarities, since our framework does not impose stationarity assumptions on the underlying process. It is worth mentioning that non-stationarity and high trial-to-trial variability may increase variance estimates if the model does not include a rich enough set of covariates to capture the source of the heterogeneity across trial baselines. However, this is a feature of our framework, rather than a bug, as it properly conveys to the analyst that high unaccounted for variability in the signal may result in high model uncertainty. Finally, mixed effects modeling provides a transparent, statistically reasonable, and flexible approach to account for between-session, and between-trial variability, a type of non-stationarity. We agree with the reviewer that this should be more explicitly discussed in the paper, and will do so.
Finally, what is the rationale for not using non-linear analysis methods? Following the paper's logic, non-linear analysis can capture more information that is diluted by linear methods.
Functional data analysis assumes that the function varies smoothly along the functional domain (i.e., across trial timepoints). It is a type of non-linear modeling technique over the functional domain since we do not assume a linear model (straight line). Therefore, our functional data analysis approach is able to capture more information that is diluted by linear models. While the basic form of our model assumes a linear change in the signal at a fixed trial timepoint, across trials/sessions, our package allows one to easily model changes with non-linear functions of covariates using splines or other basis functions. One must consider, however, the tradeoff between flexibility and interpretability when specifying potentially complex models.
Reviewer 2
Strengths:
The open-source package in R using a similar syntax as the lme4 package for the implementation of this framework on photometry data enhances the accessibility, and usage by other researchers. Moreover, the decreased fitting time of the model in comparison with a similar package on simulated data, has the potential to be more easily adopted.
The reanalysis of two studies using summary statistics on photometry data (Jeong et al., 2022; Coddington et al., 2023) highlights how trial-by-trial analysis at each time-point on the trial can reveal information obscured by averaging across trials. Furthermore, this work also exemplifies how session and subject variability can lead to opposite conclusions when not considered.
Thank you for the positive assessment of our work!
Weaknesses:
Although this work has reanalyzed previous work that used summary statistics, it does not compare with other studies that use trial-by-trial photometry data across time-points in a trial.
As described by the authors, fitting pointwise linear mixed models and performing t-test and Benjamini-Hochberg correction as performed in Lee et al. (2019) has some caveats. Using joint confidence intervals has the potential to improve statistical robustness, however, this is not directly shown with temporal data in this work. Furthermore, it is unclear how FLMM differs from the pointwise linear mixed modeling used in this work.
We agree with the reviewers that providing more detail about the drawbacks of the approach applied in Lee et al., 2019 will strengthen the paper. We will add an example analysis applying the method proposed by Lee et al., 2019 to show how the set of timepoints at which coefficient estimates reach statistical significance can vary dramatically depending on the sampling rate one subsamples their data at, a highly undesirable property of this strategy. Our approach is robust to this, and still provides a multiple comparisons correction through the joint confidence intervals.
In this work, FLMM usages included only one or two covariates. However, in complex behavioral experiments, where variables are correlated, more than two may be needed (see Simpson et al. (2023), Engelhard et al. (2019); Blanco-Pozo et al. (2024)). It is not clear from this work, how feasible computationally would be to fit such complex models, which would also include more complex random effects.
This is a good point. In our experience, the code is still quite fast (often taking seconds to tens of seconds in our experience) on a standard laptop when fitting complex models that include, for example, 10 covariates, or complex random effect specifications on dataset sizes common in fiber photometry. In the manuscript, we included results from simpler models with few covariates in an attempt to show results from the FLMM versions of the standard analyses (e.g., correlations, t-tests) applied in Jeong et al., 2022. Our goal was to show that our method reveals effects obscured by standard analyses even in simple cases. Some of our models did, however, include complex nested random effects (e.g., the models described in Section 4.5.2).
Like other mixed-model based analyses, our method becomes slower when the number of observations in the dataset is on the order of tens of thousands of observations. However, we coded the methods to be memory efficient so that even these larger analyses can be run on standard laptops. We thank the reviewer for this point, as we worked extremely hard to scale the method to be able to efficiently fit models commonly applied in neuroscience. Indeed, challenges with scalability were one of the main motivations for applying the estimation procedure that we did; in the appendix we show that the fit time of our approach is much faster than existing FLMM software such as the refund package function pffr(), especially for large sample sizes. While pffr() appears to scale exponentially with the number of clusters (e.g., animals), our method appears to scale linearly. We will more explicitly emphasize the scalability in the revision, since we agree this will strengthen the final manuscript.
Reviewer #3
Strengths:
The statistical framework described provides a powerful way to analyze photometry data and potentially other similar signals. The provided package makes this methodology easy to implement and the extensively worked examples of reanalysis provide a useful guide to others on how to correctly specify models.
Modeling the entire trial (function regression) removes the need to choose appropriate summary statistics, removing the opportunity to introduce bias, for example in searching for optimal windows in which to calculate the AUC. This is demonstrated in the re-analysis of Jeong et al., 2022, in which the AUC measures presented masked important details about how the photometry signal was changing.
Meanwhile, using linear mixed methods allows for the estimation of random effects, which are an important consideration given the repeated-measures design of most photometry studies.
Thank you for the positive assessment of our work!
Weaknesses:
While the availability of the software package (fastFMM), the provided code, and worked examples used in the paper are undoubtedly helpful to those wanting to use these methods, some concepts could be explained more thoroughly for a general neuroscience audience.
We appreciate this and, to address your and other reviewers’ comments, we are creating a series of vignettes walking users through how to analyze photometry data with our package. We will include algebraic illustrations to help users gain familiarity with the regression modeling here.
While the methodology is sound and the discussion of its benefits is good, the interpretation and discussion of the re-analyzed results are poor:
In section 2.3, the authors use FLMM to identify an instance of Simpson's Paradox in the analysis of Jeong et al. (2022). While this phenomenon is evident in the original authors' metrics (replotted in Figure 5A), FLMM provides a convenient method to identify these effects while illustrating the deficiencies of the original authors' approach of concatenating a different number of sessions for each animal and ignoring potential within-session effects. The discussion of this result is muddled. Having identified the paradox, there is some appropriate speculation as to what is causing these opposing effects, particularly the decrease in sessions. In the discussion and appendices, the authors identify (1) changes in satiation/habitation/motivation, (2) the predictability of the rewards (presumably by the click of a solenoid valve) and (3) photobleaching as potential explanations of the decrease within days. Having identified these effects, but without strong evidence to rule all three out, the discussion of whether RPE or ANCCR matches these results is probably moot. In particular, the hypotheses developed by Jeong et al., were for a random (unpredictable) rewards experiment, whereas the evidence points to the rewards being sometimes predictable. The learning of that predictability (e.g. over sessions) and variation in predictability (e.g. by attention level to sounds of each mouse) significantly complicate the analysis. The FLMM analysis reveals the complexity of analyzing what is apparently a straightforward task design.
While we are disappointed to hear the reviewer felt our initial interpretations and discussion were poor, the reviewer brings up an excellent point that we had not considered. They have convinced us that acknowledging and elaborating on this alternative perspective will strengthen the paper. We agree that the ANCCR/RPE model predictions were made for unpredictable rewards and, as the reviewer rightly points out, there is evidence that the animals sense the reward delivery. Regardless of the learning theory one adopts (RPE, ANCCR or others), we agree that this (potentially) learned predictability alone could account for the increase in signal magnitude across sessions.
After reading the reviewer’s comments, we consulted with a number of researchers in this area, and several felt that a CS+ can serve as a reward, within itself. From this perspective, the rewards in the Jeong et al., 2022 experiment might still be considered unexpected. After discussing extensively with the authors of Jeong et al., 2022, it is clear that they went to enormous trouble to prevent the inadvertent generation of a CS+, and it is likely changes in pressure from the solenoid (rather than a sound) that served as a cue. This underscores the difficulty of preventing perception of reward delivery in practice. As this paper is focused on analysis approaches, we feel that we can contribute most thoughtfully to the dopamine–learning theory conversation by presenting both sides.
Overall, we agree with the reviewer that future experiments will be needed for testing the accuracy of the models’ predictions for random (unpredicted) rewards. While we understand that our attempt to document our conversations with the Jeong et al., 2022 authors may have room for improvement, we hope the reviewer can appreciate that this was done with the best of intentions. We wish to emphasize that we also consulted with several other researchers in the field when crafting the discussion. The Jeong et al., 2022 authors could easily have avoided acknowledging the potential incompleteness of their theory, by claiming that our results do not invalidate their predictions for a random reward, as the reward was not unpredicted in the experiment (as a result of the inadvertent solenoid CS+). Instead, they went out of their way to emphasize that their experiment did test a random reward, and that our results do present problems for their theory. We think that engagement with re-analyses of one’s data, even when findings are inconvenient, is a good demonstration of open science practice. For that reason as well, we feel providing readers with a perspective on the entire discussion will contribute to the scientific discourse in this area.
Finally, we would like to reiterate that this conversation is happening because our method, by analyzing the signal at every trial timepoint, revealed a neural signal that appears to indicate that the animals sense reward delivery. Ultimately, this was what we set out to do: help researchers ask questions of their data that they could not ask before. We believe that having a demonstration that we can indeed do this for a “live” issue is the most appropriate way of demonstrating the usefulness of the method.
It is clear the reviewer put a lot of time into understanding what we did, and was very thoughtful about the feedback. We would like to thank the reviewer again for taking such care in reviewing our paper.
If this paper is not trying to arbitrate between RPE and ANCCR, as stated in the text, the post hoc reasoning of the authors of Jeong et al 2022 provided in the discussion is not germane.
While we appreciate that the post hoc reasoning of the authors of Jeong et al., 2022 may not seem germane, we would like to provide some context for its inclusion. As statisticians and computer scientists, our role is to create methods, and this often requires using open source data and recreating past analyses. This usually involves extensive conversation with authors about their data and analysis choices because, if we cannot reproduce their findings using their analysis methods, we cannot verify that results from our own methods are valid. As such, we prefer to conduct method development in a collaborative fashion, and we strive to constructively, and respectfully, discuss our results with the original authors. We feel that giving them the opportunity to suggest analyses, and express their point of view if our results conflict with their original conclusions, is important, and we do not want to discourage authors from making their datasets public. As such, we conducted numerous analyses at the suggestion of Jeong et al., 2022 and discussed the results over the course of many months. Indeed the analyses in the Appendix that the reviewer is referring to were conducted at the suggestion of the authors of Jeong et al., 2022, in an attempt to rule out alternative explanations. We nevertheless appreciate that our interpretations of these results can include some of the caveats suggested by the reviewer, and we will strive to improve these sections.
Arbitrating between the models likely requires new experimental designs (removing the sound of the solenoid, satiety controls) or more complex models (e.g. with session effects, measures of predictability) that address the identified issues.
We agree with the reviewer that the results suggest that new experimental designs will likely be necessary to adjudicate between models. It is our hope that, by weighing the different issues and interpretations, our paper might provide useful suggestions into what experimental designs would be most beneficial to rule out competing hypotheses in future data collection efforts. We believe that our methodology will strengthen our capacity to design new experiments and analyses. We will make the reviewer’s suggestions more explicit in the discussion by emphasizing the limitations of the original data.
Of the three potential causes of within-session decreases, the photobleaching arguments advanced in the discussion and expanded greatly in the appendices are not convincing. The data being modeled is a processed signal (ΔF/F) with smoothing and baseline correction and this does not seem to have been considered in the argument.
We are disappointed to hear that this extensive set of analyses, much of which was conducted at the suggestion of Jeong et al., 2022, was not convincing. We agree that acknowledging any pre-processing would provide useful context for the reader. We do wish to clarify that we analyzed the data that were made available online (raw data was not available). Moreover, for comparison with the authors’ results, we felt it was important to maintain the same pre-processing steps as they did. These conditions were held constant across analysis approaches; therefore, we think that the changes within-trial are likely not influenced substantially by these pre-processing choices. While we cannot speak definitively to the impact any of the processing conducted by the authors had on the results, we believe that it was likely minor, given that the timing of signals at other points in the trial, and in other experiments, were as expected (e.g., the signal rose rapidly after cue onset in Pavlovian tasks).
Furthermore, the photometry readout is also a convolution of the actual concentration changes over time, influenced by the on-off kinetics of the sensor, which makes the interpretation of timing effects of photobleaching less obvious than presented here and more complex than the dyes considered in the cited reference used as a foundation for this line of reasoning.
We appreciate the nuance of this point, and we will add it to our discussion. In response to your criticism, we have consulted with more experts in the field regarding the potential for bleaching in this data, and it is not clear to us why photobleaching would be visible in one time-window of a trial, but not at another (less than a second away), despite high dF/F magnitudes in both time-windows. We do wish to point out that, at the request of the authors, we analyzed many experiments from the same animals and in most cases did not observe other indications of photobleaching. Hence, it is not clear to us why this particular set of experiments would garner additional skepticism regarding the potential for photobleaching to invalidate results. While the role of photobleaching may be more complicated with this sensor than others in the references, that citation was included, at the suggestion of Jeong et al., 2022 simply as a way of acknowledging that non-linearities in photobleaching can occur.
Within this discussion of photobleaching, the characterization of the background reward experiments used in part to consider photobleaching (appendix 7.3.2) is incorrect. In this experiment (Jeong et al., 2022), background rewards were only delivered in the inter-trial-interval (i.e. not between the CS+ and predicted reward as stated in the text). Both in the authors' description and in the data, there is a 6s before cue onset where rewards are not delivered and while not described in the text, the data suggests there is a period after a predicted reward when background rewards are not delivered. This complicates the comparison of this data to the random reward experiment.
Thank you for pointing this out!! We will remove the parenthetical on page 18 of the appendix that incorrectly stated that rewards can occur between the CS+ and the predicted reward.
The discussion of the lack of evidence for backpropagation, taken as evidence for ANCCR over RPE, is also weak.
This point was meant to acknowledge that, although our method yields results that conflict with the conclusions described by Jeong et al., 2022 on data from some experiments, on other experiments our method supports their results. Again, we believe that a critical part of open science is acknowledging both areas where analyses support and conflict with those of the original authors. We agree with the reviewer that qualifying our results so as not to emphasize support for/against RPE/ANCCR will strengthen our paper, and we will make these changes.
A more useful exercise than comparing FLMM to the methods and data of Jeong et al., 2022, would be to compare against the approach of Amo et al., 2022, which identifies backpropagation (data publicly available: DOI: 10.5061/dryad.hhmgqnkjw). The replication of a positive result would be more convincing of the sensitivity of the methodology than the replication of a negative result, which could be a result of many factors in the experimental design. Given that the Amo et al. analysis relies on identifying systematic changes in the timing of a signal over time, this would be particularly useful in understanding if the smoothing steps in FLMM obscure such changes.
Thank you for this suggestion, and we agree this could be a useful analysis for the field. Your thoughtful review has convinced us that focusing on our statistical contribution will strengthen the paper, and we will make changes to further emphasize that we are not seeking to adjudicate between RPE/ANCCR. We only had space in the manuscript to include a subset of the analyses conducted on Jeong et al., 2022, and had to relegate the results from the Coddington et al., data to an appendix. Realistically, it would be hard for us to justify analyzing a third dataset. As you may surmise from the one we presented, reanalyzing a new dataset is usually very time consuming, and invariably requires extensive communication with the original authors. We did include numerous examples in our manuscript where we already replicated positive results, in a way that we believe demonstrates the sensitivity of the methodology. We have also been working with five groups at NIH and elsewhere using our approach, in experiments targeting different scientific questions. In fact, one paper that extensively applies our method and compares the results from those yielded by standard analysis of AUCs is already accepted and in press. Hence there should soon be additional demonstrations of what the method can do in less controversial settings. Finally, our forthcoming vignettes include additional analyses, not included in the manuscript, that replicate positive results. We take your point that our description of the data supporting one theory or the other should be qualified, and we will correct that. Again, your review was very thorough, and we appreciate your taking so much time to help us improve our work.
Reviewer #2 (Recommendations For The Authors):
First, I would like to commend the authors for the clarity of the paper, and for creating an open-source package that will help researchers more easily adopt this type of analysis.
Thank you!
I would suggest the authors consider adding to the manuscript, either some evidence or some intuition on how feasible would be to use FLMM for very complex model specifications, in terms of computational cost and model convergence.
This is an excellent point and we will make this suggested change in the Methods and Discussion section in the next draft.
From my understanding, this package might potentially be useful not just for photometry data but also for two-photon recordings for example. If so, I would also suggest the authors add to the discussion this potential use.
We appreciate your thinking on this point, as it would definitely help expand use of the method. We included a brief point in the Discussion that this package would be useful for other techniques, but we will expand upon this.
Reviewer #3 (Recommendations For The Authors):
The authors should define 'function' in context, as well as provide greater detail of the alternate tests that FLMM is compared to in Figure 7. Given the novelty of estimating joint CIs, the authors should be clearer about how this should be reported and how this differs from pointwise CIs (and how this has been done in the past).
Thank you, this is a very good point and will be critical for helping analysts describe and interpret results. We will add more detail to the Methods section on this point.
The authors identify that many photometry studies are complex nested longitudinal designs, using the cohort of 8 animals used in five task designs of Jeong et al. 2022 as an example. The authors miss the opportunity to illustrate how FLMM might be useful in identifying the effects of subject characteristics (e.g. sex, CS+ cue identity).
This is a great suggestion and we will add this important point to the discussion , especially in light of the factorial designs common in neuroscience experiments.
In discussing the delay-length change experiment, it would be more accurate to say that proposed versions of RPE and ANCCR do not predict the specific change.
We will make this change and agree this is a better phrasing.
-
Reviewer #3 (Public Review):
Summary:<br /> Loewinger et al., extend a previously described framework (Cui et al., 2021) to provide new methods for statistical analysis of fiber photometry data. The methodology combines functional regression with linear mixed models, allowing inference on complex study designs that are common in photometry studies. To demonstrate its utility, they reanalyze datasets from two recent fiber photometry studies into mesolimbic dopamine. Then, through simulation, they demonstrate the superiority of their approach compared to other common methods.
Strengths:<br /> The statistical framework described provides a powerful way to analyze photometry data and potentially other similar signals. The provided package makes this methodology easy to implement and the extensively worked examples of reanalysis provide a useful guide to others on how to correctly specify models.
Modeling the entire trial (function regression) removes the need to choose appropriate summary statistics, removing the opportunity to introduce bias, for example in searching for optimal windows in which to calculate the AUC. This is demonstrated in the re-analysis of Jeong et al., 2022, in which the AUC measures presented masked important details about how the photometry signal was changing.
Meanwhile, using linear mixed methods allows for the estimation of random effects, which are an important consideration given the repeated-measures design of most photometry studies.
Weaknesses:<br /> While the availability of the software package (fastFMM), the provided code, and worked examples used in the paper are undoubtedly helpful to those wanting to use these methods, some concepts could be explained more thoroughly for a general neuroscience audience.
While the methodology is sound and the discussion of its benefits is good, the interpretation and discussion of the re-analyzed results are poor:
In section 2.3, the authors use FLMM to identify an instance of Simpson's Paradox in the analysis of Jeong et al. (2022). While this phenomenon is evident in the original authors' metrics (replotted in Figure 5A), FLMM provides a convenient method to identify these effects while illustrating the deficiencies of the original authors' approach of concatenating a different number of sessions for each animal and ignoring potential within-session effects. The discussion of this result is muddled. Having identified the paradox, there is some appropriate speculation as to what is causing these opposing effects, particularly the decrease in sessions. In the discussion and appendices, the authors identify (1) changes in satiation/habitation/motivation, (2) the predictability of the rewards (presumably by the click of a solenoid valve) and (3) photobleaching as potential explanations of the decrease within days. Having identified these effects, but without strong evidence to rule all three out, the discussion of whether RPE or ANCCR matches these results is probably moot. In particular, the hypotheses developed by Jeong et al., were for a random (unpredictable) rewards experiment, whereas the evidence points to the rewards being sometimes predictable. The learning of that predictability (e.g. over sessions) and variation in predictability (e.g. by attention level to sounds of each mouse) significantly complicate the analysis. The FLMM analysis reveals the complexity of analyzing what is apparently a straightforward task design. If this paper is not trying to arbitrate between RPE and ANCCR, as stated in the text, the post hoc reasoning of the authors of Jeong et al 2022 provided in the discussion is not germane. Arbitrating between the models likely requires new experimental designs (removing the sound of the solenoid, satiety controls) or more complex models (e.g. with session effects, measures of predictability) that address the identified issues.
Of the three potential causes of within-session decreases, the photobleaching arguments advanced in the discussion and expanded greatly in the appendices are not convincing. The data being modeled is a processed signal (ΔF/F) with smoothing and baseline correction and this does not seem to have been considered in the argument. Furthermore, the photometry readout is also a convolution of the actual concentration changes over time, influenced by the on-off kinetics of the sensor, which makes the interpretation of timing effects of photobleaching less obvious than presented here and more complex than the dyes considered in the cited reference used as a foundation for this line of reasoning.
Within this discussion of photobleaching, the characterization of the background reward experiments used in part to consider photobleaching (appendix 7.3.2) is incorrect. In this experiment (Jeong et al., 2022), background rewards were only delivered in the inter-trial-interval (i.e. not between the CS+ and predicted reward as stated in the text). Both in the authors' description and in the data, there is a 6s before cue onset where rewards are not delivered and while not described in the text, the data suggests there is a period after a predicted reward when background rewards are not delivered. This complicates the comparison of this data to the random reward experiment.
The discussion of the lack of evidence for backpropagation, taken as evidence for ANCCR over RPE, is also weak. A more useful exercise than comparing FLMM to the methods and data of Jeong et al., 2022, would be to compare against the approach of Amo et al., 2022, which identifies backpropagation (data publicly available: DOI: 10.5061/dryad.hhmgqnkjw). The replication of a positive result would be more convincing of the sensitivity of the methodology than the replication of a negative result, which could be a result of many factors in the experimental design. Given that the Amo et al. analysis relies on identifying systematic changes in the timing of a signal over time, this would be particularly useful in understanding if the smoothing steps in FLMM obscure such changes.
-
-
www.sciencedirect.com www.sciencedirect.com
-
The Jackson LaboratoryStrain code: 003831
DOI: 10.1016/j.isci.2024.109248
Resource: (IMSR Cat# JAX_003831,RRID:IMSR_JAX:003831)
Curator: @evieth
SciCrunch record: RRID:IMSR_JAX:003831
-
Charles RiverStrain code: 027
DOI: 10.1016/j.isci.2024.109248
Resource: (IMSR Cat# CRL_027,RRID:IMSR_CRL:027)
Curator: @evieth
SciCrunch record: RRID:IMSR_CRL:027
-
Charles RiverStrain code: 028
DOI: 10.1016/j.isci.2024.109248
Resource: (IMSR Cat# CRL_028,RRID:IMSR_CRL:028)
Curator: @evieth
SciCrunch record: RRID:IMSR_CRL:028
-
-
www.youtube.com www.youtube.com
-
Les Menstrueuses 2023 – De nouveaux essentialismes ? Partie 1
https://www.youtube.com/watch?v=GAAwxpeX-LI
Voici un résumé des points forts de la transcription de la vidéo, avec les timecodes correspondants :
- Introduction du Festival [00'00'08] : Présentation du festival "les monstrueuses" et de son évolution au fil des années.
- Exposition sur les Menstruations [00'00'36] : Inauguration d'une exposition intitulée "Song" par Lucie IDF, abordant l'histoire des menstruations, les tabous et les maladies associées.
- Objectifs du Festival [00'01'14] : Discussion sur les buts du festival, notamment la médiation culturelle et scientifique autour des menstruations.
- Programme Culturel et Scientifique [00'01'36] : Annonce des événements scientifiques et culturels organisés par l'Université de Poitiers et l'Espace Mendès France.
- Performance Artistique [00'02'01] : Présentation d'une performance immersive par des artistes résidents travaillant sur la représentation des corps et l'image de soi.
- Soirée Festive [00'02'45] : Invitation à une soirée festive avec un code vestimentaire rouge organisée par le Confort Moderne.
- Publication d'un Ouvrage [00'04'37] : Annonce de la sortie d'un livre intitulé "Idées reçues sur les menstruations" coïncidant avec le festival.
- Déconstruction des Idées Reçues [00'05'09] : Explication de la structure du livre et de son approche pour déconstruire les idées reçues sur les menstruations.
- Disciplines Universitaires et Contributions [00'06'03] : Évocation de la diversité des disciplines et des contributeurs au livre, incluant des universitaires et des militants.
- Journée d'Étude sur les Discriminations [00'07'40] : Introduction de la journée d'étude visant à explorer les discriminations au-delà des menstruations.
- Discours Biologiques dans les Médias [00'08'16] : Analyse de la prévalence des discours biologiques et de leur impact dans les espaces médiatiques.
- Parentalité et Procréation [00'09'45] : Discussion sur les perceptions de la parentalité et de la procréation comme naturelles ou contre-nature.
- Conservatismes en Ligne [00'10'11] : Examen de la diffusion des conservatismes et des mouvements réactionnaires sur les réseaux sociaux numériques.
- Mobilisations Anti-Trans [00'11'03] : Table ronde sur les mobilisations anti-trans et le contexte actuel des droits trans.
- Conférence sur Genre, Sexe et Biologie [00'12'12] : Annonce d'une conférence publique par Thierry Hoquet sur les liens entre genre, sexe et biologie.
- Introduction Historique [00'12'25] : Présentation historique sur l'instrumentalisation des savoirs biologiques et la différenciation des genres.
Ce résumé couvre la première partie de la transcription jusqu'à [01h16min40s], mettant en lumière les principaux sujets et événements mentionnés. Voici un résumé des points forts de la transcription vidéo de la page, avec les timecodes correspondants :
-
Sciences Naturelles [00h17min26s] Les sciences naturelles, précurseurs de la biologie moderne, se sont développées en Europe du 15e au 19e siècle, se concentrant sur l'observation et la classification de la nature.
-
Procréation et Société [00h21min02s] La capacité de procréer est considérée comme essentielle pour les femmes et les hommes, avec une forte influence des croyances religieuses et médicales sur la reproduction et la sexualité.
-
Corps Féminin et Maternité [00h27min43s] Les traités médicaux du 18e et 19e siècle se concentrent sur la grossesse et la maternité comme réalisation principale des femmes, souvent en lien avec des symptômes d'hystérie chez les femmes célibataires ou sans enfants.
-
Discrimination et Biopolitique [00h32min03s] Des arguments biologiques sont utilisés pour justifier la hiérarchie sociale et la subordination des femmes et des minorités de genre, avec une intersection des questions de genre, de race et de classe sociale. Voici un résumé des points forts de la transcription de la vidéo entre [00h35min00s] et [01h16min40s], avec les timecodes correspondants :
-
Introduction par Marie Valin [00h35min00s] Marie Valin présente l'histoire des discours biologiques utilisés politiquement contre l'indifférenciation des genres, soulignant leur constance à travers le temps.
-
Sciences Naturelles et Biologie [00h37min26s] Discussion sur l'évolution des sciences naturelles et la naissance de la biologie au XIXe siècle, héritière des savoirs naturalistes.
-
Reproduction et Biopolitique [00h41min02s] Analyse de la perception de la reproduction comme un devoir incontournable et de l'impact des croyances religieuses sur les pratiques contraceptives et abortives.
-
Corps Féminin et Tempérament [00h49min21s] Exploration des idées reçues sur le tempérament féminin et l'impact supposé de la physiologie sur le désir sexuel des femmes.
Ces points mettent en lumière la manière dont les savoirs biologiques ont été et sont encore utilisés pour justifier des hiérarchies sociales et des rôles de genre spécifiques. Voici un résumé des points forts de la transcription vidéo de [00:50:00] à [01:16:40]:
- [00:50:00] Discussion sur la Frigidité Féminine: L'auteur Manger Opérationnelle aborde la perception de l'indifférence des femmes envers les plaisirs de l'amour, attribuée à l'absence de sécrétion de sperme et à un tempérament lymphatique.
- [00:58:00] Lutte contre l'Onanisme et l'Avortement: Les médecins et la religion du XIXe siècle s'opposent à l'onanisme conjugal et à l'avortement, considérés comme des crimes anti-patriotiques.
- [01:06:00] Rôles Genrés et Reproduction: La médecine et les sciences biologiques insistent sur la binarité de genre et la division sexuelle de la société pour des raisons biopolitiques.
- [01:14:00] Instrumentalisation des Savoirs Biologiques: L'historienne Marie Vallin introduit la journée d'étude en soulignant l'utilisation historique des savoirs biologiques pour justifier l'ordre genré au nom de la nature.
Ces points mettent en lumière les thèmes de la biologie, du genre, et de la reproduction dans le contexte historique et social du XIXe siècle.
Tags
Annotators
URL
-
-
www.researchsquare.com www.researchsquare.com
-
Author Response
The following is the authors’ response to the original reviews.
Reviewer 1:
Comment 1.1: The distinction of PIGS from nearby OPA, which has also been implied in navigation and ego-motion, is not as clear as it could be.
Response1.1: The main “functional” distinction between TOS/OPA and PIGS is that TOS/OPA responds preferentially to moving vs. stationary stimuli (even concentric rings), likely due to its overlap with the retinotopic motion-selective visual area V3A, for which this is a defining functional property (e.g. Tootell et al., 1997, J Neurosci). In comparison, PIGS does not show such a motion-selectivity. Instead, PIGS responds preferentially to more complex forms of motion within scenes.
Moreover, PIGS and TOS/OPA are located in differently relative to the retinotopic visual areas. Briefly, PIGS is located adjacent to areas IPS3-4 while TOS/OPA overlaps with areas V3A/B and IPS0 (V7). This point is now highlighted in the new experiment 3b and the new Figure 6. In this revision, we also tried to better highlight these point in sections 4.3, 4.4 and 4.5. (see also the response to the first comment from Reviewer #2).
Reviewer 2:
Comment 2.1: First, the scene-selective region identified appears to overlap with regions that have previously been identified in terms of their retinotopic properties. In particular, it is unclear whether this region overlaps with V7/IPS0 and/or IPS1. This is particularly important since prior work has shown that OPA often overlaps with v7/IPS0 (Silson et al, 2016, Journal of Vision). The findings would be much stronger if the authors could show how the location of PIGS relates to retinotopic areas (other than V6, which they do currently consider). I wonder if the authors have retinotopic mapping data for any of the participants included in this study. If not, the authors could always show atlas-based definitions of these areas (e.g. Wang et al, 2015, Cerebral Cortex).
Response 2.1: We thank the reviewers for reminding us to more clearly delineate this issue of possible overlap, including the information provided by Silson et al, 2016. The issue of possible overlap between area TOS/OPA and the retinotopic visual areas, both in humans and non-human primates, was also clarified by our team in 2011 (Nasr et al., 2011). As you can see in Figure 6 (newly generated), and consistent with those previous studies, TOS/OPA overlaps with visual areas V3A/B and V7. Whereas PIGS is located more dorsally close to IPS3-4. As shown here, there is no overlap between PIGS and TOS/OPA and there is no overlap between PIGS and areas V3A/B and V7.
To more directly address the reviewer’s concern, in this revision, we have added a new experiment (Experiment 3b) in which we have shown the relative position of PIGS and the retinotopic areas in two individual subjects (Figure 6). All the relevant points are also discussed in section 4.3.
Comment 2.2: Second, recent studies have reported a region anterior to OPA that seems to be involved in scene memory (Steel et al, 2021, Nature Communications; Steel et al, 2023, The Journal of Neuroscience; Steel et al, 2023, biorXiv). Is this region distinct from PIGS? Based on the figures in those papers, the scene memory-related region is inferior to V7/IPS0, so characterizing the location of PIGS to V7/IPS0 as suggested above would be very helpful here as well. If PIGS overlaps with either of V7/IPS0 or the scene memory-related area described by Steel and colleagues, then arguably it is not a newly defined region (although the characterization provided here still provides new information).
Response 2.2: The lateral-place memory area (LPMA) is located on the lateral brain surface, anterior relative to the IPS (see Figure 1 from Steel et al., 2021 and Figure 3 from Steel et al., 2023). In contrast, PIGS is located on the posterior brain surface, also posterior relative to the IPS. In other words, they are located on two different sides of a major brain sulcus. In this revision we have clarified this point, including the citations by Steel and colleagues in section 4.3.
Comments 2.3: Another reason that it would be helpful to relate PIGS to this scene memory area is that this scene memory area has been shown to have activity related to the amount of visuospatial context (Steel et al, 2023, The Journal of Neuroscience). The conditions used to show the sensitivity of PIGS to ego-motion also differ in the visuospatial context that can be accessed from the stimuli. Even if PIGS appears distinct from the scene memory area, the degree of visuospatial context is an alternative account of what might be represented in PIGS.
Response 2.3: The reviewer raises an interesting point. One minor confusion is that we may be inadvertently referring to two slightly different types of “visuospatial context”. Specifically, the stimuli used in the ego-motion experiment here (i.e. coherently vs. incoherently changing scenes) represent the same scenes, and the only difference between the two conditions is the sequence of images across the experimental blocks. In that sense, the two experimental conditions may be considered to have the same visuospatial “context”. However, it could be also argued that the coherently changing scenes provide more information about the environmental layout. In that case, considering the previous reports that PPA/TPA and RSC/MPA may also be involved in layout encoding (Epstein and Kanwisher 1998; Wolbers et al. 2011), we expected to see more activity within those regions in response to coherently compared incoherently changing scenes. These issues are now more explicitly discussed in the revised article (section 4.6).
Reviewer 3:
Comment 3.1: There are few weaknesses in this work. If pressed, I might say that the stimuli depicting ego-motion do not, strictly speaking, depict motion, but only apparent motion between 2s apart photographs. However, this choice was made to equate frame rates and motion contrast between the 'ego-motion' and a control condition, which is a useful and valid approach to the problem. Some choices for visualization of the results might be made differently; for example, outlines of the regions might be shown in more plots for easier comparison of activation locations, but this is a minor issue.
Response 3.1: We thank the reviewer for these constructive suggestions, and we agree with their comment that the ego-motion stimuli are not smooth, even though they were refreshed every 100 ms. However, the stimuli were nevertheless coherent enough to activate areas V6 and MT, two major areas known to respond preferentially to coherent compared to incoherent motion.
Reviewer #1 (Recommendations For The Authors):
I enjoyed reading this article. I have a few suggestions for improvement:
(1) Delineation from OPA: The OPA has been described in quite similar terms as PIGS, with its involvement in ego-motion (e.g., crawling, walking) and navigation in general (e.g., Dilks' recent work; Bonner and Epstein). The authors address the distinction in section 4.4. Unlike Kamps et al. (2016) and Jones et al. (2023), the authors found weak or no evidence for ego-motion in OPA. They explain this discrepancy with differences in refresh rates and different levels of spatial smoothing of the fMRI data. It is not clear why these fairly small methodological differences would lead to different findings of ego-motion in the OPA. Arguably, the OPA is the closest of the "established" scene areas to PIGS, both in anatomical location and in function. I would therefore appreciate a more detailed discussion of the differences between these two areas.
Response: Jones et al. have also shown that ego-motion TOS/OPA activity when compared to scrambled scenes. This is fundamentally different than what we have shown here, which coherently vs. incoherently changing scenes (i.e. not a small difference). Also, Kamps et al. used static scenes as a control which, considering TOS/OPA motion-selectivity, have a large impact on TOS/OPA response.
(2) Random effects analysis: The authors mention using a "random effects analysis" for several of their experiments. I would ask them to provide more details on what statistical models were used here. Were they purely random-effects models or actually mixed-effects models? What were the factors that entered into the analysis? Providing more detail would make the analysis techniques more transparent.
Response: This point is now clarified in the Methods section.
(3) Data and code availability: The authors write that data and code "are ready to be shared upon request." (section 2.5) In the spirit of transparency and openness, I strongly encourage the authors to make the data publicly available, e.g., on OSF or OpenNeuro. In particular, having probabilistic maps of PIGS available will allow other researchers to include PIGS in their analysis pipelines, making the current work more impactful.
Response: We have made the probabilistic labels available to the public. This point is now highlighted in section 2.5.
(4) Minor comments on the writing that caught my eye while reading the article:
- Line 27: "in the human brain".
Response: Done.
-Line 30: I don't agree with the characterization of the previous model of scene perception as "simplistic." Adding one additional ROI makes it no less simplistic. Perhaps the authors can rephrase to make this slightly less antagonistic?
Response: Done.
- Line 71: it is not clear why NHPs are relevant here.
Response: We decided to keep the text intact.
- Line 138" "were randomized".
Response: Done.
- Line 152: "consisting".
Response: Done.
- Line 155: "sets" (plural).
Response: Done.
- Lines 253-255: Why were the 3T spatially smoothed but not the 7T data? This seems odd.
Response: We kept the text intact.
- Line 481: "we found strong motion selectivity" (remove "a").
Response: Done.
- Line 564: a word is missing, probably: "a stronger effect of ego-motion".
Response: Done.
- Line 591: "controlling spatial attention" (remove "the").
Response: Done.
- Line 591 and 594: Both sentences start with "However". I think the first of these should not because it is setting up the contrast for the second sentence.
Response: Done.
- Line 607: "higher-level" (hyphen).
Response: Done.
- Throughout the manuscript: adverbial phrases such as "(in)coherently changing" or "probabilistically localized" do not get a hyphen.
Response: Done.
Reviewer #2 (Recommendations For The Authors):
The authors state that "All data, codes and stimuli are ready to be shared upon request". Ideally, these materials should be deposited in appropriate repositories (e.g. OpenMRI, GitHub) and not require readers to contact the authors to obtain such materials.
Other Comments:
(a) The title ("A previously undescribed scene-selective site is the key to encoding ego-motion in natural environments") is potentially misleading - the work was not conducted in a natural environment. At best, you could say they are 'naturalistic stimuli'. Also, in what sense is PIGS "key" to encoding ego-motion - the study just shows sensitivity to this factor.
Response: We changed the title to “naturalistic environments”.
(b) Figure 1 - I'm not sure what point the authors are trying to make with Figure 1. The comparison is between a highly smoothed, group fixed-effects analysis and a less-smoothed individual subject analysis. The differences between the two could reflect group vs. individual, highly-smoothed (5 mm) versus less-smoothed (2 mm), or differences in thresholding. If the thresholding were lower for the group analysis, it would probably start to look more similar to the individual subject. As it stands, this figure isn't particularly informative, it seems redundant with Figure 2, and Figure 1A is not even referenced in the main text. Further, fixed effects analyses are relatively uncommon in the recent literature, so their inclusion is unusual.
Response: Figure 1A is a replication of the data/method used in Nasr et al., 2011 and it will help the readers see the difference between the “traditional” scene-selectivity maps generated based on group-averaging” vs. data from individual subjects. In this case, we decided not to change the Figure.
(c) Figure 3 - why are the two sets of maps shown at different thresholds? For 3B given the larger sample size, it is expected that the extent of the significant activations will increase. Currently the higher threshold for 3B and the smaller range for 3A is making the sets of maps look more comparable.
Response: As the reviewer noticed, the number of subjects is larger in Figure 3B compared to 3A. The main point of this figure is to show that the PIGS activity center does not vary across populations. Considering this point, we decided not to change this figure.
(d) Figure 10 - why is the threshold lower than used for other figures? It would be helpful if there was consistent thresholding across figures.
Response: Experiment 6 and Experiment 1 are based on different stimuli (see Methods). Also, among those subjects who participated in Experiment 1, two subjects did not participate in Experiment 6. These points are already highlighted in the text.
(e) Figures - how about the AFNI approach of thresholding and showing sub-threshold data at the same time? (Taylor et al, 2023, Neuroimage).
Response: We highly appreciate the methodology suggested by Taylor and colleagues. However, our main point here is to show the center of PIGS activity. In this condition, showing an unthresholded activity map doesn’t have any advantage over the current maps. Considering these points, we decided not to change the figures.
(f) Coherent versus incoherent scenes - there are many differences between the coherent and incoherent scenes. Arguing that it must be ego-motion seems a little premature without further investigation. Activity anterior to OPA has been associated with the construction of an internal representation of a spatial environment (Steel et al., 2023, The Journal of Neuroscience). Could it be that this is the key effect, not really the ego-motion?
Response: In this revision, we discussed the study by Steel et al., 2021 and 2023 in section 4.3.
Reviewer #3 (Recommendations For The Authors):
Overall, I think this is already an excellent contribution. The suggestions I have are minor and may help with the clarity of the results.
(1) My main request of the authors would be to provide more points of reference in some of the figures with cortical maps. In many cases, the authors use arrows to point to the locations of activations of interest. However, the arrows in adjacent figures are often not placed in exactly the same places on maps that are meant to be compared. It would very much help the viewer to compare activations if the arrows pointing to activations or regions of interest were placed in identical locations for the same brains appearing in different sub-panels (e.g. in panels A and B of Figure 1). The underlying folds of the cortical surface provide some points of reference, but these are often occluded to different extents by data in figures that are meant to be compared.
Response: To address the reviewer’s concern, we regenerated Figure 8 (Figure 7 in the previous submission) and we tried to put arrowheads in identical locations, as much as possible. Especially for PIGS, this point was also considered in Figures 2 and 3.
(2) Outlines (such as those in Figure 5) are also very useful, and I would encourage broader use of them in other figures (e.g. Figures 7, 10, and 12). Figures 10 and 12 are on the fsaverage surface, so the same outlines could be used for them as for Figure 5.
To be clear, it's possible to apprehend the results with the figures as they are, but I think a few small changes could help a lot.
Response: In this revision, we added outlines to Figures 11 and 13 (Figure 10 and 12 in the previous submission). We did not add the outline to Figure 8 because it made it hard to see PIGS. Rather we used arrows (see the previous comment).
Other minor points:
In the method for Experiment 4, the authors write: "Other details of the experiment were similar to those in Experiment 1.". Similar or the same? The authors should clarify this statement, e.g. "the number of images per block, the number of blocks, the number of runs were the same as Experiment 1" - with any differences noted.
Response: This point is now addressed in the Methods section.
In Figure 8, it would be better to have the panel labels (A, B, C, D) in the upper left of each panel rather than the lower left.
Response: We tried to keep the panels arrangement consistent across the figures. That is why letters are positioned like this.
A final gentle suggestion: pycortex (http://github.com/gallantlab/pycortex) provides a means to visualize the flattened fsaveage surface with outlines for localized regions of interest and overlaid lines for major sulci. Though it is by no means necessary for publication, It would be lovely to see these results on that surface, which is freely available and downloadable via a pycortex command (surface here: https://figshare.com/articles/dataset/fsaverage_subject_for_pycortex/9916166)
Response: We thank the reviewer for bringing pycortex to our attention. We will consider using it in our future studies.
-
-
-
Follow the steps below or see the full source code.
You can find the full source code [here].
Tags
Annotators
URL
-
-
-
If you see this result, you’ve set up everything correctly and are ready to begin learning how to read data in Lesson 3.
I think it would be super useful to explain "Running a code selection" as well on this page (with screenshots). Otherwise, they'll have create a new file for each query.
-
-
underactuated.mit.edu underactuated.mit.edu
-
we can apply this same algorithm to any number of dynamical systems virtually without modification
I am a little confused as to how DP is able to get the optimal policy for the double integrator and the pendulum problem. Is it trivial to code this (value iteration) to work on the double integrator and pendulum problem from scratch? Grid world is easy because the mapping from actions to states is easy. But for the integrator and pendulum problem, since you discretize the input, how do we know which state is the next state if we use the inputs in range linspace(-1, 1,9) (integrator prob.). I looked into the actual FittedValueIteration function, it seems there is some barycentric interpolation taking place. Is that the only way to obtain this? I was thinking of implementing it from scratch but actually got a little confused as to how to achieve the action-state mapping to access the correct value for the next state. Am I missing something very basic? I apologize that the question is too long and please let me know if it is not possible to answer it here or where I could look for an answer. Thank you for the lectures!!
-
-
www.biorxiv.org www.biorxiv.org
-
Reviewer #2 (Public Review):
Summary:
The goal of this project was to test the hypothesis that a common neuroanatomic substrate in the left inferior parietal lobule (area PF) underlies reasoning about the physical properties of actions and objects. Four functional MRI (fMRI) experiments were created to test this hypothesis. Group contrast maps were then obtained for each task, and overlap among the tasks was computed at the voxel level. The principal finding is that the left PF exhibited differentially greater BOLD response in tasks requiring participants to reason about the physical properties of actions and objects (referred to as technical reasoning). In contrast, there was no differential BOLD response in the left PF when participants engaged in fMRI variant of the Raven's progressive matrices to assess fluid cognition.
Strengths:
This is a well-written manuscript that builds from extensive prior work from this group mapping the brain areas and cognitive mechanisms underlying object manipulation, technical reasoning, and problem-solving. Major strengths of this manuscript include the use of control conditions to demonstrate there are differentially greater BOLD responses in area PF over and above the baseline condition of each task. Another strength is the demonstration that area PF is not responsive in tasks assessing fluid cognition - e.g., it may just be that PF responds to a greater extent in a harder condition relative to an easy condition of a task. The analysis of data from Task 3 rules out this alternative interpretation. The methods and analysis are sufficiently written for others to replicate the study, and the materials and code for data analysis are publicly available.
Weaknesses:
The first weakness is that the conclusions of the manuscript rely on there being overlap among group-level contrast maps presented in Figure 2. The problem with this conclusion is that different participants engaged in different tasks. Never is an analysis performed to demonstrate that the PF region identified in e.g., participant 1 in Task 2 is the same PF region identified in Participant 1 in Task 4.
A second weakness is that there is a variance in accuracy between tasks that are not addressed. It is clear from the plots in the supplemental materials that some participants score below chance (~ 50%). This means that half (or more) of the fMRI trials of some participants are incorrect. The methods section does not mention how inaccurate trials were handled. Moreover, if 50% is chance, it suggests that some participants did not understand task instructions and were systematically selecting the incorrect item.
A third weakness is related to the fluid cognition task. In the fMRI task developed here, the participant must press a left or right button to select between 2 rows of 3 stimuli while only one of the 3 stimuli is the correct target. This means that within a 10-second window, the participant must identify the pattern in the 3x3 grid and then separately discriminate among 6 possible shapes to find the matching stimulus. This is a hard task that is qualitatively different from the other tasks in terms of the content being manipulated and the time constraints.
In sum, this is an interesting study that tests a neuro-cognitive model whereby the left PF forms a key node in a network of brain regions supporting technical reasoning for tool and non-tool-based tasks. Localizing area PF at the level of single participants and managing variance in accuracy is critically important before testing the proposed hypotheses.
-
-
www.biorxiv.org www.biorxiv.org
-
Reviewer #2 (Public Review):
Summary:
The manuscript presents a valuable investigation into the use of Fisher Kernels for extracting representations from temporal models of brain activity, with the aim of improving regression and classification applications. The authors provide solid evidence through extensive benchmarks and simulations that demonstrate the potential of Fisher Kernels to enhance the accuracy and robustness of regression and classification performance in the context of functional magnetic resonance imaging (fMRI) data. This is an important achievement for the neuroimaging community interested in predictive modeling from brain dynamics and, in particular, state-space models.
Strengths:
(1) The study's main contribution is the innovative application of Fisher Kernels to temporal brain activity models, which represents a valuable advancement in the field of human cognitive neuroimaging.
(2) The evidence presented is solid, supported by extensive benchmarks that showcase the method's effectiveness in various scenarios.
(3) Model inspection and simulations provide important insights into the nature of the signal picked up by the method, highlighting the importance of state rather than transition probabilities.
(4) The documentation and description of the methods are solid including sufficient mathematical details and availability of source code, ensuring that the study can be replicated and extended by other researchers.
Weaknesses:
(1) The generalizability of the findings is currently limited to the young and healthy population represented in the Human Connectome Project (HCP) dataset. The potential of the method for other populations and modalities remains to be investigated.
(2) The possibility of positivity bias in the HMM, due to the use of a population model before cross-validation, needs to be addressed to confirm the robustness of the results.
(3) The statistical significance testing might be compromised by incorrect assumptions about the independence between cross-validation distributions, which warrants further examination or clearer documentation.
(4) The inclusion of the R^2 score, sensitive to scale, would provide a more comprehensive understanding of the method's performance, as the Pearson correlation coefficient alone is not standard in machine learning and may not be sufficient (even if it is common practice in applied machine learning studies in human neuroimaging).
(5) The process for hyperparameter tuning is not clearly documented in the methods section, both for kernel methods and the elastic net.
(6) For the time-averaged benchmarks, a comparison with kernel methods using metrics defined on the Riemannian SPD manifold, such as employing the Frobenius norm of the logarithm map within a Gaussian kernel, would strengthen the analysis, cf. Jayasumana (https://arxiv.org/abs/1412.4172) Table 1, log-euclidean metric.
(7) A more nuanced and explicit discussion of the limitations, including the reliance on HCP data, lack of clinical focus, and the context of tasks for which performance is expected to be on the low end (e.g. cognitive scores), is crucial for framing the findings within the appropriate context.
(8) While further benchmarks could enhance the study, the authors should provide a critical appraisal of the current findings and outline directions for future research, considering the scope and budget constraints of the work.
-
-
mutabit.com mutabit.com
-
As I mentioned in this book's introduction, these include Data & Society, the AI Now Institute, and the Digital Equity Lab in New York City; the new Data Justice Lab in Cardiff; and the Public Data Lab.111 Coding Rights, led by hacker, lawyer, and feminist Joana Varon, works across Latin America to make complex issues of data and human rights much more accessible for broader publics, engage in policy debates, and help produce consent culture for the digital environment. They do this through projects like Chupadados (“the data sucker”).112 Others groups include Fair Algorithms, the Data Active group, the Center for Civic Media at MIT; the Digital Justice Lab, recently launched by Nasma Ahmed in Toronto; Building Consentful Tech, by the design studio And Also Too in Toronto; the Our Data Bodies Project; and the FemTechNet network.113 There is also a growing number of conferences and convenings dedicated to related themes; besides FAT*, 2018 saw the Data4BlackLives conference, the 2018 Data Justice Conference in Cardiff, and the AI and Inclusion conference in Rio de Janeiro, organized by the Berkman-Klein Center for Internet & Society, ITS Rio, and the Network of Centers; as well as the third design justice track at the Allied Media Conference in Detroit.114 Regardless of the design domain, design justice explicitly urges designers to adopt social justice values, to work against the unequal distribution of design's benefits and burdens, and to attempt to understand and counter white supremacy, cisheteropatriarchy, capitalism, ableism, and settler colonialism, or what Black feminist thought terms the matrix of domination. Design justice is interested in how to hard-code the liberatory values of intersectional feminism at every level of designed objects and systems, including the interface, the database, the algorithm, and sociotechnical practices “in the wild.” What's more, this approach is interested not only in designed objects and systems, but in all stages of design, from the framing and scoping of design problems (chapter 3) to designing and evaluating particular affordances (as we explored in this chapter) to the sites where we do design work (chapter 4). The next chapter (chapter 2) unpacks the implications of design justice for the question, “Who gets to be a designer?”
Esta cartografía, y las que están por hacer, sería un trabajo muy interesante para conectar los puntos en el campo, 1 Métodos de investigación a nivel doctoral. 2. cómo son sus metodologías de trabajo. 3 qué podemos aprender de estas. 4. Una cartografía posible nos puede ayudar a pensar el problema. Cuáles son los principios del feminismo negro interseccional que pueden aplicarse a la investigación: Ahmed, bell hooks.
https://chupadados.codingrights.org. https:// href="http://datasociety.net/">datasociety.net, https:// href="http://ainowinstitute.org/">ainowinstitute.org, https://www.newschool.edu/digital-equity-lab, https://datajusticelab.org, and https:// href="http://publicdatalab.org/">publicdatalab.org. https://www.fatml.org, https://datasociety.net, https://civic.mit.edu, https:// datajusticelab.org, http:// href="http://www.communitysolutionsva.org/files/Building_Consentful_Tech_zine.pdf">www.communitysolutionsva.org/files/Building_Consentful_Tech_zine.pdf, https://www.odbproject.org, and http://femtechnet.org/about/the-network. https:// href="http://alliedmedia.org/amc2018/design-justice-track">alliedmedia.org/amc2018/design-justice-track. formatted by Mar
-
-
hub.wsu.edu hub.wsu.edu
-
335-4831
This is in area code 509
-
-
www.cs.princeton.edu www.cs.princeton.edu
-
Its performance is not very different from the system versions of grep, which shows that the recursive technique is not too costly and that it's not worth trying to tune the code.
-
The occurrence of a do-while instead of a while should always raise a question: why isn't the loop termination condition being tested at the beginning
-
-
theconversation.com theconversation.com
-
Le télétravail a été introduit dans le Code du travail à l’article 1222-9 par la loi du 23 mars 2012 (l’article 46 de la loi dite Warsmann définit le télétravail). Cette loi prévoit des mesures de protection des données et de préservation de la vie privée. L’Accord National Interprofessionnel du 19 juillet 2005 dans son article premier donne du télétravail la définition suivante :
L'auteur s'appuie pour la seconde fois sur la dimension légale pour asseoir sa future argumentation. Il montre ainsi les récentes évolutions légales en lien avec la question du télétravail, définissant ainsi son contour grâce à des données épistémiques.
-
-
social-media-ethics-automation.github.io social-media-ethics-automation.github.io
-
The reason few non-English programming languages exist is due to the network effect, which we mentioned last chapter. Once English became the standard language for programming, people who learn programming learn English (or enough to program with it). Attempts to create a non-English programming language face an uphill battle, since even those that know that language would still have to re-learn all their programming terms in the non-English language.
In my opinion, English is often the language of international business, and as software development is a global industry, English serves as a common ground for collaboration. English technical terms are widely adopted even in non-English speaking countries, so even non-native English speakers are familiar with the necessary technical English to code.
-
Now, since many people do speak other languages, you can often find comments, variable names, and even sometimes coding libraries which use non-English languages, but the core coding terms (e.g., for, if, etc.), are still almost always in English.
I am now taking a CS class so I understand. All the coding language we are learning right now is in english. I think the reason why almost no one is using other language to code is because even if you are a foreigner, when you first learn coding there aren't even an option you can choose. So even if there are other languages coding program that exist, it is still really unlikely for people to learn it again.
-
-
peerherholz.github.io peerherholz.github.io
-
Live Share Extension Pack
I can't find the "Live Share Extension Pack" in the visual studio code. Is there other substitution? Thanks.
-
-
social-media-ethics-automation.github.io social-media-ethics-automation.github.io
-
If there is an opportunity for even more profit by making worse movies, then that is what business leaders are obligated to do:
I think that this goes back to the unethical decisions that the entertainment industry enforces. I wonder what kind of moral code they have within the companies.
-
-
coolshell.cn coolshell.cn
-
Sharing Guideline Please follow the following sharing protocols Understand Sharing Sharing is the hard way to learn knowledge. The presenter gains the biggest advantages. not audience. 分享是学习知识的最难的方式。分享者获得的好处最最多的,而不是观众。 Sharing can open the knowledge door for the audience, but you have to walk to knowledge by yourself. 分享可以为听众打开知识的大门,但你能不能获得知识还要靠你自己。 Best Practices To perform a great sharing, please follow the below practices. Do not share a big topic, a small topic is better. A big topic could make the audience lose focus. Remember, Less is More! Sharing time less than 60 mins is the best. English language for slides is preferred. While prepare the sharing contents, it’s better to discuss with the senior people to help you to see the whole picture, understand the good side and bad side, know what you don’t know … etc. Strong Recommend Materials Outlines What’s the Problem? How to Solve the Problem? The Best Solution or Practice. The Mechanism, Key Techniques, and Source Code Pros/Cons References (Further reading) For example, if you want to sharing a topic about Docker. the following outlines would be good one: What’s the major problems need to solve. (Provision, Environment, Isolation etc.) The Alternative solutions. (Puppet/Chef/Ansible, VM, LXC etc.) The Best Solution – Docker. Why? Docker’s key techniques – image, cgroup, union fs, namespace… Docker’s Pros/Cons Further reading list.

-
-
www.biorxiv.org www.biorxiv.org
-
Note: This response was posted by the corresponding author to Review Commons. The content has not been altered except for formatting.
Learn more at Review Commons
Reply to the reviewers
*Reviewer #1 (Evidence, reproducibility and clarity (Required)):
Deletion of CerS4 in the entire mouse epidermis throughout development via the K14-Cre results in enlarged sebaceous glands and perturbed HFSC molecular phenotype. There is low or no expression of CD34, a known marker of the HFSCs along with apparent reduction of several other HFSC markers and acquisition of a more differentiated cell phenotype in these cells. Interestingly, skin and hair follicles seem to remain normal otherwise up to advanced age, though this contradicts the notion that HFSC were indeed affected at the functional level. The data does not demonstrate 'gradual decline' in the HFSC compartment, as claimed by the authors, but rather seem to indicate that the adult HFSC compartment is not properly established in its molecular signatures. Organoid cultures document defects in HFSC, which included reduced proliferation in the CerS4 KO cells. Lipid composition in plasma membranes was also affected by the CerS4 KO. Associated with this, Wnt signal transduction is also affected according to experiments that enhance the strength of wnt signals via a specific small molecular agonist of the pathway. Finally, the authors discover a resemblance of the mouse KO immune-phenotype, with human atopic dermatitis. The study is likely of interest to a specialized readership in skin biology and dermatology and adds to previous studies on CerS4 in skin that erroneously placed its role in the sebaceous gland. [The authors here demonstrate that deletion of CerS4 in the sebaceous glands via SCD3-Cre led to no phenotype, contradicting the previous assessment that CerS4 is important in sebaceous glands.] The study would need to be corrected in a few of its interpretations regarding stem cells to better match the data, as indicated below. *
We thank the reviewer for the constructive comments that will help us to improve the manuscript. In particular, it is clear that we have not been sufficiently clear in the data presentation.
Firstly, contrary to what the reviewer states, the CerS4epi-/- mice have a very strong hair follicle phenotype that results in complete hair loss. Also the epidermis is not normal as an inflammatory phenotype develops later, after the hair follicle architecture and function has been disrupted. Thus, there are clear functional consequences to the hair follicle and epidermis that arise from the dysfunction of the HFSC compartment. We will edit the manuscript and add photodocumentation of the macroscopic phenotype to ensure clarity.
We fully agree with the reviewer that the initial phenotype is inability to establish the adult hair follicle stem cell niche, as shown by the single cell sequencing data and as also stated in the manuscript title. We will further edit the manuscript to clarify this conclusion. Importantly, however, some hair follicle stem cells are generated but these become gradually depleted. So there is a dual phenotype: an inability to efficiently establish and maintain the hair follicle stem cell population. We will clarify this in the text.
Finally, we want to emphasize that the main finding of this manuscript is that hair follicle stem cells contain a unique lipid profile and perturbing this profile by deleting CerS4 leads to profound defects in stem cell fate regulation through Wnt. This is a completely new finding that has implications far beyond dermatology.
Major revisions: Fig1B - the data seems to simply shows that bulge cells express less or no CD34 and not that ' CerS4epi-/- mice showed reduced HFSC numbers'; the primary FACS data should be shown somewhere too.
Outer bulge hair follicle stem cells are defined as a population of cells that expresses CD34 and integrin-a6. The quantifications in Fig 1B show the quantitative FACS analyses of the size of this population and indicate less CD34+/integrin-a6+ cells in CerS4epi-/- epidermis. The mean fluorescence intensity of CD34 and integrin-a6 was not reduced in these CerS4epi-/- stem cells. This FACS analysis therefore allows the conclusion that there are less CD34+/integrin-a6+ cells in CerS4epi-/- epidermis. We will include the original FACS plot data to support this notion and the quantifications.
The conclusion that stemness is affected, and HFSCs lose their normal gene expression signature is at more convincing after looking at other HFSC markers down the road in the paper. However, in the absence of functional assays that would demonstrate stem cell function is lacking and seeing that hair follicles are maintained and grow in long-term, the notion that stem cells are lacking in these conditions is not supported by the data.
We appreciate that the reviewer finds the marker gene analysis convincing. To assay stem cell functionality, we have used the organoid assays (spheroid formation is classical, widely used assay for stemness). Using these functional assays we observe impaired self-renewal of stem cells (Fig. 3D), enhanced differentiation (Fig. 3I), and altered Wnt responsiveness (Fig. 5 E, F), all indicative of stem cell dysfunction and explaining the in vivo phenotypes of altered stem cell differentiation and inability to establish and maintain the stem cell population.
In the revised manuscript we will also include measurements of stem cell self-renewal in vivo using BrdU incorporation and provide more detailed description on the hair loss phenotype of the mice to further strengthen this conclusion.
*The conclusion after figure 2: "Collectively, these data indicate that CerS4-deficiency triggers ... ... gradual depletion of the quiescent HFSC compartment." There is no data showing gradual depletion of the quiescent HFSC compartment. We would need to see a gradual activation of HFSCs with over proliferation to conclude this. There is some data albeit not always convincing (see NFAC1 staining in Fig. 5C) indicating loss of markers associated with quiescence but there is no data indicating 'gradual' loss of markers. *
We agree with the reviewer that showing gradual activation of the HFSCs in vivo is important to conclude loss of quiescence. We will include in situ stainings of in vivo BrdU labeling and quantify proliferation in the hair follicle bulge stem cell region. Preliminary data of P47 mice already shows a clear increase in BrdU+ cell in the stem cell compartment in CerS4epi-/- skin . Further analysis at P21 will be carried out during the revision.
*Minor revisions: *
- Figure 1C legend - please spell out what are the abbreviations for the different subpopulations; please show these populations as % as opposed to absolute numbers. *
We will edit the Figure 1C legend for clarity and express the populations as %.
*
*
*Figure 1D - please make it clear in the cartoon what the different sub-populations listed are; *
We will edit the cartoon for clarity.
Is OB1 a CD34- HFSC population?
The outer bulge 1 (OB1) is a population of cells that expresses hair follicle stem cell markers, including CD34. We will clarify this in the legend.
Fig. 3B - the colors of the legend do not match the colors in the data so it is confusing as to which one is which!
We will alter the colors to match the data and thank the reviewer for pointing this out.
Fig 5C - the differences in NFATc1 are not visible in the images shown
We apologize for the suboptimal quality of these images and will replace them with higher resolution images to more clearly demonstrate the difference.
*
Reviewer #2 (Evidence, reproducibility and clarity (Required)):
The manuscript authored by Peters et al. titled "Sphingolipid metabolism orchestrates the establishment of the adult hair follicle stem cell niche to control skin homeostasis" elucidates the critical role of ceramide synthase 4 (CerS4) in the epidermal stem cell niche, particularly in regulating hair follicle bulge stem cells (HFSCs). Using epidermal specific CerS4 knockout mice as an in vivo model and hair follicle organoid culture as an ex vivo model, the authors conducted a comprehensive analysis, which includes cutting edge approaches such as scRNA-seq, proteomics, and lipidomics. The results highlight CerS4's function in the establishment/maintenance of the HFSC niche, as absence of CerS4 changes HFSCs' number and differentiation state. Potential underlying mechanisms identified include altered membrane lipid profiles and Wnt signaling responsiveness. Possible link to a chronic inflammatory skin disease, atopic dermatitis, is also implicated. The data presented are generally of high quality, and the work is significant as it uncovers a new regulator of HFSC fate with mechanistic connection to lipid metabolism. *
We thank the reviewer for the positive assessment of our work and finding it to be of high quality and significance. We further appreciate the constructive comments that will further help us to improve the manuscript.
*However, some issues were identified, most of them having to do with in vivo characterization and data interpretation:
*
*Major: 1. The in vivo HFSC phenotype can be better characterized. "Collective, these data show that CerS4 in HFSCs is essential to establish the adult stem cell compartment and to assure lineage fidelity." - this statement premature based on order of the data shown. Also the trajectory difference shown in Figure 2A is not striking. Subclustering out the relative cell subsets and redo the analysis might help to tease out the difference. Additional experiments such as lineage tracing would be useful to support the notion that there is lineage fidelity issue in the mutant - though it is understood that this is quite involved and may lie outside the scope of the current study. Are bulge cells in the mutant proliferative? - the authors should consider in vivo Edu labelling experiment or the like to assess the quiescence/proliferation of the bulge cells. Finally, analyzing hair follicles at earlier stages might help to clarify when and where the bulge and sebaceous gland changes start - is possible that aberrant divergence of bulge/sebaceous fates occur prior to the establishment of a stable bulge fate? *
We thank the reviewer for suggesting additional analyses of the single cell sequencing data. We have performed subclustering of the relevant populations for the trajectory analyses to more clearly demonstrate the altered lineage trajectories. We will include these new analyses in the manuscript. Importantly, the in vitro organoids show abnormal differentiation (Fig. 3H, 3I, Supplementary Fig. 2G), closely resembling the in vivo phenotypes, thereby strengthening the conclusion of cell-autonomously altered lineage trajectories of the hair follicle stem cells.
We will further preform in vivo BrdU labeling as suggested. These experiments have already been initiated and preliminary data show increased proliferation in the bulge stem cell region of CerS4epi-/- mice in older mice (P47). The data will be included to emphasize long term loss of quiescence in the stem cell compartment in CerS4epi-/- mice.
Understanding the early development of bulge and sebaceous fates is indeed an interesting question. This will be addressed by detailed analyses of stem cell fate at early stages (P17-P21) using key markers of stem cell state and sebaceous linages (CD34, Krt15, Lhx2, Nfatc1, SCD1 and FASN).
Finally, we have initiated lineage tracing experiments using the stem cell-specific Lgr5-Cre to conclusively demonstrate that Cers4-deletion leads to altered routing of hair follicle stem cells into upper hair follicle and sebaceous gland fates. This notion is supported by the preliminary analyses of these experiments. We will finalize these analyses and include them in the manuscript.
*2. The exclusion of IFE contribution is not backed up by data. Figure 6D - model emphasizes HFSC involvement in atopic dermatitis, but this could be due to epidermal barrier defect. Barrier defect could already be present even though IFE morphology appears normal. Maybe TEWL is measured at the time of analysis and shows no change - if so, this data should be included. HFSC changes might contribute but the involvement of IFE cannot be excluded. The conclusion that "CerS4 expression was restricted to the hair follicle" is not supported by data. IFE expression is apparent in Figure S1C. Along this line, there is also an apparent expansion of IFE basal II in the mutant (Figure 1C). *
We acknowledge that we have not been clear enough with the evidence that allowed us to exclude the involvement of an IFE-mediated barrier defect in the early skin inflammation phenotype. To address a potential barrier defect early on, we have performed careful analysis of TEWL. In Peters at al., 2020 we demonstrate no changes in TEWL at P0, a reduced TEWL at P21 and an increased TEWL in adult CerS4epi-/- mice starting only at P33. The reduction of the TEWL in adolescent CerS4epi-/- mice (P21) is likely linked to an increased production of sebaceous lipids lubricating the skin surface at this time point (Peters et al., 2020). Thus, defects in the hair follicle stem cell compartment, present at adolescence (P21) arise prior to defects in the adult (P33) IFE barrier function. We will clarify this in the manuscript.
Cers4 expression is overall low in skin, as is typical for enzymes. In situ stainings of Cers4mRNA (Fig.S1C) indeed show a sparse signal also in the IFE. This signal is also detected in CerS4-/- sections, although the KO skin cannot be conclusively used to control background as these mice were generated by deletion of exon 3 only, and Cers4 RNAscope probes might detect remnant Cers4 RNA in these mice. Importantly, our data on FACS sorted basal cells of the IFE shows no substantial Cers4 mRNA expression in IFE progenitors (Fig. S1D) and no mRNA is detected in the IFE in the single cell sequencing. Thus, while we cannot fully exclude low levels of CerS4 expression in the IFE, the levels are substantially lower than in the HFSC and SG compartments, and the phenotype, including the slight expansion of the IFE basal II population, is very minor compared to the hair follicle phenotype. However, to avoid overinterpreting our data, we will carefully edit the conclusions to be less strong on the involvement of the IFE. Furthermore, we will perform hair follicle stem cell lineage tracing experiments as outlined in resspose to the previous point to strengthen the conclusion on the hair follicle stem cell-autonomous phenotype.
3. Figure 1 - single cell analysis was done using only 2 pairs of mice, and data in E lack statistical assessment. At the very least, data for individual pairs should be shown in supplemental data to ensure that changes are consistent in both mutant mice rather than being dominated by dramatic alteration in only 1 mutant mouse.
We naturally have rigorously analyzed the replicates to ensure that the phenotype is consistently present in both. We will include the separate analysis of the mice to document this and include statistical analysis.
Minor: 1. CerS4SCD3-/+ nomenclature is mis-leading.
We will edit this for clarity
-
- Figure 2- "Furthermore, we observed expansion of the inner bulge identity marker Krt6 protein expression into outer bulge stem cells and along the infundibulum in CerS4epi-/- hair follicles, whereas in control mice Krt6 was restricted to the inner bulge (Fig. 2C)." - Krt6 staining is presented in Fig 2D, not 2C. *
We thank the reviewer for pointing out this mistake that will correct.
-
- Figure 3C - size of the organoids should be quantified with statistics. The images shown do not support the statement that "Strikingly, CerS4epi-/- organoids showed altered morphology characterized by smaller size and loss of cohesion of peripheral cells from the organoid clusters (Fig. 3C), ...".*
We will include quantifications.
-
- Section titled "CerS4 regulates HFSC differentiation in a stem cell autonomous manner": "CD34- integrin- a6+ cells, which based on extensive transcriptome and marker expression analyses represent a mixture of HFSCs, hair follicle outer root sheath (ORS) cells and inner bulge cells (collectively termed non-HFSCs)." - shouldn't the CD34- integrin- a6+ population also contain IFE stem/progenitor cells? Are hair follicles micro-dissected out for FACS? *
The hair follicles are not micro-dissected out for FACS, and the entire basal cell population is initially isolated. However the organoid culture conditions speficically promote the expansion of the hair follicle linage, whereas cells of the IFE are not expanded and long term maintained as extensively documented in previous publications using this organoid system (see for example Kim et al., Cell Metabolism 2020; Chacon-Martinez EMBOJ 2016).
*5. Figure 5D - please provide the working concentration of Chir99021. *
We will provide the working concentration.
*6. Figure 5F - explain what arrows mean in legends. *
We will define the arrows.
-
- Figure 6A - no significant changes in Th2 and ILC2 were observed at a 95% confidence interval. Increasing mouse number will help to increase statistical power*.
We agree with the reviewer and acknowledge that this experiment was unfortunately underpowered. We will repeat it with a larger cohort.
-
- Additional Wnt target genes such as Axin 2 should be looked at.*
We thank the reviewer for this suggestion, we will include analyses of additional Wnt target genes.
-
- The increased BMP signaling and decreased Nfatc1 expression are seemingly contradictory.*
We apologize for the lack of clarity here. Single cell sequencing showed increased BMP-signaling of outer bulge cell cells to inner bulge cells on mRNA level (Figure S4A). No alteration in BMP signaling was detected within the outer bulge stem cell compartment (Figure 5A). Nfatc1 protein expression was analyzed in the upper bugle (Figure 5C). The data indicate no differential gene expression of ligand receptor pairs mediating BMP-signaling within the outer bulge. A decrease in Nfact1 protein expression (Fig. 5C) together with an increased proliferation (see above) and loss of label retention (Peters et al., 2015) indicates loss of quiescence in this compartment. This data does not contradict an increased BMP signaling in the inner bulge (Figure S4A). An increase in BMP signaling in the inner bulge is in line with reduced inner bulge cell cluster detected in CerS4epi-/- skin via single cell sequencing, likely contributing to the hair loss observed. We will edit this paragraph to make this more clear.
-
- Paragraph starting with "It is interesting to note that ceramide availability was shown to regulate Wnt signaling in Drosophila through strong effects on recycling endocytosis of the receptors (Pepperl et al., 2013)." Is redundant in the manuscript.*
We apologize for the accidental duplication of this paragraph and thank the reviewer for noticing this mistake.
*
Reviewer #3 (Evidence, reproducibility and clarity (Required)):
The authors created CerS4 mutant mice to test the role of sphingolipids in hair follicle stem cells (HFSCs) and the hair cycle. This work extends previous studies that show that loss of this enzyme leads to defects in the hair cycle and eventually hair loss. In this study the authors look early on in the course of the deletion in an attempt to understand why loss of this enzyme leads to the phenotype described previously. They use single cell profiling, proteomics, and in situ imaging to pinpoint issues in the stem cell niche that drive phenotypes and propose novel interactions between sphingolipid metabolism, Wnt signaling, and inflammation in regulation of HFSC homeostasis. The data are nicely presented, and the text is well written. The conclusions are clearly defined.*
We thank the reviewer for the positive assessment of our work and finding it well written and presented. We further appreciate the constructive comments that will further help us to improve the manuscript.
*While the data are clearly presented, there are numerous issues that are confusing to this reviewer. In addition, some of the phenotypes described are subtle, and thus do not make a convincing case.
1, In figure 1C, the cell proportion analysis suggests there are no OBII or SG in WT. I am not sure how this could be possible. In addition, there appears to be almost no sebaceous cells in either, but the mutant supposedly has much larger sebaceous glands (in Fig 2). In Fig S1I, there is no change in bulge cells? In Fig 1B, there is less HFSCs in the mutant than in the WT, but in 1C, there is more OBI in the mutant. The results in Fig 1B and C are confusing. Also, the schematic in Fig 1 is hard to read, the authors should color code the text with the image.*
It is important to emphasize that the single cell RNA sequencing was carried out at P19 when the bulge stem cell compartment only starts to be established. This explains why only few bulge stem cells are detected at this point. Nevertheless, the OBII and SG cluster is visible also in the wt in Figure 1D. We will include subclustering of the relevant subpopulations to make these populations more clearly visible also in the wt. We will also edit the labels for clarity.
Mature sebocytes are very large cells, and inherent to the single cell sequencing workflows these large cells are excluded from the sequencing libraries. Importantly, we do not detect a change in bulge stem cells in a mouse line in which CerS4 was specifically deleted only in sebocytes (Figure S1I). This analysis was carried out to exclude a sebocyte intrinsic effect on the hair follicle stem cell state and fate. The data does not contradict Fig. 1, as data in Fig. 1 was generated using a different mouse line in which CerS4 is deleted in the entire epidermal stem cell population using K14Cre. We will edit the manuscript to make this more clear.
Data presented in Fig 1B and C focus on two different aspects. Fig 1B shows the inefficient establishment and maintenance of CD34+/integrin-a6+ bulge hair follicle stem cells. The quantification is based on FACS analyses of cells expressing these cell surface molecules/stem cell markers. Fig 1C shows the quantification of the various cell states based on single cell RNA expression and subsequent clustering of the control and CerS4epi-/- epidermal cells together. The “outer bulge” cluster was annotated based on these cells expressing hair follicle stem cell markers. While the CerS4epi-/- epidermis shows increased number of cells in this cluster, the expression of all key stem cell genes (CD34, Sox9, Krt15, Lhx2) is reduced in CerS4epi-/- outer bulge 1 compartment compared to control. Thus, while this “outer bulge” population is expanded in the KO, the stem cell properties of this population are clearly attenuated, as defined by decreased expression of key stem cell transcription factors and increased expression of differentiation genes. We will clarify this in the revised version of the manuscript and also rename this cluster “outer bulge-like” to highlight that these cells are not necessarily bona fide stem cells and might not express high levels of CD34+/integrin-a6+ protein.
*2, In Figure 5, the signaling chart shows a strong upregulation of non-canonical Wnt signaling in the mutant bulge. Canonical Wnt signaling appears to be unchanged between wt and ko. Thus, it is not clear why the authors came to the conclusion that Wnt signaling is induced in the mutant. They further show expression of Lef1 and Nfatc1, but these are not typical markers used to denote canonical wnt activation, as implied. In fact, the data in Fig S4B suggest the induction of Lef1 and Tcf4 is actually very subtle. Instead, the authors should use nuclear b-catenin or transcriptional targets such as Axin or CyclinD. The authors should in fact explore the observation of Wnt5, as that appears to be the most dramatic change. In addition, the authors should use an ontological analysis with the single cell data from the tissue in the same manner that they did for organoids to take another look at molecular consequences of loss of CerS4. *
We agree with the reviewer that further analysis of canonical and non-canonical Wnt signaling will strengthen this conclusion. In our experience, nuclear b-catenin is very difficult to detect in the skin even when Wnt is highly active, but we will investigate Axin2 and CyclinD1 expression. We will also investigate Wnt5a signaling by analyzing its expression as well as its downstream target genes. We will further perform additional ontological analyses from the single cell sequencing data to strengthen the conclusions on the signaling alterations.
* 3, The authors suggest that much of the phenotype is due to inflammation. In Fig 6A, they showed analysis of CD45 cells in the skin. However, the only change was a very subtle change in Th2 cells, while no other CD45+ cells were altered.*
We agree with the reviewer and acknowledge that this experiment was unfortunately underpowered. We will repeat these analyses with a larger cohort.
4, The authors showed upregulation of Immune response in Fig 6C, but then in Fig S2, the genes downregulated are also related to immune response...how do the authors reconcile this?
We apologize for this confusion. Importantly, keratinocyte-intrinsic downregulation of homeostatic immune modulating activity is a key driver of allergic disorders, like atopic dermatitis. This barrier intrinsic immune modulation is distinct from immune cell-mediated inflammation. There is a strong overlap of genes constituting the term “Inflammatory abnormality of the skin” (Human phenotype ontology terms) Fig 6C and “Immune system process” (GOBP terms) Fig S2F. To name some, i.e. Adam-, ALOX-, ASXL- family members are annotated by both terms. Mutations in these genes are known to cause skin diseases associated with immune dysregulation but are likewise known to regulate immune responses.
Data in Figure 6C shows enrichment of this term from both up and downregulated proteins in the CerS4epi-/- condition compared to control, indicating that proteins involved in “Inflammatory abnormality of the skin” are dysregulated in CerS4epi-/- organoids. Data in Figure S2F shows the downregulation of these proteins in CerS4epi-/- organoids compared to control. We will clarify this in the text and figure legends.
5, The author propose that the phenotype in CerS4 null mice is due to disruption of the stem cell Niche. However, the authors have not shown evidence for such an effect through any in situ analysis. The single cell approaches are valuable, but in that case the niche is dissociated. The organoid work is also nice, but not exactly a stem cell niche either. The authors should instead test their hypothesis through an in situ analysis.
We have used the term niche to describe the cellular interactions between stem cells and the other niche resident cells such as the Krt6+ inner bulge cells that have been analyzed here. We will edit the conclusions for clarity. We will further include additional immunofluorescence analyses of the bulge compartment in situ, as suggested (including markers for quiescence and activation).
-
Note: This preprint has been reviewed by subject experts for Review Commons. Content has not been altered except for formatting.
Learn more at Review Commons
Referee #3
Evidence, reproducibility and clarity
The authors created CerS4 mutant mice to test the role of sphingolipids in hair follicle stem cells (HFSCs) and the hair cycle. This work extends previous studies that show that loss of this enzyme leads to defects in the hair cycle and eventually hair loss. In this study the authors look early on in the course of the deletion in an attempt to understand why loss of this enzyme leads to the phenotype described previously. They use single cell profiling, proteomics, and in situ imaging to pinpoint issues in the stem cell niche that drive phenotypes and propose novel interactions between sphingolipid metabolism, Wnt signaling, and inflammation in regulation of HFSC homeostasis. The data are nicely presented, and the text is well written. The conclusions are clearly defined.
While the data are clearly presented, there are numerous issues that are confusing to this reviewer. In addition, some of the phenotypes described are subtle, and thus do not make a convincing case.
- In figure 1C, the cell proportion analysis suggests there are no OBII or SG in WT. I am not sure how this could be possible. In addition, there appears to be almost no sebaceous cells in either, but the mutant supposedly has much larger sebaceous glands (in Fig 2). In Fig S1I, there is no change in bulge cells? In Fig 1B, there is less HFSCs in the mutant than in the WT, but in 1C, there is more OBI in the mutant. The results in Fig 1B and C are confusing. Also, the schematic in Fig 1 is hard to read, the authors should color code the text with the image.
- In Figure 5, the signaling chart shows a strong upregulation of non-canonical Wnt signaling in the mutant bulge. Canonical Wnt signaling appears to be unchanged between wt and ko. Thus, it is not clear why the authors came to the conclusion that Wnt signaling is induced in the mutant. They further show expression of Lef1 and Nfatc1, but these are not typical markers used to denote canonical wnt activation, as implied. In fact, the data in Fig S4B suggest the induction of Lef1 and Tcf4 is actually very subtle. Instead, the authors should use nuclear b-catenin or transcriptional targets such as Axin or CyclinD. The authors should in fact explore the observation of Wnt5, as that appears to be the most dramatic change. In addition, the authors should use an ontological analysis with the single cell data from the tissue in the same manner that they did for organoids to take another look at molecular consequences of loss of CerS4.
- The authors suggest that much of the phenotype is due to inflammation. In Fig 6A, they showed analysis of CD45 cells in the skin. However, the only change was a very subtle change in Th2 cells, while no other CD45+ cells were altered.
- The authors showed upregulation of Immune response in Fig 6C, but then in Fig S2, the genes downregulated are also related to immune response...how do the authors reconcile this?
- The author propose that the phenotype in CerS4 null mice is due to disruption of the stem cell Niche. However, the authors have not shown evidence for such an effect through any in situ analysis. The single cell approaches are valuable, but in that case the niche is dissociated. The organoid work is also nice, but not exactly a stem cell niche either. The authors should instead test their hypothesis through an in situ analysis.
Significance
The authors created CerS4 mutant mice to test the role of sphingolipids in hair follicle stem cells (HFSCs) and the hair cycle. This work extends previous studies that show that loss of this enzyme leads to defects in the hair cycle and eventually hair loss. In this study the authors look early on in the course of the deletion in an attempt to understand why loss of this enzyme leads to the phenotype described previously. They use single cell profiling, proteomics, and in situ imaging to pinpoint issues in the stem cell niche that drive phenotypes and propose novel interactions between sphingolipid metabolism, Wnt signaling, and inflammation in regulation of HFSC homeostasis. The data are nicely presented, and the text is well written. The conclusions are clearly defined.
-
-
localhost:4743 localhost:4743
-
h executable code.
So what do you think of this
-
-
engl490spr24.commons.gc.cuny.edu engl490spr24.commons.gc.cuny.edu
-
reading and writing were equally class privileges: Rhetoric, the great literary code of that time, taught writing (even if what was ordinarily produced were discourses
Reading and writing has been proven to increase intelligence and has become something everyone must learn to do.
-
-
media.dltj.org media.dltj.org
-
Actually, ChatGPT is INCREDIBLY Useful (15 Surprising Examples) by ThioJoe on YouTube, 8-Feb-2024
- 0:00 - Intro
- 0:28 - An Important Point
- 1:26 - What If It's Wrong?
- 1:54 - Explain Command Line Parameters
- 2:36 - Ask What Command to Use
- 3:04 - Parse Unformatted Data
- 4:54 - Use As A Reverse Dictionary
- 6:16 - Finding Hard-To-Search Information
- 7:48 - Finding TV Show Episodes
- 8:20 - A Quick Note
- 8:37 - Multi-Language Translations
- 9:21 - Figuring Out the Correct Software Version
- 9:58 - Adding Code Comments
- 10:18 - Adding Debug Print Statements
- 10:42 - Calculate Subscription Break-Even
- 11:40 - Programmatic Data Processing
-
-
www.portlandoregon.gov www.portlandoregon.gov
-
victim
Of vandalism or city code? Either way, good news for a graffiti removal business.
-
-
mutabit.com mutabit.com
-
Bloques For this part a Basic understanding of Basic Types is recommended. If you have not done that part yet, please go to that subtree and run the code there Ahora hablemos de los bloques. Piensa en los bloques como una manera de 'congelar' código que luego podremos ejecutar por demanda, cuando queramos y/o las condiciones para su ejecución se cumplan. Los bloques son métodos anónimos, lo cual quiere decir que no es necesario ponerles nombres para invocarlos (como ocurre con las operaciones sobre objetos que hemos visto hasta el momento) y pueden pueden ser almacenados en variables. Los bloques están delimitados por paréntesis cuadrados: []
-
Métodos anónimos: Los bloques no tienen nombres asociados y pueden ser creados y utilizados sobre la marcha sin necesidad de definir un nombre específico para ellos.
-
Almacenamiento en variables: Los bloques pueden ser asignados a variables y pasados como argumentos a otros métodos o bloques.
-
Delimitados por paréntesis cuadrados: Los bloques en Pharo están delimitados por paréntesis cuadrados [ ].
-
Captura de contexto: Los bloques pueden capturar variables definidas en el contexto en el que se crean, lo que les permite acceder y manipular esos valores
-
-
La concatenación de String usa el operador coma: ['PharoTutorial', ' is cool']. "versión cambiada" 'Pharo tutorial ', ' is cool', ' when i active the code '
La concatenación de cadenas (strings) es el proceso de unir dos o más cadenas para formar una sola cadena más larga y se realiza utilizando el operador coma
Ejemplo: 'Pharo tutorial ', ' is cool', ' when i active the code '

-
-
yarnpkg.com yarnpkg.com
-
when projects want to keep strict boundaries within their code and avoid becoming an entangled monolith. This is for example the case for Yarn itself, or many enterprise codebases.
-
-
-
By jumping into unfamiliar areas of code, even if you do not "solve" the bug, you can learn new areas of the code, tricks for getting up to speed quickly, and debugging techniques.
Building a mental model of the codebase, as Jennifer Moore says over at Jennifer++:
The fundamental task of software development is not writing out the syntax that will execute a program. The task is to build a mental model of that complex system, make sense of it, and manage it over time.
-
Thinking about how you will observe whether things are working correctly or not ahead of time can also have a big impact on the quality of the code you write.
YES. This feel similar to the way that TDD can also improve the code that you write, but with a broader/more comprehensive outlook.
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
Reviewer #1 (Public Review):
Major Concerns:
(1) An important point that the authors should clarify in this study is whether mice are detecting qualitative or quantitative differences between fresh and old cat saliva. Do the environmental conditions in which the old saliva was maintained cause degradation of Fel d 4, the main protein known for inducing a defensive response in rodents? (see Papes et al, 2010 again). If that is the case, one would expect that a lower concentration of Fel d 4 in the old saliva after protein degradation would result in reduced antipredator responses. Alternatively, if the authors believe that different proteins that are absent in the old saliva are contributing to the increased defensive responses observed with the fresh saliva, further protein quantification experiments should be performed. An important experiment to differentiate qualitative versus quantitative differences between the two types of saliva would be diluting the fresh saliva to verify if the amount of protein, rather than the type of protein, is the main factor regulating the behavioral differences.
We thank the reviewer for their important suggestions. We agree that both the quality and quantity of molecular components in saliva undergo changes after the saliva is kept at room temperature for 4 hours. Our findings indicate that mice detect these changes through the VNO and adjust their defensive response patterns accordingly. For instance, freezing behavior is reduced in response to 4-hour-old saliva compared to fresh saliva. On the other hand, the duration of interaction with saliva (investigation behavior) remains low, and the stress hormone ACTH level is upregulated in both cases. A future study ought to identify the specific molecules—most likely proteins or peptides—in cat saliva responsible for these distinct defensive responses in mice. While Fel d 4 stands as one of the potential candidates as it has been shown to induce a form of defensive behavior in mice (Papes et al., 2010), there exists a possibility of a different molecule or a combination of multiple molecules playing a role. Once the molecules are identified, it is imperative to investigate how their quantity and quality change over time and how these factors correlate with freezing behavior in mice. Such an exploration will provide answers to this ethologically significant question raised by the reviewer. We added a paragraph in Discussion under the “The VNO as the sensor of predator cues that induce fear-related behavior” section to clarify this.
(2) The authors claim that fresh saliva is recognized as an immediate danger by rodents, whereas old saliva is recognized as a trace of danger. However, the study lacks empirical tests to support this interpretation. With the current experimental tests, the behavioral differences between animals exposed to fresh vs. old saliva could be uniquely due to the reduced amount of the exact same protein (e.g., Fel d 4) in the two samples of saliva.
As mentioned in response to comment 1, we agree with the alterations in both the quality and quantity of molecules within saliva after 4 hours. What we would like to emphasize in our current study is that mice detect these time-dependent changes through the VNO and subsequently adjust their defensive response patterns. Identifying the specific molecules responsible for inducing behavioral changes and investigating their time-dependent alterations is crucial in the next step. We added a paragraph in the Discussion under the 'The VNO as the sensor of predator cues that induce fear-related behavior' section to clarify this.
(3) In Figure 4H, the authors state that there were no significant differences in the number of cFos-positive cells between the two saliva-exposed groups. However, this result disagrees with the next result section showing that fresh and old saliva differentially activate the VMH. It is unclear why cFos quantification and behavioral correlations were not performed in other upstream areas that connect the VNO to the VMH (e.g., BNST, MeA, and PMCo). That would provide a better understanding of how brain activity correlates with the different types of behaviors reported with the fresh vs. old saliva.
We greatly appreciate this valuable advice. We added c-Fos immunoreactivity (IR) data in the BNST, MeApv, and PAG, together with the data for VMH as shown in new Figure 4G-J. Upon exposure to both fresh and old saliva, we observed an upregulation trend of cFos in the MeApv, VMH, and dPAG, but not in the BNST, compared to the control stimulus.
Moreover, we conducted correlation analyses between the numbers of cFos-positive neurons and the duration of freezing behavior in those neural substrates, which have been added to new Figure 5. The numbers of cFos-IR signals in neurons in the BNST and dPAG did not correlate with the duration of freezing behavior in any of the exposure groups (Figure 5C, F). However, in addition to a significant positive correlation in the VMH for the fresh saliva-exposed group (R2 = 0.5708, 95% CI [-0.1449, 0.9714], p = 0.0412) (Figure 5E), we observed a similar positive correlation trend in the MeApv (R2 = 0.3854, 95% CI [0.3845, 0.9525], p = 0.0942), although it was not statistically significant possibly due to low sample numbers (Figure 5D).
Based on these results, our current circuit model is as follows: different numbers of the VNO sensory neurons activated by fresh and old saliva result in differential excitation levels in mitral cells in the AOB. This, in turn, leads to the differential activation of targeting neural substrates, possibly MeApv, resulting in the differential activation of VMH neurons. This model is depicted in Figure 7 and discussed under the section of 'Differential processing of fresh and old saliva signals in the VNO-to-VMH pathway' in the Discussion."
(4) The interpretation that fresh and old saliva activates different subpopulations of neurons in the VMH based on the observation that cFos positively correlates with freezing responses only with the fresh saliva lacks empirical evidence. To address this question, the authors should use two neuronal activity markers to track the response of the same population of VHM cells within the same animals during exposure to fresh vs. old saliva. Alternatively, they could use single-cell electrophysiology or imaging tools to demonstrate that cat saliva of distinct freshness activates different subpopulations of cells in the VMH. Any interpretation without a direct within-subject comparison or the use of cell-type markers would become merely speculative. Furthermore, the authors assume that differential activations of mitral cells between fresh and old saliva result in the differential activation of VMH subpopulations (page 13, line 3). However, there are intermediate structures between the mitral cells and the VMH, which are completely ignored in this study (e.g., BNST, medial amygdala).
We appreciate this important feedback. We agree that performing a same-animal comparison for fresh and old saliva exposure will offer direct evidence of the differential activation of a sub-population of VMH neurons. However, there is technical difficulties. We have stimulated the same animal with the same or different types of swabs (e.g., Freshcontrol, fresh-fresh, fresh-old, or old-fresh) and observed that once mice were exposed to a saliva-containing swab and exhibited freezing behavior, they no longer made contact with the second swab within the timeframe when two different types of neuroactivity markers can be analyzed. As shown in Figure 2A, direct contact with the saliva swab is necessary for triggering saliva-elicited freezing behavior. Therefore, we concur that conducting further investigations into real-time neural activation responses to both fresh and old saliva within the same subjects, using an appropriate stimulus delivery method into the VNO, as demonstrated in (Bansal et al., 2021; Ben-Shaul et al., 2010; Bergan et al., 2014), would be useful to strengthen our argument.
For the second part of the comment regarding the intermediate structures between the mitral cells and the VMH, please refer to our comment above in response to comment 3.
(5) The authors incorrectly cited the Papes et al., 2010 article on several occasions across the manuscript. In the introduction, the authors cited the Papes et al 2010 study to make reference to the response of rodents to chemical cues, but the Papes et al. study did not use any of the chemical cues listed by the authors (e.g., fox feces, snake skin, cat fur, and cat collars). Instead, the Papes et al. 2010 article used the same chemical cue as the present study: cat saliva. The Papes et al. 2010 article was miscited again in the results section where the authors cited the study to make reference to other sources of cat odor that differ from the cat saliva such as cat fur and cat collars. Because the Papes et al. 2010 article has previously shown the involvement of Trpc2 receptors in the VNO for the detection of cat saliva and the subsequent expression of defensive behaviors by using Trpc2-KO mice, the authors should properly cite this study in the introduction and across the manuscript when making reference to their findings.
The study conducted by Papes et al. in 2010 (Papes et al., 2010) explored mouse defensive responses triggered by native odors derived from three natural mouse predator species: cat, snake, and rat. These odors were derived from neck fur swabs, shed skin, and urine, respectively. Notably, all three types of samples induced defensive risk assessment and avoidance behaviors in mice. These responses were significantly diminished in Trpc2 knock-out (KO) mice, which lack the Trpc2 transduction channel in their vomeronasal sensory neurons, resulting in an impairment in transmitting sensory signals to the brain. Moreover, Papes et al. (2010) mentioned that, 'we did find cat saliva, a potential source of fur chemosignals, sufficient to induce c-Fos expression in the AOB and initiate defensive behavior.' While Papes et al. reported c-Fos expression in the AOB as well as behavioral responses induced by cat saliva in C57BL/6 mice, they did not provide information regarding the c-Fos expression or the defensive behavioral responses to cat saliva in Trpc2KO mice. Overall, we highly value these findings and explicitly state in the results section of our study that ‘Cat saliva has been considered as a source of predator cues found on cat fur and collars, which induce defensive behaviors in rodents (Engelke et al., 2021; Papes et al., 2010),’ providing the rationale for our utilization of cat saliva in our experimental design.
(6) In the introduction, the authors hypothesized that the VNO detects predator cues and sends sensory signals to the VMH to trigger defensive behavioral decisions and stated that direct evidence to support this hypothesis is still missing. However, the evidence that cat saliva activates the VMH and that activity in the VMH is necessary for the expression of antipredator defensive response in rodents has been previously demonstrated in a study by Engelke et al., 2021 (PMID: 33947849), which was entirely omitted by the authors.
We appreciate this insightful comment. Our original sentence meant that the direct evidence was missing for the hypothesis that the mouse VNO detects predator cues and sends sensory signals to the VMH, triggering appropriate defensive behavioral decisions. To clarify this, we altered the sentence (the last sentence of the second last paragraph in Introduction) to “However, how the sensory signals detected through the VNO-to-VMH circuitry modulate behavioral decisions in specific contexts remains elusive.
The study in Engelke et al., 2021(Engelke et al., 2021) has shown that cat saliva activates the VMH and that activity in the VMH is necessary for the expression of antipredator defensive response, including freezing behavior, in rats. This important paper is now cited at multiple locations; page 4 line 16, page 9 line 8, and page 14 line 17. Interestingly, the vomeronasal receptor genes expressed in cat saliva-responsive VNO neurons, V2R-A4 subfamily genes, seem to have expanded independently within mice and rats, lacking direct V2R-A4 orthologues between mice and rats (Rocha et al. submitted). Therefore, exploring the sensory mechanism behind the induction of defensive behavioral responses in rats by cat saliva would be highly intriguing. Comparing the mechanism operating in rats with that observed in mice could offer valuable insights into understanding how the divergent sensory signaling pathways lead to the VMH-mediated defensive behavioral responses across different species.
(7) In the discussion, the authors stated that their findings suggest that the induction of robust freezing behavior is mediated by a distinct subpopulation of VMH neurons. The authors should cite the study by Kennedy et al., 2020 (PMID: 32939094) that shows the involvement of VMH in the regulation of persistent internal states of fear, which may provide an alternative explanation for why distinct concentrations of saliva could result in different behavioral outcomes.
We appreciate this valuable advice to cite this important paper. It is now cited at page 14 line 17 in the Discussion under “Differential activation of VMH neurons potentially underlying distinct intensities of freezing behavior.” We agree that it is intriguing to hypothesize that different freshness of cat saliva induces different degree of persistence of neural activity in a subpopulation of VMH neurons, which regulates the freezing behavior intensity.
(8) The anatomical connectivity between the olfactory system and the ventromedial hypothalamus (VMH) in the abstract is unclear. The authors should clarify that the VMH does not receive direct inputs from the vomeronasal organ (VNO) nor the accessory olfactory bulb (AOB) as it seems in the current text.
We apologize for the confusion caused by our statement in the abstract. The reviewer is correct that the VMH does not receive direct inputs from the VNO and AOB. The abstract now states: 'The vomeronasal organ (VNO) is one of the major sensory input channels through which predator cues are detected with ascending inputs to the medial hypothalamic nuclei, especially to the ventromedial hypothalamus (VMH), through the medial amygdala (MeA) and bed nucleus of the stria terminalis (BNST).’
Reviewer #2 (Public Review):
Weakness:
The findings are relatively preliminary. The identities of the receptor and the ligand in the cat saliva that induces the behavior remain unclear. The identity of VMH cells that are activated by the cat saliva remains unclear. There is a lack of targeted functional manipulation to demonstrate the role of V2R-A4 or VMH cells in the behavioral response to cat saliva.
We concur with the reviewer’s comments and agree with the necessity to explore the behavioral response to cat saliva in mice with V2R-A4 receptor(s) knocked out, alongside those with targeted functional manipulations in the VMH. These future studies will allow us to further elucidate the molecular and neural mechanisms underlying this sensory-tohypothalamic circuit.
Reviewer #3 (Public Review):
Weaknesses:
(1) It is unclear if fresh and old saliva indeed alter the perceived imminence predation, as claimed by the authors. Prior work indicates that lower imminence induces anxiety-related actions, such as re-organization of meal patterns and avoidance of open spaces, while slightly higher imminence produces freezing. Here, the authors show that fresh and old predator saliva only provoke different amounts of freezing, rather than changing the topography of defensive behaviors, as explained above. Another prediction of predatory imminence theory would be that lower imminence induced by old saliva should produce stronger cortical activation, while fresh saliva would activate the amygdala, if these stimuli indeed correspond to significantly different levels of predation imminence.
We thank the reviewer for this valuable insight. In our current study, we exclusively compared defensive behavioral responses to 15-minute-old and 4-hour-old cat saliva in mice within their home cages. In future studies, it would be intriguing to expand this investigation by examining behavioral changes in response to saliva collected at additional time points across diverse behavioral settings. Additionally, exploring neural activity in various brain regions in future studies would complement our understanding of these responses.
(2) It is known that predator odors activate and require AOB, VNO, and VMH, thus replications of these findings are not novel, decreasing the impact of this work.
We acknowledge the previous findings mentioned by the reviewer. Our finding in this paper is that cat saliva samples with different freshness predominantly activate different numbers of VNO sensory neurons expressing the same subfamily of sensory receptors, which results in differential activation of the downstream circuit to modulate behavioral outputs.
(3) There is a lack of standard circuit dissection methods, such as characterizing the behavioral effects of increasing and decreasing the neural activity of relevant cell bodies and axonal projections, significantly decreasing the mechanistic insights generated by this work.
We thank the reviewer for the valuable comments. We acknowledge that exploring the behavioral effects through the manipulation of specific cell types within defined neural substrates, along with characterizing circuit connectivity, is crucial to understand this circuit more thoroughly in future studies.
(4) The correlation shown in Figure 5c may be spurious. It appears that the correlation is primarily driven by a single point (the green square point near the bottom left corner). All correlations should be calculated using Spearman correlation, which is non-parametric and less likely to show a large correlation due to a small number of outliers. Regardless of the correlation method used, there are too few points in Figure 5c to establish a reliable correlation. Please add more points to 5c.
We thank the reviewer for this important suggestion. We assessed normality of the data using the Shapiro-Wilk and Kolmogorov-Smirnov tests, confirming that the dataset is parametric. We anticipate employing a larger sample size in future studies to further examine rigorous correlation patterns.
(5) Some of the findings are disconnected from the story. For example, the authors show that V2R-A4-expressing cells are activated by predator odors. Are these cells more likely to be connected to the rest of the predatory defense circuit than other VNO cells?
Yes, our hypothesis posits that V2R-A4-expressing VNO sensory neurons serve as receptor neurons for predator cues present in cat saliva. Additionally, we assume that these specific sensory neurons have stronger anatomical connections with the defensive circuit compared to VNO sensory neurons expressing other receptor subfamilies. In our modified Discussion section, we discussed this point under “V2R-A4 subfamily as the receptor for predator cues in cat saliva.”
(6) Were there other behavioral differences induced by fresh compared to old saliva? Do they provoke differences in stretch-attend risk evaluation postures, number of approaches, the average distance to odor stimulus, the velocity of movements towards and away from the odor stimulus, etc?
We appreciate the reviewer's valuable comments. We have now incorporated an analysis of stretch-sniff risk assessment behavior, presented in new Figure 1F (graph) and Supplemental Figure 1B (raster plot). Mice exhibited stretch-sniff risk assessment behavior, which remained consistent across control, fresh saliva, and old saliva swabs. Additionally, we have also included a raster plot for direct investigation, previously noted as ‘interaction’ in the original manuscript (Supplemental Figure 1C). Mice exposed to a swab containing either fresh or old saliva significantly avoided directly investigating the swab. In contrast, mice exposed to a clean control swab spent a significant amount of time directly investigating the swab, engaging in behaviors such as sniffing and chewing (Figure 1G). A comparison of temporal behavioral patterns revealed a slightly higher frequency of direct investigation behavior toward old saliva compared to fresh saliva at the beginning of the exposure period (Supplemental Figure 1C).
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
(A) In the discussion (page 13, line 13), the authors proposed approaches to isolate receptors among the V2R-A4 subfamily that could be responsible for the detection of predator cues in cat saliva such as mRNA profiling from cells isolated from VNO GCaMP imaging. However, the authors argue that this method can lead to false positive results. The authors should clarify what they mean by this exactly.
We meant that pairing of kairomones and their cognate vomeronasal receptors is overall challenging, and subsequent confirmations by performing loss-of-function, as well as gainof-function studies, are necessary to avoid false positive receptor-ligand pairings. We modified the sentence in the discussion as follows: “…. as well as receptor mRNA profiling from isolated single cells activated by cat saliva in GcaMP imaging using the VNO slices in vitro (Haga-Yamanaka et al., 2014; Wong et al., 2020). Receptor candidates identified using either of the methods can be further confirmed by examining necessity and sufficiency for detecting cat saliva using genetically modified mouse lines.”
(B) In the discussion, the authors mention that imminent predator cues present in the cat saliva activate a specific population of VMN neurons. However, the authors have not demonstrated that imminent predator cues exist and the differences between fresh and old saliva are not simply a matter of concentration and integrity of the same protein (see a similar concern in item 2 above).
In alignment with our responses to the reviewer’s public comments 1 and 2, we acknowledge the changes in both the quality and quantity of molecules in cat saliva when kept at room temperature for 4 hours. Our findings demonstrate that mice detect this timedependent alteration through the VNO, leading to subsequent adjustments in their defensive response patterns. The identification of specific molecules responsible for inducing behavioral changes and an exploration of their time-dependent alterations are crucial steps in our ongoing research. To provide further clarification, we have added a paragraph in the discussion section under 'The VNO as the sensor of predator cues that induce fear-related behavior.’
(C) In the introduction, the authors cite several studies and reviews that investigated sensory neural circuits that mediate behavioral responses to chemical predator cues in mice. However, the majority of these studies used rats. Therefore, it is recommended to instead indicate that these studies focus on using rodent models.
We appreciate this insightful comment. We have now replaced the term 'mice/mouse' with 'rodents' in corresponding parts of the manuscript.
(D)The description of the extended amygdala is unclear and gives the impression that the posteroventral part of the medial amygdala is also part of the extended amygdala (page 3, line 25).
We appreciate the reviewer’s important feedback. We have removed the phrase 'the extended amygdala consisting of' from the text.
(E) The authors should justify why they have focused on the role of V2R-A4 in cat saliva detection. As shown in the Figure 3A schematic, many other receptors within the V2R family could have been evaluated. Additionally, the authors should indicate how many mice were used for calculating the ratio for each receptor in Figure 3C, and a group comparison should be performed.
As shown in Supplemental Figure 2 and Figure 3C, our initial investigation involved assessing the co-localization of pS6 signals with signals derived from in situ hybridization probes for all V2R subfamilies. Each probe was designed to recognize all the receptor genes within the subfamily under the tested conditions. This examination led to the identification of V2R-A4, whose probe signals overlap with pS6 signals induced by exposure to cat saliva. In Figure 3C, the percentage of total overlap between the in situ probe and pS6 signals in VNO sections was examined from n=3-6 animals, which is now mentioned in the modified figure legend.
(F) The authors should make it clear to readers at the very beginning of the manuscript that the behavioral differences between fresh and old saliva are not caused by the inefficiency of the old cat saliva to induce defensive responses. Thus, other antipredator behavioral responses should be also quantified (e.g., avoidance time, number and time of investigations to the cat saliva source, risk-assessment, etc.)
We appreciate this valuable comment from the reviewer. In the original version of our manuscript, we used the term 'interaction' to indicate 'direct interaction with the swab for investigation.' We have now replaced the term 'interaction' with 'direct investigation' and added the temporal patterns of these behavioral episodes in Supplemental Figure 1C. Our observations indicate that mice avoid directly investigating both fresh and old saliva compared to the control (Figure 1G). However, there is a slight increase in investigation behavior toward old saliva at the beginning of exposure compared to fresh saliva (Supplemental Figure 1C). Furthermore, we have included the duration (Figure 1F) and temporal patterns (Supplemental Figure 1B) of stretch-sniff risk assessment behavior. Notably, stretch-sniff behavior did not differ towards control, fresh, and old saliva swabs.
(G) The selected representative images for Gαo- and pS6-labeled neurons in Figure 2 should have similar levels of DAPI labeling. Further, the plot depicting the duration of freezing as a function of pS6-IR signals in the VNO (Figure 2H) is difficult to follow. The authors should indicate on the graph which data points represent fresh or old cat saliva exposure, similar to the style used in Figure 5 plots.
We have replaced the representative image in Figure 2E to align the DAPI intensity. Additionally, we updated the data points in Figure 2H and introduced a color code to indicate saliva types.
(H) The schematic in Figure 4 is misleading because the AOB does not directly project to the VMH. The authors should explain which regions are conveying indirect predator information from AOB to VMH (see a similar concern in item 7 above).
We thank the reviewer’s important feedback. We modified the image in Figure 4A to show the entire defensive behavior circuit initiated from the VNO.
Reviewer #2 (Recommendations For The Authors):
(1) This result suggests that V2R-A4 may be the dominant VR for mice to detect cat saliva.
Future studies should determine the identity of the receptor and the ligand in the cat saliva. Additionally, the functional importance of V2R-A4 remains unclear. It is important to knockout the receptor and test changes in cat saliva-induced freezing.
We concur with the reviewer’s comments and recognize the necessity of exploring the behavioral response to cat saliva in mice with V2R-A4 receptor(s) knocked out. Moreover, the identification of the ligand in cat saliva is critical for a deeper understanding of the molecular mechanisms in future studies.
(2) AOB does not project to VMH directly. Other known important nodes for the predator defense circuit include MeApv, BNST, PMd, AHN, and PAG. It will be helpful to provide c-Fos data in those regions (especially MEA and BNST as they are between AOB and VMH) to provide a complete picture of how the brain processes cat saliva to induce the behavior change.
We appreciate this important feedback by the reviewer. We have now added c-Fos expression analysis data in the BNST, MeApv, and PAG, in addition to the VMH. Upon exposure to fresh and old saliva, we observed the upregulation of cFos in the MeApv, VMH, and dPAG, but not in the BNST, compared to the control stimulus. The data are now shown in Figure 4G-J. Moreover, we also added correlation analyses between the numbers of cFospositive neurons and the duration of freezing behavior in those neural substrates to Figure 5. The numbers of cFos-IR signals in neurons in the BNST and dPAG, did not correlate with the duration of freezing behavior in any of the exposure groups (Figure 5C, F). However, in addition to a significant positive correlation in the fresh saliva-exposed group in the VMH (R2 = 0.5708, 95% CI [-0.1449, 0.9714], p = 0.0412) (Figure 5E), we observed a similar positive correlation trend in the MeApv (R2 = 0.3854, 95% CI [-0.3845, 0.9525], p = 0.0942), although it was not statistically significant possibly due to low sample numbers (Figure 5D). Based on these results, our current circuit model is as follows: different numbers of the VNO sensory neurons activated by fresh and old saliva result in differential excitation levels in mitral cells in the AOB. Differential excitation of mitral cells leads to the differential activation of targeting neural substrates, possibly MeApv, which results in differential activation of VMH neurons. This model is depicted in Figure 7 and discussed under the section of “Differential processing of fresh and old saliva signals in the VNO-toVMH pathway” in Discussion.
(3) It is interesting that activation level difference in the VNO by old and fresh cat saliva does not transfer to AOB. It could be informative to examine the correlation between VNO and AOB p6/c-Fos cell number and AOB and VMH c-Fos cell number across animals to understand whether the activation levels across those regions are related. If they are not correlated, it could be helpful to add a discussion regarding potential reasons, e.g. neuromodulatory inputs to the AOB.
We agree that analyzing the number of pS6/cFos-positive cells from all the regions in the same animals are ideal; however, due to technical difficulties, we were unable to collect the entire set of neural substrates from the same animals.
(4) Please indicate n in all figure plots and specify what individual dots mean. In Figure 4h, there are 7 dots in the old saliva group, presumably indicating 7 animals. In Figure 6b, there appear to be more than 7 dots for the old cat saliva group. Are there more than 7 animals? If so, why are they not included in Figure 4h? If not, what does each dot mean? Note that each dot should represent an independent sample. One animal should not contribute more than one dot.
We apologize for the confusion about Figure 6b. Each of these dots indicates the number of cFos signals in a single VMH hemisphere sample. The data used for this analysis were the same as the ones for the VMH used in Figure 4. This is now clarified in the figure legends.
(5) The identification of a cluster of VMHdm cells uniquely activated by fresh cat saliva urine is interesting. It will be important to identify the molecular handle of the cells to facilitate further investigation. This could be achieved using either activity-dependent RNAseq or double in situ of saliva-induced c-Fos and candidate genes (candidate gene may be identified based on the known gene expression pattern).
We agree that these experiments are very valuable. We would like to perform those experiments in future studies.
Reviewer #3 (Recommendations For The Authors):
(1) Please cite recent relevant papers showing VMH activity induced by predators, such as https://pubmed.ncbi.nlm.nih.gov/33115925/ and https://pubmed.ncbi.nlm.nih.gov/36788059/
We thank the reviewer’s suggestion to cite these important papers. https://pubmed.ncbi.nlm.nih.gov/33115925/ (Esteban Masferrer et al., 2020) and https://pubmed.ncbi.nlm.nih.gov/36788059/ (Tobias et al., 2023) are now cited at page 14 line 17 in the Discussion under “Differential activation of VMH neurons potentially underlying distinct intensities of freezing behavior.”
(2) Add complete statistical information in the figure legends of all figures, which should include n, name of test used, and exact p values.
We included statistical analysis results in figure legends; for Figure 6B, we provided statistical analysis results in Supplemental Table 1.
(3) Please paste all figure legends directly below their corresponding figure to make the manuscript easier to read.
We have added figure legends directly below their corresponding figures.
Editor's note:
Should you choose to revise your manuscript, please include full statistical reporting including exact p-values wherever possible alongside the summary statistics (test statistic and df) and 95% confidence intervals. These should be reported for all key questions and not only when the p-value is less than 0.05.
Statistics analysis results have been included in figure legends and supplemental table 1.
References
Bansal R, Nagel M, Stopkova R, Sofer Y, Kimchi T, Stopka P, Spehr M, Ben-Shaul Y. 2021. Do all mice smell the same? Chemosensory cues from inbred and wild mouse strains elicit stereotypic sensory representations in the accessory olfactory bulb. BMC Biol 19:133.
Ben-Shaul Y, Katz LC, Mooney R, Dulac C. 2010. In vivo vomeronasal stimulation reveals sensory encoding of conspeciic and allospeciic cues by the mouse accessory olfactory bulb. Proc Natl Acad Sci U S A 107:5172‒5177.
Bergan JF, Ben-Shaul Y, Dulac C. 2014. Sex-speciic processing of social cues in the medial amygdala. Elife 3:e02743.
Engelke DS, Zhang XO, OʼMalley JJ, Fernandez-Leon JA, Li S, Kirouac GJ, Beierlein M, Do-Monte FH. 2021. A hypothalamic-thalamostriatal circuit that controls approachavoidance conlict in rats. Nat Commun 12:2517.
Esteban Masferrer M, Silva BA, Nomoto K, Lima SQ, Gross CT. 2020. Differential Encoding of Predator Fear in the Ventromedial Hypothalamus and Periaqueductal Grey. J Neurosci 40:9283‒9292.
Papes F, Logan DW, Stowers L. 2010. The vomeronasal organ mediates interspecies defensive behaviors through detection of protein pheromone homologs. Cell 141:692‒703.
Tobias BC, Schuette PJ, Maesta-Pereira S, Torossian A, Wang W, Sethi E, Adhikari A. 2023. Characterization of ventromedial hypothalamus activity during exposure to innate and conditioned threats. Eur J Neurosci 57:1053‒1067.
-
-
-
Author Response
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public Review):
In this manuscript, Lee et al. compared encoding of odor identity and value by calcium signaling from neurons in the ventral pallidum (VP) in comparison to D1 and D2 neurons in the olfactory tubercle (OT).
Strengths:
They utilize a strong comparative approach, which allows the comparison of signals in two directly connected regions. First, they demonstrate that both D1 and D2 OT neurons project strongly to the VP, but not the VTA or other examined regions, in contrast to accumbal D1 neurons which project strongly to the VTA as well as the VP. They examine single unit calcium activity in a robust olfactory cue conditioning paradigm that allows them to differentiate encoding of olfactory identity versus value, by incorporating two different sucrose, neutral and air puff cues with different chemical characteristics. They then use multiple analytical approaches to demonstrate strong, low-dimensional encoding of cue value in the VP, and more robust, high-dimensional encoding of odor identity by both D1 and D2 OT neurons, though D1 OT neurons are still somewhat modulated by reward contingency/value. Finally, they utilize a modified conditioning paradigm that dissociates reward probability and lick vigor to demonstrate that VP encoding of cue value is not dependent on encoding of lick vigor during sucrose cues, and that separable populations of VP neurons encode cue value/sucrose probability and lick vigor.
Weaknesses:
The conclusions of the data are mostly well supported by the analyses, but the statistical analysis is somewhat limited and needs to be clarified and extended.
(1) The manuscript includes limited direct statistical comparison of the neural populations, and many of the comparisons between the subregions are descriptive, including descriptions of the percentage of neurons having specific response types, or differences in effect sizes or differing "levels" of significance. An additional direct comparison of data from each subpopulation would help to confirm whether the differences reported are statistically meaningful.
Response: We thank the reviewer for their helpful suggestions. As the reviewer noted, the first version of our manuscript had limited direct comparisons of single-neuron metrics across subpopulations. These analyses were also limited to the supplementary figures: 1) {SK vs. XK} and {SK vs. ST} decoder auROC (S10F), 2) Valence scores (S10G), and 3) S-cue confusion after MNR classification (S11D). We have now included the following statistical comparisons of single-neuron metrics across subpopulation: 1) % of neurons that respond to both S cues (Tables S10, S11), 2) % of neurons that have auROC >0.75 for {SK vs. XK}, {SK vs. PK}, and {SK vs. ST} (Tables S12-S17), 3) response magnitudes to S cues (Table S38), and 4) valence scores (Tables S44-46).
(2) When hypothesis tests are conducted between the neural populations, it is not clear whether the authors have accounted for the random effect of the subject, or whether individual units were treated as fully independent. For instance, pairwise differences are reported in Figures 4I, 5G/I/L, and others, but the statistical methods are unclear. Assessment of the statistics is further limited by the lack of reporting of degrees of freedom. If the individual neurons are treated as independent in these analyses, it could increase the likelihood of
Response: We have clarified when statistical analyses are comparing individual neurons vs. simultaneously recorded populations. Per the reviewer’s recommendation, we have also incorporated linear mixed-effects models when statistically analyzing individual neurons. Lastly, to further clarify the statistical analyses used, we have added multiple supplementary tables that better describe the statistical tests used and the relevant outputs.
Reviewer #2 (Public Review):
Summary:
This work is interesting since the authors provide an in vivo analysis into how odor-associations may change as represented at the level of olfactory tubercle (presynaptic) and next at the level of the ventral pallidum (postsynaptic). First the authors start-off with a seemingly careful characterization of the anterograde and retrograde connectivity of dopamine 1 receptor (D1) and dopamine 2 receptor (D2) expressing medium spiny neurons in the olfactory tubercle and neurons in the ventral pallidum. From this work they claim that regardless of D1 or D2 expression, tubercle neurons mainly project to the lateral portion of the ventral pallidum. Next, to compare how odor-associated neuronal activity in the ventral pallidum and the olfactory tubercle (D1 vs D2 MSNs) transforms across association learning, the authors performed 2photon calcium imaging while mice engaged in a lick / no-lick task wherein two odors are associated with reward, two odors are associated with no outcome, and two odors are associated with an air puff.
This manuscript builds off of prior work by several groups indicating that the olfactory tubercle neurons form flexible learned associations to odors by looking at outputs into the pallidum (but without looking specifically at palladial neurons that truly get input from tubercle I should highlight) and with that, this work is novel. We appreciated the use of a straight-forward odoroutcome behavioral paradigm and the careful computational methods and analyses utilized to disentangle the contributions of single neurons vs population level responses to behavior. With one exception from the Murthy lab, 2P imaging in the tubercle is a new frontier and that is appreciated - as is the 2P imaging in the pallidum which was well-supported by the histology. The anatomical work is also well presented.
Overall the approach and methods are superb. The issues come when considering how the authors present the story and what conclusions are made from these data. Several key points before going into specifics about each are: 1) The authors can not conclude that their results are contradictory to prior results, 2) The authors over-interpret the results and do not discuss several key methodological issues. We were concerned with the ability to make strong claims regarding the circuitry presented, especially given how much the presented claims contradict prior work. There were also issues with the interpretability of neuronal encoding of value vs valence based on the present behavior (in which a distinction between the air puff and neutral trial types was not clear) and the imaging methodology (in which the neuronal populations analyzed were not clearly defined). In addition to toning down and rectifying some of the language and interpretations, we suggest including a study limitations section where these methodological and interpretation issues are discussed. Over-interpreting and playing up the significance of this work is unnecessary, especially given eLife's new review and publication policy. Readers should be given a sufficiently detailed and nuanced presentation of these thought-provoking results, and from there allowed to interpret the results as they want.
Strengths:
State-of-the-art approaches (as detailed above)
Possible conceptual innovation in terms of looking into output from the olfactory tubercle which has yet to be investigated in this avenue.
Weaknesses:
On the first point regarding the authors repeated and unsupported claims that their results are contradictory. There are papers by numerous groups, in respected journals including this one, all together which used 5 different methods (cfos, photometry, 2P, units, fMRI), in animals ranging from humans to mice, which support that tubercle neurons reflect the emotional association of an odor, whether spontaneous or learned. With that, it is on the authors to not claim that their results contradict as if the other papers are suspect, but instead, from our standpoint it is on the authors to explain how and why their results differ from these other papers versus just simply saying they found something different [which at present is framed in a way that is 'correct' due to primacy if nothing else].
Response: We acknowledge that the first version of the manuscript contained unnecessary disagreeing language. We do not think that our results are broadly in disagreement with the existing literature, but we do come to different conclusions about what the OT is representing. Namely, our comparison of valence encoding in OT to that in the VP strongly indicates that the anteromedial OT has a less robust representation of valence, and we argue that this reflects either an intermediate form of valence representation or potentially might not be important for valence representation at all. We have toned down our conclusions, made clear that we are only recording from one domain of the OT, limited our speculation to the discussion and added a “speculations” section.
Second, onto the points of interpretation of results, there are several specific areas where this should be rectified. As is, the authors overinterpret their results and draw too far-reaching conclusions. This needs to be corrected.
In particular, the claims that D1 and D2 neurons of the olfactory tubercle nearly exclusively send projections to the ventral pallidum must be interpreted with caution given that the authors injected an anterograde AAV into the anteromedial olfactory tubercle, and did not examine the projections from either the posterior or lateral portions of the olfactory tubercle. This is especially significant since the retrograde tracing performed from the ventral pallidum indicates that the lateral olfactory tubercle, not the medial olfactory tubercle, primarily projects to the ventral pallidum (Fig 1D-F), however this may be due to leakage into the nucleus accumbens, as seen in the supplementary figure, S1G.
Response: We thank the reviewer for the point of caution. We have now made it clear that our conclusions are limited to the anteromedial portion of the OT, and other areas may have other projections.
The same caution must be advised when interpreting the retrograde tracing performed in Fig 1G-I, since the neuronal tracer used and the laterality and rostral-caudal injection site within the VTA could result in different projection patterns and under- or over-labelling. Additionally, the metric used, %Fiber Density (Figure 1C), as in the percentage of 16-bit pixels within the region of interest with an intensity greater than 200, is semi-quantitative, and is more applicable for examining axonal fibers that pass through a region rather than the synaptic terminals (like with a synaptophysin fusion protein-based tracing paradigm) found within a region (puncta). The statements made in contrast to prior studies should therefore be softened, and these concerns should be addressed in the introduction, discussion, and the limitations section if added.
Response: We have added statements to address these limitations.
The other major concern is whether the behavioral data generated is indicative of the full spectrum of valence. The authors appropriately state that the mice "perceive" the air puff, yet based on their data the mice did not clearly experience the puff-associated odor as emotionally aversive (viz., negative valence). The way the authors describe these results, it seems they agree with this. With that, the authors can't say the puff is aversive without data to show such - that is an assumption which, while seemingly intuitive, is not supported by the data unfortunately. To elaborate more since this is important to the messaging of the paper: The authors utilized a simple behavioral design, wherein two molecular classes of odors were included in either a sucrose rewarded, neutral no outcome, or air puff punished trial type. The odor-outcome pairs were switched after three days, allowing the authors to compare neuronal responses on the basis of odor identity and the later associated outcome. While the mice showed clear learning of the rewarded trial types by an increase in anticipatory licking during the odor, they did not show any significant changes in behavior that indicated learning of the air puff trial type (change in running velocity or % maximal eye size), especially in contrast to the neutral trial type. This brings up the concern that either the odor-air puff aversive associations (to odors) were not learned, or that the neutral trial types, in which a reward was omitted, were just as aversive as the air puff to the rear, despite the lack of startle response - perhaps due to stimulus generalization between neutral and air puff odor. The possibility of lack of learning is addressed in the paragraph starting at line 578, but does not account for the possibility that the lack of reward is also sufficiently punishing. The authors also address the possibility that laterality in the VP contributed to the lack of neural responsivity observed, but should also include a statement regarding laterality in the olfactory tubercle, as described in https://doi.org/10.7554/eLife.25423 and https://doi.org/10.1523/JNEUROSCI.0073-15.2015, since the effects of modulating the lateral portion of the olfactory tubercle are not yet reported. Lastly, use of the term "reward processing" should be avoided/omitted since the authors did not specifically study the processing of reinforcers.
Response: As the reviewer points out, we tried to be cautious interpreting the “aversive” odor response, and focused mainly on the reward association. This was discussed in the discussion. We don’t see the need to further add a redundent statement to a “limitations section”. We have also added a note about the previously identified laterality of the OT, which might account for lack of aversive responsive neurons in the OT. The reviewer makes an interesting suggestion that behavioral responses to airpuff-associated odors are not significantly different from un-associated because the lack of reward in this context is already aversive. We note that the walking velocity between reward- and puff-associated odor is significantly different, but not that to unassociated. This is in agreement with the suggestion, and we have added a statement to reflect this.
Also, I would appreciate justification of the term "value". How specifically does the assay used assess value versus a more simplistic learned association which influences perceived hedonics or valence of the odors.
Response: We have removed the term “value” with the exception of areas where we cite the work of others. We acknowledge that the word value is complicated in the incentive learning field and appreciate the suggestion. Our experimental design was meant to investigate learned association for positive and negative stimuli, thus valence is more appropriate and we have used this term.
More information is needed regarding how neurons are identified day-to-day, both in textual additions to the Methods and also in terms of elaborating more in the results and/or figure legends about what neurons are included:
(a) The ROI maps for identifying/indicating cells in the FOVs are nice to see and at the same time raise some concerns about how cells are identified and/or borders for those specific ROIs drawn. For instance, Figure 4, A & D, ROI #13 (cell #13) between those two panels is VERY different in shape/size. Also see ROIs 15 and 4. Why was an ROI map not made on day 1 and then that same map applied and registered to frames from consecutive imaging days in that same mouse? As it is new ROIs are drawn, smaller for some "cells" and larger for others. And at least in ROI #13 above, one ROI is about twice as large as the other. This inconsistency in the work flow and definition of the ROIs is needing to be addressed in Methods. Also, the authors should address if and how this could possibly impact their results.
Response: We have added details and clarified the methods section to make this more clear. We note that we extracted calcium transients from the raw data with the the widely used Constrained Nonnegative Matrix Factorization (CNMF) algorithm. This processing algorithm simultaneously identifies spatial and temporal components using modeled kinetics of calcium transients and pre-trained CNN classifiers. Using 2-photon microscopy the optical resolution in the z plane is narrow and we may not always capture components of a neuron that look like “neurons”, but all ROIs were confirmed manually to ensure they were not artifacts.
(b) Also, more details are needed in results and/or figure legends regarding the changes in cell numbers over days that are directly compared in the results. Some days there are 10% or more or less cells. Why? It is not the same population being compared in this case and so some Discussion of this is needed.
Response: The shapes of the spatial components can vary across days due to nonrigid motion in the brain and/or miniscule differences in the imaging angle across days. Although we visually verified that we are imaging approximately the same z plane across days, we cannot (and do not) claim to image identical populations of neurons across days.
Reviewer #3 (Public Review):
Summary:
This manuscript describes a study of the olfactory tubercle in the context of reward representation in the brain. The authors do so by studying the responses of OT neurons to odors with various reward contingencies and compare systematically to the ventral pallidum. Through careful tracing, they present convincing anatomical evidence that the projection from the olfactory tubercle is restricted to the lateral portion of the ventral pallidum.
Using a clever behavioral paradigm, the authors then investigate how D1 receptor- vs. D2 receptor-expressing neurons of the OT respond to odors as mice learn different contingencies. The authors find that, while the D1-expressing OT neurons are modulated marginally more by the rewarded odor than the D2-expressing OT neurons as mice learn the contingencies, this modulation is significantly less than is observed for the ventral pallidum. In addition, neither of the OT neuron classes shows significant modulation by the reward itself. In contrast, the OT neurons contained information that could distinguish odor identities. These observations have led the authors to conclude that the primary feature represented in the OT is not reward.
Strengths:
The highly localized projection pattern from olfactory tubercle to ventral pallidum is a valuable finding and suggests that studying this connection may give unique insights into the transformation of odor by reward association.
Comparison of olfactory tubercle vs. ventral pallidum is a good strategy to further clarify the olfactory tubercle's position in value representation in the brain.
Weaknesses:
The authors' interpretation of the physiologic results - that a novel framework is needed to interpret the OT's role - requires more careful treatment.
Response: We thank the reviewer for their recommendation. We have toned down the conclusiveness of our language in the discussion. Additionally, we have removed several speculative sentences from the concluding paragraph.
Reviewer recommendations for Authors:
We thank the reviewers for this helpful list of recommended changes to the manuscript.<br /> Regrettably, a few of the recommendations were overlooked in the revision, as indicated below.<br /> We do agree with the suggestions and plan to add appropriate changes to the version of record.
Reviewer #1 (Recommendations For The Authors):
If the comparisons mentioned in point 2 in the public review do not account for the lack of independence of individual neurons, I suggest the authors do so by either running linear mixed effects models with a random effect for subject, or one-way ANOVAs with a random effect of subject, where appropriate. The authors could also run analyses on summarized individual subject data (averages, % of neurons, etc.), though the authors would lose substantial power when assessing whether average changes differ between subjects in each recording group.
We have clarified when statistical analyses are comparing individual neurons vs. simultaneously recorded populations. Per the reviewer’s recommendation, we have also incorporated linear mixed-effects models when statistically analyzing individual neurons. Lastly, to further clarify the statistical analyses used, we have added supplementary tables for every statistical test that better describe the parameters used and the relevant outputs.
Reviewer #2 (Recommendations For The Authors):
Of minor note, there are some symbols/special characters that did not translate in the figure caption for Figure 6C, repeated text between lines 700-705 and 707-712, and some other small grammatical errors. Additionally, the source of the anterograde tracing virus (AAV9-phSyn1FLEX-tdTomato-T2A-SypEGFP-WPRE) needs to be stated.
Thank you for pointing these out. We have added description to the figure legend, and deleted the repeated lines and fixed grammatical errors. During the revision, we Regrettably overlooked the request to provide the source for the AAV9-phSyn1-FLEX-tdTomato-T2A-SypEGFP-WPRE. We agree that this small detail is important and will add it before publication of the version of record. This viral vector was purchased from The Salk Institute GT3 Core.
Reviewer #3 (Recommendations For The Authors):
The authors' interpretation of the physiologic results - that a novel framework is needed to interpret the OT's role - requires more careful treatment. As the authors note, there is rewardcontingency modulation in OT, especially when D1 neurons are compared against D2, as shown in Fig. 3D,E, Fig. 4I, and Fig. F,J. Though small in effect size, presumably, these modulations cannot be explained by the odor identity. These observations, to this reviewer, suggest the D1 neurons of OT have a component of cue-reward representation. In other words, rather than developing an entirely new framework, an alternative possibility that D1 neurons of OT occupy an intermediate stage in associating cues with reward (i.e., under the same framework, but occupying a different position in the emergence of value representation) should be considered.
We thank the reviewer for this thoughtful comment. We have eliminated the statement that “novel framework is needed” and have been more conservative in our interpretations. We have also acknowledged that our results are not necessarily in conflict with existing literature, but we do draw different conclusions, namely that the anteromedial OT is not a robust valence encoding population in comparison to that in the VP. We appreciate the suggestion of the term “intermediate stage” in reward association and have now included this in the discussion. Lastly, we have limited broader speculation to a “speculation” section of the discussion.
Related to the above point, have the authors analyzed if the similarities in the chemical structures correspond to perceptual and neural similarities? In the data presented in Figure S4, there are greater similarities in the population patterns within the same rewarding condition than within chemical groups. A comparison of the reward vs. chemical group (a simpler version of Fig. 5B) may be beneficial and take full advantage of the experimental design.
This comparison already exists in 5B and lines 285-289 of results. In VP populations, the distribution was structured such that intervalence pairwise comparisons between sucrose-paired and not sucrose-paired odors (e.g. ||SK-PK|| and ||SK-XK||) were larger than intravalence pairwise comparisons (e.g. ||SK-ST||, or ||XK-XT||). OTD1 populations showed an intermediate trend where most intravalence pairwise distances were smaller than intervalence pairwise distances with the exception of ||SK-ST||.
Related to the point about chemical similarities - is the smaller effect size (amount of modulation associated with reward contingency) in this study, compared to the study by Martiros et al, explained by the similarities of odorants used?
This is an interesting point. Although the odorants we use are different from those in Martiros et al, we think it is unlikely to the basis of smaller effect size due to reward modulation. If OT represents odor in a population code, whereby identity is encoded in unique ensembles of activity, then variation in the expression of D1R between OT neurons could account for different effects in different ensembles. However, there is no evidence for such varied expression and it doesn’t seem like an ideal mechanism for the OT to broadly associate odor with reward. Moreover, we do not observe any differences in effect size of reward association between the different odorants used in our study. Rather, we think the difference between our findings is more likely to result from recording in different populations of neurons, which is addressed in lines 522-535.
Regarding the data presented in Fig. 3I - the rewarded odor responses (Sk) are compared against neutral ones (Xk responses), but an S vs. P comparison may be informative, too. Even though the authors mention that the effect of air puff is subtle, the behavioral data presented in Fig. 2F and G suggest that these serve as aversive stimuli. For example, on day 4, the first day after the reward contingency switch, the licking levels seem the lowest for the P odors.
We have added the S vs P comparison. Indeed, we had originally omitted this because the neural and behavioral response to puff cues was not robust. This is discussed in the discussion (lines 563-579), and our conclusions about aversive conditioning are cautious.
Regarding the data presented in Fig. 4G: it is difficult to interpret the data when the data for day 1 reward period and day 3 reward cue period are combined. Or do the authors mean day 1 S cue and day 3 S cue?
These data were based on an observation that some neurons in the VP only responded to sucrose (not odor) on day 1, but later became responsive to the associated odor on day 4. To quantify this, Fig. 4G shows the percentage of these neurons by reporting the percentage that were both responsive to sucrose (not odor) on day 1 and also rewarded odor on day 3. This is described in lines 260-274.
Figure 6 presentation would benefit from a revision. For example, it is unclear if the water port becomes available for the "N" odors with 100% or 50% chance of reward delivery, and if so, how that happens. There are some errors e.g., colormap used for panel G; odors listed may be wrong in line 752 etc. It was unfortunately not possible to understand what was presented.
We have added a schematic (Fig 6B) to better describe the movement of the port and details to the methods. The color scale was indeed inverted in panel G (now H), and it has been corrected. We have verified that the odors listed in the methods are correct. Although not included in the revision, in the version of record we will also add corresponding descriptors (e.g., LHi & Lx) to the odors in the methods for easier comparison.
Minor comments
For Figure 2H, an alternative description in the legend may be beneficial, as the phrasing is not intuitive. A suggested alternative is "licks in response to sugar-associated odors expressed as fraction of all odors".
We appreciate the suggestion and have changed this to “licks during either sucrose cue expressed as a fraction of all licks during any odor.”
Figure 2H: please explain the color code for crosses in the legend and the statistical comparison shown in the figure.
We have added a legend to explain the color code and included a statement about the statistics in the legend with a link to a supplemental table for statistical parameters.
Figure 3D: may contain mislabeling in the legend - the legend for 3D does not match the plot (legend refers to bar graph while plot shows line graphs)
Unclear what is meant. 3D legend says: “Percentage of total neurons that were significantly excited or inhibited by each odor (Bonferroni- adjusted FDR < 0.05) as a function of time relative to odor. Lines represent the mean across biological replicates and the shaded area reflects the mean ± SEM.” This is not a bar plot and is not referred to as one. 3E does show bar plots and is correctly described in the legend.
Figure 3M: uses letters to refer to cell populations that are identical to the roman numerals used in Fig 3 A-C as well as colours similar to the ones in Fig 3C. However, the cell groups are unrelated; splitting the figures or using a different nomenclature might help
We have adapted a different color code that we think makes this more distinct.
Figure 4I: statistical comparison shown in figure not explained (neither in main text nor legend)
We have added a statement about the statistical comparison and referenced a supplementary table.
Figure 5 D: color code appears to have a different range than the values shown (i.e. lower limit is 0.7 while the plot shows values below 0.7)
We confirm this is not a mistake but a stylistic choice. The displayed color scale does only show values to lower limit of 0.7, while the lower limit of values is 0.67. Although the color for 0.67 is not shown in the scale it is approximately the same as the lower limit. The values are reported for full transparency and accuracy.
Figure 5 G, I, & L: statistical comparison shown in figure not explained
The comparisons have been explained in supplemental tables (S22-29) and referenced in the legend.
Figure 5 I: meaning of symbols overlayed over bars not explained
“Markers represent the mean across biological replicates” has been added.
Figure 5 J&K: please state if error bars show SEM or SD; also please describe individual thinner lines in the legend
This has been added to describe 5I. The same format applies to J&K.
Figure 5L: please describe the individual crosses overlayed over bars in the legend
Described in 5I.
Figure S6A-C: please mention the odors used.
S6A-C shows kinetics for the odor a-terpinene, which is now indicated in the legend.
Line 129: mentions a 70 psi airpuff but methods say 75 psi - please clarify This has been corrected. 70 psi is the correct value.
Line 134 typo: SP should be PK
This has been corrected.
Line 428: typo; should be cluster 3, not 2
This has been corrected.
Line 474 (and figure 6O): please explain what "P" is
“P” is probability, used as P(S), as in probability of sucrose. This is defined in in line 466.
Line 692: please describe the staining protocol in the methods (rather than just listing the antibodies and concentrations)
We have added more details (lines 692-699).
Line 707-712: duplicate text (identical to Line 700-705)
This has been deleted.
-
-
www.biorxiv.org www.biorxiv.org
-
Reviewer #1 (Public Review):
Summary:<br /> The authors have created a system for designing and running experimental pipelines to control and coordinate different programs and devices during an experiment, called Heron. Heron is based around a graphical tool for creating a Knowledge Graph made up of nodes connected by edges, with each node representing a separate Python script, and each edge being a communication pathway connecting a specific output from one node to an input on another. Each node also has parameters that can be set by the user during setup and runtime, and all of this behavior is concisely specified in the code that defines each node. This tool tries to marry the ease of use, clarity, and self-documentation of a purely graphical system like Bonsai with the flexibility and power of a purely code-based system like Robot Operating System (ROS).
Strengths:<br /> The underlying idea behind Heron, of combining a graphical design and execution tool with nodes that are made as straightforward Python scripts seems like a great way to get the relative strengths of each approach. The graphical design side is clear, self-explanatory, and self-documenting, as described in the paper. The underlying code for each node tends to also be relatively simple and straightforward, with a lot of the complex communication architecture successfully abstracted away from the user. This makes it easy to develop new nodes, without needing to understand the underlying communications between them. The authors also provide useful and well-documented templates for each type of node to further facilitate this process. Overall this seems like it could be a great tool for designing and running a wide variety of experiments, without requiring too much advanced technical knowledge from the users.
The system was relatively easy to download and get running, following the directions and already has a significant amount of documentation available to explain how to use it and expand its capabilities. Heron has also been built from the ground up to easily incorporate nodes stored in separate Git repositories and to thus become a large community-driven platform, with different nodes written and shared by different groups. This gives Heron a wide scope for future utility and usefulness, as more groups use it, write new nodes, and share them with the community. With any system of this sort, the overall strength of the system is thus somewhat dependent on how widely it is used and contributed to, but the authors did a good job of making this easy and accessible for people who are interested. I could certainly see Heron growing into a versatile and popular system for designing and running many types of experiments.
Weaknesses:<br /> The number one thing that was missing from the paper was any kind of quantification of the performance of Heron in different circumstances. Several useful and illustrative examples were discussed in depth to show the strengths and flexibility of Heron, but there was no discussion or quantification of performance, timing, or latency for any of these examples. These seem like very important metrics to measure and discuss when creating a new experimental system.
After downloading and running Heron with some basic test Nodes, I noticed that many of the nodes were each using a full CPU core on their own. Given that this basic test experiment was just waiting for a keypress, triggering a random number generator, and displaying the result, I was quite surprised to see over 50% of my 8-core CPU fully utilized. I don't think that Heron needs to be perfectly efficient to accomplish its intended purpose, but I do think that some level of efficiency is required. Some optimization of the codebase should be done so that basic tests like this can run with minimal CPU utilization. This would then inspire confidence that Heron could deal with a real experiment that was significantly more complex without running out of CPU power and thus slowing down.
I was also surprised to see that, despite being meant specifically to run on and connect diverse types of computer operating systems and being written purely in Python, the Heron Editor and GUI must be run on Windows. This seems like an unfortunate and unnecessary restriction, and it would be great to see the codebase adjusted to make it fully cross-platform-compatible.
Lastly, when I was running test experiments, sometimes one of the nodes, or part of the Heron editor itself would throw an exception or otherwise crash. Sometimes this left the Heron editor in a zombie state where some aspects of the GUI were responsive and others were not. It would be good to see a more graceful full shutdown of the program when part of it crashes or throws an exception, especially as this is likely to be common as people learn to use it. More problematically, in some of these cases, after closing or force quitting Heron, the TCP ports were not properly relinquished, and thus restarting Heron would run into an "address in use" error. Finding and killing the processes that were still using the ports is not something that is obvious, especially to a beginner, and it would be great to see Heron deal with this better. Ideally, code would be introduced to carefully avoid leaving ports occupied during a hard shutdown, and furthermore, when the address in use error comes up, it would be great to give the user some idea of what to do about it.
Overall I think that, with these improvements, this could be the beginning of a powerful and versatile new system that would enable flexible experiment design with a relatively low technical barrier to entry. I could see this system being useful to many different labs and fields.
-
Reviewer #2 (Public Review):
Summary:<br /> The authors provide an open-source graphic user interface (GUI) called Heron, implemented in Python, that is designed to help experimentalists to<br /> (1) design experimental pipelines and implement them in a way that is closely aligned with their mental schemata of the experiments,<br /> (2) execute and control the experimental pipelines with numerous interconnected hardware and software on a network.
The former is achieved by representing an experimental pipeline using a Knowledge Graph and visually representing this graph in the GUI. The latter is accomplished by using an actor model to govern the interaction among interconnected nodes through messaging, implemented using ZeroMQ. The nodes themselves execute user-supplied code in, but not limited to, Python.
Using three showcases of behavioral experiments on rats, the authors highlighted three benefits of their software design:<br /> (1) the knowledge graph serves as a self-documentation of the logic of the experiment, enhancing the readability and reproducibility of the experiment,<br /> (2) the experiment can be executed in a distributed fashion across multiple machines that each has a different operating system or computing environment, such that the experiment can take advantage of hardware that sometimes can only work on a specific computer/OS, a commonly seen issue nowadays,<br /> (3) the users supply their own Python code for node execution that is supposed to be more friendly to those who do not have a strong programming background.
Strengths:<br /> (1) The software is light-weight and open-source, provides a clean and easy-to-use GUI,<br /> (2) The software answers the need of experimentalists, particularly in the field of behavioral science, to deal with the diversity of hardware that becomes restricted to run on dedicated systems.<br /> (3) The software has a solid design that seems to be functionally reliable and useful under many conditions, demonstrated by a number of sophisticated experimental setups.<br /> (4) The software is well documented. The authors pay special attention to documenting the usage of the software and setting up experiments using this software.
Weaknesses:<br /> (1) While the software implementation is solid and has proven effective in designing the experiment showcased in the paper, the novelty of the design is not made clear in the manuscript. Conceptually, both the use of graphs and visual experimental flow design have been key features in many widely used softwares as suggested in the background section of the manuscript. In particular, contrary to the authors' claim that only pre-defined elements can be used in Simulink or LabView, Simulink introduced MATLAB Function Block back in 2011, and Python code can be used in LabView since 2018. Such customization of nodes is akin to what the authors presented.
(2) The authors claim that the knowledge graph can be considered as a self-documentation of an experiment. I found it to be true to some extent. Conceptually it's a welcoming feature and the fact that the same visualization of the knowledge graph can be used to run and control experiments is highly desirable (but see point 1 about novelty). However, I found it largely inadequate for a person to understand an experiment from the knowledge graph as visualized in the GUI alone. While the information flow is clear, and it seems easier to navigate a codebase for an experiment using this method, the design of the GUI does not make it a one-stop place to understand the experiment. Take the Knowledge Graph in Supplementary Figure 2B as an example, it is associated with the first showcase in the result section highlighting this self-documentation capability. I can see what the basic flow is through the disjoint graph where 1) one needs to press a key to start a trial, and 2) camera frames are saved into an avi file presumably using FFMPEG. Unfortunately, it is not clear what the parameters are and what each block is trying to accomplish without the explanation from the authors in the main text. Neither is it clear about what the experiment protocol is without the help of Supplementary Figure 2A.
In my opinion, text/figures are still key to documenting an experiment, including its goals and protocols, but the authors could take advantage of the fact that they are designing a GUI where this information, with properly designed API, could be easily displayed, perhaps through user interaction. For example, in Local Network -> Edit IPs/ports in the GUI configuration, there is a good tooltip displaying additional information for the "password" entry. The GUI for the knowledge graph nodes can very well utilize these tooltips to show additional information about the meaning of the parameters, what a node does, etc, if the API also enforces users to provide this information in the form of, e.g., Python docstrings in their node template. Similarly, this can be applied to edges to make it clear what messages/data are communicated between the nodes. This could greatly enhance the representation of the experiment from the Knowledge graph.
(3) The design of Heron was primarily with behavioral experiments in mind, in which highly accurate timing is not a strong requirement. Experiments in some other areas that this software is also hoping to expand to, for example, electrophysiology, may need very strong synchronization between apparatus, for example, the record timing and stimulus delivery should be synced. The communication mechanism implemented in Heron is asynchronous, as I understand it, and the code for each node is executed once upon receiving an event at one or more of its inputs. The paper, however, does not include a discussion, or example, about how Heron could be used to address issues that could arise in this type of communication. There is also a lack of information about, for example, how nodes handle inputs when their ability to execute their work function cannot keep up with the frequency of input events. Does the publication/subscription handle the queue intrinsically? Will it create problems in real-time experiments that make multiple nodes run out of sync? The reader could benefit from a discussion about this if they already exist, and if not, the software could benefit from implementing additional mechanisms such that it can meet the requirements from more types of experiments.
(4) The authors mentioned in "Heron GUI's multiple uses" that the GUI can be used as an experimental control panel where the user can update the parameters of the different Nodes on the fly. This is a very useful feature, but it was not demonstrated in the three showcases. A demonstration could greatly help to support this claim.
(5) The API for node scripts can benefit from having a better structure as well as having additional utilities to help users navigate the requirements, and provide more guidance to users in creating new nodes. A more standard practice in the field is to create three abstract Python classes, Source, Sink, and Transform that dictate the requirements for initialisation, work_function, and on_end_of_life, and provide additional utility methods to help users connect between their code and the communication mechanism. They can be properly docstringed, along with templates. In this way, the com and worker scripts can be merged into a single unified API. A simple example that can cause confusion in the worker script is the "worker_object", which is passed into the initialise function. It is unclear what this object this variable should be, and what attributes are available without looking into the source code. As the software is also targeting those who are less experienced in programming, setting up more guidance in the API can be really helpful. In addition, the self-documentation aspect of the GUI can also benefit from a better structured API as discussed in point 2 above.
(6) The authors should provide more pre-defined elements. Even though the ability for users to run arbitrary code is the main feature, the initial adoption of a codebase by a community, in which many members are not so experienced with programming, is the ability for them to use off-the-shelf components as much as possible. I believe the software could benefit from a suite of commonly used Nodes.
(7) It is not clear to me if there is any capability or utilities for testing individual nodes without invoking a full system execution. This would be critical when designing new experiments and testing out each component.
-
Reviewer #3 (Public Review):
Summary:<br /> The authors present a Python tool, Heron, that provides a framework for defining and running experiments in a lab setting (e.g. in behavioural neuroscience). It consists of a graphical editor for defining the pipeline (interconnected nodes with parameters that can pass data between them), an API for defining the nodes of these pipelines, and a framework based on ZeroMQ, responsible for the overall control and data exchange between nodes. Since nodes run independently and only communicate via network messages, an experiment can make use of nodes running on several machines and in separate environments, including on different operating systems.
Strengths:<br /> As the authors correctly identify, lab experiments often require a hodgepodge of separate hardware and software tools working together. A single, unified interface for defining these connections and running/supervising the experiment, together with flexibility in defining the individual subtasks (nodes) is therefore a very welcome approach. The GUI editor seems fairly intuitive, and Python as an accessible programming environment is a very sensible choice. By basing the communication on the widely used ZeroMQ framework, they have a solid base for the required non-trivial coordination and communication. Potential users reading the paper will have a good idea of how to use the software and whether it would be helpful for their own work. The presented experiments convincingly demonstrate the usefulness of the tool for realistic scientific applications.
Weaknesses:<br /> In my opinion, the authors somewhat oversell the reproducibility and "self-documentation" aspect of their solution. While it is certainly true that the graph representation gives a useful high-level overview of an experiment, it can also suffer from the same shortcomings as a "pure code" description of a model - if a user gives their nodes and parameters generic/unhelpful names, reading the graph will not help much. Making the link between the nodes and the actual code is also not straightforward, since the code for the nodes is spread out over several directories (or potentially even machines), and not directly accessible from within the GUI. The authors state that "[Heron's approach] confers obvious benefits to the exchange and reproducibility of experiments", but the paper does not discuss how one would actually exchange an experiment and its parameters, given that the graph (and its json representation) contains user-specific absolute filenames, machine IP addresses, etc, and the parameter values that were used are stored in general data frames, potentially separate from the results. Neither does it address how a user could keep track of which versions of files were used (including Heron itself).
Another limitation that in my opinion is not sufficiently addressed is the communication between the nodes, and the effect of passing all communications via the host machine and SSH. What does this mean for the resulting throughput and latency - in particular in comparison to software such as Bonsai or Autopilot? The paper also states that "Heron is designed to have no message buffering, thus automatically dropping any messages that come into a Node's inputs while the Node's worker function is still running."- it seems to be up to the user to debug and handle this manually?
As a final comment, I have to admit that I was a bit confused by the use of the term "Knowledge Graph" in the title and elsewhere. In my opinion, the Heron software describes "pipelines" or "data workflows", not knowledge graphs - I'd understand a knowledge graph to be about entities and their relationships. As the authors state, it is usually meant to make it possible to "test propositions against the knowledge and also create novel propositions" - how would this apply here?
-
-
www.biorxiv.org www.biorxiv.org
-
eLife assessment
This study presents valuable empirical work and simulations that are relevant for the evolution of genetic load linked to self-incompatibility alleles in Arabidopsis. The evidence supporting the findings is solid but could be improved by a more detailed quantitative assessment of the impacts of deleterious alleles and their dominance. The simulation results are somewhat incomplete, as details of the approach and code do not appear to be available, but this could be easily remedied. The work will be of relevance to geneticists interested in the evolution of allelic diversity in similar systems.
-
Reviewer #2 (Public Review):
Summary:
This study looks into the complex dominance patterns of S-allele incompatibilities in Brassicaceae, through which it attempts to learn more about the sheltering of deleterious load. I found several weak points in the analyses that diminished my excitement about the results. In particular, the way in which deleterious mutations were classified lacked the ability to distinguish the severity of the mutations and thus their expected associated dominance. Furthermore, the simulation approach could have provided this exact sort of insight but was not designed to do so, making this comparison to the empirical data also less than exciting for me.
Major and minor comments:
I think the introduction (or somewhere before we dive into it in the results) of the dominance hierarchy for the S-alleles needs a more in-depth explanation. Not being familiar with this beforehand really made this paper inaccessible to me until I then went to find out more before continuing. I would expect this paper to be broad enough that self-contained information makes it accessible to all readers. For example, lines 110-115 could be in the Introduction.
Along with my above comment, perhaps it is not my place to comment, but I find the paper not of a broad enough scope to be of interest to a broad readership. This S-allele dominance system is more than simple balancing selection, it is a very complex and specific form of dominance between several haplotypes, and the mechanism of dominance does not seem to be genetic. I am not sure that it thus extrapolates to broad comments on general dominance and balancing selection, e.g. it would not be the same as considering inversions and this form of balancing selection where we also expect recessive deleterious mutations to accumulate.
It would have been particularly interesting, or a nice addition, to see deleterious mutations classed by something like SNPeff or GERP where you can have different classes of moderate to severe deleterious variants, which we would expect also to be more recessive the more deleterious they are. In line with my next comment on the simulations, I think relative differences between mutations expected to be more or less dominant may be even more insightful into the process of sheltering which may or may not be going on here.
In the simulations, h=0 and s=0.01 (as in Figure 5) for all deleterious mutations seems overly simplistic, and at the convenient end for realistic dominance. I think besides recessive lethals which we expect to be close to h=0 would have a much larger selection coefficient, and other deleterious mutations would only be partially recessive at such an s value. I expect this would change some of the simulation results seen, though to what degree I am not certain. It would be nice to at least check the same exact results for h=0.3 or 0.2 (or additionally also for recessive lethals, e.g. h=0 and s=-0.9). I would also disagree with the statement in line 677, many studies have shown, particularly those on balancing selection, that partially recessive deleterious mutations are not eliminated by natural selection and do play a role in population genetic dynamics. I am also not surprised that extinction was found for higher s values when the mutation rate for such mutations was very high and the distribution of s values was constant. An influx of such highly deleterious mutations is unlikely to ever let a population survive, yet that does NOT mean that in nature, the rare influx of such mutations does lead to them being sheltered. I find overall that the simulation results contribute very little, to none, to this paper, as without something more realistic, like a simultaneous distribution of s and h values, you cannot say which, if any class of these mutations are the ones expected to accumulate because of S-allele dominance. Rather they only show the disappointing or less exciting result that fully recessive, weakly deleterious mutations (which I again think do not even exist in nature as I said above) have minor, to no effect across the classes of S-allele dominance. They provide no insight into whether any type of recessive deleterious mutation can accumulate under the S-allele dominance hierarchy, and that is the interesting question at hand. I would either remove these simulations or redo them in another approach. The authors never mention what simulation approach was used, so I can only assume this is custom, in-house code. Yet I do not find that code provided on the github page. I do not know if the lack of a distribution for h and s values is then a choice or a programming limitation, but I see it as one that should be overcome if these simulations are meant to be meaningful to the results of the study.
-
-
www.biorxiv.org www.biorxiv.org
-
Data Accessibility
Do the authors plan to share the code that underpins these analyses at a future date?
-
- Feb 2024
-
happinessatwork.eu happinessatwork.eu
-
Talking about happiness at work is different from talking than talking about performance, commitment or sick leave
As software engineers, I think this is even more important because of how draining the work can be at times. It is easy to forget about the enjoyment of your work when you are caught up in a problem with your code which is why this is important.
-
-
emojipedia.org emojipedia.org
-
Man Technologist
emoji 👨💻 =
for - tech - tech.mix - Indy0.Net - dream.code - dreaming in code
-
-
emojipedia.org emojipedia.org
-
Thought Balloon
💭
emoji.for - thought - dreaming - dreaming in code
A large cloud-like shape more commonly known as a thought bubble.
Used to represent thinking, or thoughts, and commonly used in comics to display the thoughts of a drawn character. May also be used to represent a dream, or daydream.
-
-
www.semanticscholar.org www.semanticscholar.org
-
C
Not used in code for AR. Just for reference
-
-
docs.kadaster.nl docs.kadaster.nl
-
3.3.6.3.1 Referentielijst GNSS-ApparatuurType
Hier heb ik wer even een opfris nodig :-) code ?? igsNaam: LEIAR25.R3 LEIM of LEICA GR50 merknaam: Leica of Leica GR50?, wat is het verchil met 3.3.6.3.2 TypeGNSS-apparatuur?
-
-
www.biorxiv.org www.biorxiv.org
-
Reviewer #2 (Public Review):
The purpose of this study is to develop a tool that serves as a starting point for investigating and uncovering genes and pathways associated with aging. The tool utilizes information from the GTEx public database, which contains post-mortem human data. It focuses on identifying age-related gene expression changes across different age range, biological sexes, and medical histories, with a focus on specific tissues.
Additionally, the authors envision the platform as continuously evolving, with ongoing development and expansion to include new data and features, ensuring it remains a cutting-edge resource for researchers studying aging.
voyAGEr presents a tool for exploring gene expression changes across multiple tissues in the context of aging. One of the main strengths of the tool is its intuitive and user-friendly interface, which allows for easy navigation and exploration of gene expression patterns for biologists. Users can explore changes in gene expression of single genes across multiple tissues, enabling them to identify genes of interest that can be further investigated.
A particularly noteworthy strength of the tool is its ability to show tissue-specific gene expression patterns. This feature is essential for elucidating the paradigm of tissue-specific asynchronous aging and provides a unique and valuable resource for the aging community.
However, the choice of the R shiny platform for visualization may not be the most conducive to extensibility and open-source collaboration, owing to its lack of modularity. Alternatives like Flask or FastAPI, which are more production-oriented, could be more appropriate. Additionally, despite using preprocessed data and functioning primarily as a visualization platform, the tool occasionally experiences lag, indicating room for performance improvement. These aspects are worth considering for future versions of the tool.
Overall, voyAGEr offers an entry point for further investigation of genes involved in aging, and its ability to show tissue-specific gene expression patterns provides a unique and valuable resource for the scientific community.
Finally, the tool is complemented by a comprehensive tutorial that elucidates each functionality and includes examples. The authors have shared the code for preprocessing and the tool itself. They also acknowledge the limitations of the statistical inference tests and their interpretation in the manuscript, contributing to its transparency.
-
-
ovanisound.com ovanisound.com
-
This is for those who purchased our Humble Bundle at the Tier 1 level ($1). First, click add to cart. Do not remove this item from your order - it will also be discounted to $0.Once this is added to the cart, it will automatically add the appropriate products. Enter the coupon you were given in your Humble Bundle receipt to get 100% off. Be sure to use normal checkout. PayPal, Apple Pay, and Google Pay will not work with the coupon code. On the checkout page, scroll all of the way down and the coupon code field is on the bottom right. You will not have to enter any card information as the coupon code you were given from Humble Bundle takes 100% off and removes the card information fields. If you are asked to enter card information, something was done incorrectly.
-
-
www.sciencedirect.com www.sciencedirect.com
-
Jackson LaboratoriesStrian Code: 003770
DOI: 10.1016/j.medj.2024.02.002
Resource: (IMSR Cat# JAX_003770,RRID:IMSR_JAX:003770)
Curator: @abever99
SciCrunch record: RRID:IMSR_JAX:003770
-
Charles RiverStrain Code: 028
DOI: 10.1016/j.medj.2024.02.002
Resource: (IMSR Cat# CRL_028,RRID:IMSR_CRL:028)
Curator: @abever99
SciCrunch record: RRID:IMSR_CRL:028
-
Jackson LaboratoriesStrain Code: 000664
DOI: 10.1016/j.medj.2024.02.002
Resource: (IMSR Cat# JAX_000664,RRID:IMSR_JAX:000664)
Curator: @abever99
SciCrunch record: RRID:IMSR_JAX:000664
-
-
unix.stackexchange.com unix.stackexchange.com
-
but in a different situation target_utility "${@}" could represent more complex code
-
Duplicating non-trivial code in both the if and in the else could create unnecessary issues.
-
-
stackoverflow.com stackoverflow.com
-
For anyone else having the same issue with the integrity, adding the following code to my config.yml file solved the problem.
这个有效
-
-
phoenixnap.com phoenixnap.com
-
Master (master/main) branch. The default production branch in a Git repository that needs to be permanently stable. Developers can merge changes into the master branch only after code review and testing. All collaborators on a project must keep the master branch stable and updated.
-
-
www.tedmouw.info www.tedmouw.info
-
key variables
(#37)
Let's inspect the code for the plm FE model here.
(McKenna) In the HW6 code, what does “index=c(“id”, “year”) refer to?
Response: This tells R the structure of the (longitudinal) data; “id” is the i subscript and “year” is the j subscript. I.e., the model will calculate level 2 means based on id. Level 1 is id and year.
-
-
www.biorxiv.org www.biorxiv.org
-
eLife assessment
This study provides useful initial information on how humans represent two-dimensional abstract spaces in relation to the social traits of warmth and competence. While the study poses an interesting question, the evidence for a grid-like code at present is incomplete. This study will be of interest to researchers working in the field of spatial navigation as well as the navigation of conceptual abstract space.
-
Reviewer #2 (Public Review):
Summary:<br /> In this work, Liang et al. investigate whether an abstract social space is neurally represented by a grid-like code. They trained participants to 'navigate' around a two-dimensional space of social agents characterized by the traits warmth and competence, then measured neural activity as participants imagined navigating through this space. The primary neural analysis consisted of three procedures: 1) identifying brain regions exhibiting the hexagonal modulation characteristic of a grid-like code, 2) estimating the orientation of each region's grid, and 3) testing whether the strength of the univariate neural signal increases when a participant is navigating in a direction aligned with the grid, compared to a direction that is misaligned with the grid. From these analyses, the authors find the clearest evidence of a grid-like code in the prefrontal cortex and weaker evidence in the entorhinal cortex.
Strengths:<br /> The work demonstrates the existence of a grid-like neural code for a socially-relevant task, providing evidence that such coding schemes may be relevant for a variety of two-dimensional task spaces.
Weaknesses:<br /> In the revised manuscript, the authors soften their claims about finding a grid code in the entorhinal cortex and provide additional caveats about limitations in their findings. It seems that the authors and reviewers are in agreement about the following weaknesses, which were part of my original review: Claims about a grid code in the entorhinal cortex are not well-supported by the analyses presented. The whole-brain analysis does not suggest that the entorhinal cortex exhibits hexagonal modulation; the strength of the entorhinal BOLD signal does not track the putative alignment of the grid code there; multivariate analyses do not reveal any evidence of a grid-like representational geometry.
In the authors' response to reviews, they provide additional clarification about their exploratory analyses examining whether behavior (i.e., reaction times) and individual difference measures (i.e., social anxiety and avoidance) can be predicted by the hexagonal modulation strength in some region X, conditional on region X having a similar estimated grid alignment with some other region Y. My guess is that readers would find it useful if some of this language were included in the main text, especially with regard to an explanation regarding the rationale for these exploratory studies.
-
Reviewer #3 (Public Review):
Liang and colleagues set out to test whether the human brain uses distance and grid-like codes in social knowledge using a design where participants had to navigate in a two-dimensional social space based on competence and warmth during an fMRI scan. They showed that participants were able to navigate the social space and found distance-based codes as well as grid-like codes in various brain regions, and the grid-like code correlated with behavior (reaction times).
On the whole, the experiment is designed appropriately for testing for distant-based and grid-like codes, and is relatively well powered for this type of study, with a large amount of behavioral training per participant. They revealed that a number of brain regions correlated positively or negatively with distance in the social space, and found grid-like codes in the frontal polar cortex and posterior medial entorhinal cortex, the latter in line with prior findings on grid-like activity in entorhinal cortex. The current paper seems quite similar conceptually and in design to previous work, most notably Park et al., 2021, Nature Neuroscience.
(1) The authors claim that this study provides evidence that humans use a spatial / grid code for abstract knowledge like social knowledge.
This data does specifically not add anything new to this argument. As with almost all studies that test for a grid code in a similar "conceptual" space (not only the current study), the problem is that, when the space is not a uniform, square/circular space, and 2-dimensional then there is no reason the code will be perfectly grid like, i.e., show six-fold symmetry. In real world scenarios of social space (as well as navigation, semantic concepts), it must be higher dimensional - or at least more than two dimensional. It is unclear if this generalizes to larger spaces where not all part of the space is relevant. Modelling work from Tim Behrens' lab (e.g., Whittington et al., 2020) and Bradley Love's lab (e.g., Mok & Love, 2019) have shown/argued this to be the case. In experimental work, like in mazes from the Mosers' labs (e.g., Derdikman et al., 2009), or trapezoid environments from the O'Keefe lab (Krupic et al., 2015), there are distortions in mEC cells, and would not pass as grid cells in terms of the six-fold symmetry criterion.
The authors briefly discuss the limitations of this at the very end but do not really say how this speaks to the goal of their study and the claim that social space or knowledge is organized as a grid code and if it is in fact used in the brain in their study and beyond. This issue deserves to be discussed in more depth, possibly referring to prior work that addressed this, and raise the issue for future work to address the problem - or if the authors think it is a problem at all.
-
-
hypothes.is hypothes.is
-
Don Quixote's moral code arises from a narcissistic pursuit kleos based on the outdated notion of chivalry. Here, Don Quixote assumes that the farmer will abide by his orders because of an outdated notion of knightly chivalry. However, as soon as Don Quixote leaves, the farmer continues abusing Andres–even intensifying it to spite Don Quixote. This shows that simply abiding by a moral system, individually, doesn't mean that others follow that system which makes Don Quixote's pursuit of justice futile seeing as people will just continue their previous behavior as soon as Don Quixote leaves. This is an example of Don Quixote's narcissism since he believes that everyone abides by knightly chivalry just because he does, and he assumes that his influence is so strong that others will automatically abide by his instructions out of fear. However, that moral system is not compatible with the society Don Quixote lives in, and his reputation is simply not large enough to force compliance with his orders, as shown with this farmer.
I totally agree. My reading of DQ so far is that his misunderstanding of the situations he finds himself in, be they hallucinations or poor judgement, reflect a greater personification of attitudes toward shifts in European societal structures and the almost melancholy undertones of change. Don Quixote is at once an individualist and someone seeking public recognition. However nuclear his views, he propounds a far simpler perspective that is no doubt appealing to men who feel their only outlet of power is to gain kleos through physical prowess and courtly love.
-
-
softwareengineering.stackexchange.com softwareengineering.stackexchange.com
-
The increment-after-release model makes sense for branching too. Suppose you have a mainline development branch, and you create maintenance branches for releases. The moment you create your release branch, your development branch is no longer linked to that release's version number. The development branch contains code that is part of the next release, so the version should reflect that.
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
First, we would like to thank you and all the reviewers for acknowledging the meaningful contribution of our manuscript to the field. Your useful comments helped us improve the manuscript's quality. We understood the key issues of the manuscript were the quantification of inference accuracy and applicability to methylome data. We here therefore present a revised version of the manuscript addressing all major comments.
For each demographic inference we have added the root mean square error as demanded by the reviewers. These results confirm the previous interpretation of the graphs especially in recent times. We also added TMRCA inference analysis as requested by one reviewer as a proof of principle that integrating multiple markers can improve ARG inference.
The discussion was rewritten to further discuss the challenges of application to empirical methylation data. We clarify that in the case epimutations are well understood and modelled, they can be integrated into a SMC framework to improve the approaches accuracy. When epimutations are not well understood, our approach can help understand the epimutations process through generations at the evolutionary time scale along the genome. Hence, in both cases our approach can be used to unveil marker evolution processes through generations, and/or deepen our understanding of the population past history. We hope our discussion underlies better how our approach is designed and can be used.
eLife assessment
This important study advances existing approaches for demographic inference by incorporating rapidly mutating markers such as switches in methylation state. The authors provide a solid comparison of their approach to existing methods, although the work would benefit from some additional consideration of the challenges in the empirical use of methylation data. The work will be of broad interest to population geneticists, both in terms of the novel approach and the statistical inference proposed.
Public Reviews:
Reviewer #1 (Public Review):
The authors developed an extension to the pairwise sequentially Markov coalecent model that allows to simultaneously analyse multiple types of polymorphism data. In this paper, they focus on SNPs and DNA methylation data. Since methylation markers mutate at a much faster rate than SNPs, this potentially gives the method better power to infer size history in the recent past. Additionally, they explored a model where there are both local and regional epimutational processes.
Integrating additional types of heritable markers into SMC is a nice idea which I like in principle. However, a major caveat to this approach seems to be a strong dependence on knowing the epimutation rate. In Fig. 6 it is seen that, when the epimutation rate is known, inferences do indeed look better; but this is not necessarily true when the rate is not known. A roughly similar pattern emerges in Supp. Figs. 4-7; in general, results when the rates have to be estimated don't seem that much better than when focusing on SNPs alone. This carries over to the real data analysis too: the interpretation in Fig. 7 appears to hinge on whether the rates are known or estimated, and the estimated rates differ by a large amount from earlier published ones.
Overall, this is an interesting research direction, and I think the method may hold more promise as we get more and better epigenetic data, and in particular better knowledge of the epigenetic mutational process. At the same time, I would be careful about placing too much emphasis on new findings that emerge solely by switching to SNP+SMP analysis.
Answer: We thank the reviewer 1 for his positive comments and acknowledging the future promises of our method as better and more reliable data will be available in different species. We appreciate the reviewer noticing the complete set of work undertaken here to integrate local and regional effects of methylation into a model containing as much knowledge of the epigenetics mutational processes as possible. Note that in Figure 2 of the manuscript we observed a gain of accuracy even when the rates are unknown. Our results thus suggests that the accuracy gain of additional marker with unknown rates is also possible, although it is most likely be scenario and rate dependent.
At last, as noticed and highlighted by the very recent work of the Johannes lab (Yao et al. Science 2023) using phylogenetic methods, knowing the epimutation rate is essential at short time scale to avoid confounding effects of homoplasy. In our estimation of the coalescent trees, the same applies, though our model considers finite site markers. We now provide additional evidence for the potential gain of power to infer the TMRCA (Supplementary Table S7) when knowing or not the epimutation rates and revised the discussion to clarify the potential shortcomings/caveats for the analysis of real data.
Reviewer #2 (Public Review):
A limitation in using SNPs to understand recent histories of genomes is their low mutation frequency. Tellier et al. explore the possibility of adding hypermutable markers to SNP based methods for better resolution over short time frames. In particular, they hypothesize that epimutations (CG methylation and demethylation) could provide a useful marker for this purpose. Individual CGs in Arabidopsis tends to be either close to 100% methylated or close to 0%, and are inherited stably enough across generations that they can be treated as genetic markers. Small regions containing multiple CGs can also be treated as genetic markers based on their cumulative methylation level. In this manuscript, Tellier et al develop computational methods to use CG methylation as a hypermutable genetic marker and test them on theoretical and real data sets. They do this both for individual CGs and small regions. My review is limited to the simple question of whether using CG methylation for this purpose makes sense at a conceptual level, not at the level of evaluating specific details of the methods. I have a small concern in that it is not clear that CG methylation measurements are nearly as binary in other plants and other eukaryotes as they are in Arabidopsis. However, I see no reason why the concept of this work is not conceptually sound. Especially in the future as new sequencing technologies provide both base calling and methylating calling capabilities, using CG methylation in addition to SNPs could become a useful and feasible tool for population genetics in situations where SNPs are insufficient.
Answer: We thank the reviewer 2 for his positive comments. Indeed, surveys of CG methylation in other plant species show that its distribution is clearly bimodal (i.e. binary). This is not the case for non-CG methylation, such as CHG and CHH (where H=C,T,A). However, these later types of methylation contexts are also not heritable across generations and can therefore not be used as heritable molecular markers.
Reviewer #3 (Public Review):
I very much like this approach and the idea of incorporating hypervariable markers. The method is intriguing, and the ability to e.g. estimate recombination rates, the size of DMRs, etc. is a really nice plus. I am not able to comment on the details of the statistical inference, but from what I can evaluate it seems sound and reasonable. This is an exciting new avenue for thinking about inference from genomic data. I have a few concerns about the presentation and then also questions about the use of empirical methylation data sets.
I think a more detailed description of demographic accuracy is warranted. For example, in L245 MSMC2 identifies the bottleneck (albeit smoothed) and only slightly overestimates recent size. In the same analysis the authors' approach with unknown mu infers a nonexistent population increase by an order of magnitude that is not mentioned.
Answer: We thank the reviewer 3 for his positive comments and refer to our answer to reviewer 1 above. We added RMSE (Root Mean Square Error) analyses to quantify the inference accuracy. We apologize for not mentioning this last point. Thank you for pointing this out and we have now fixed it (line 245-253).
Similarly, it seems problematic that (L556) the approach requiring estimation of site and region parameters (as would presumably be needed in most empirical systems like endangered nonmodel species mentioned in the introduction) does no better than using only SNPs. Overall, I think a more objective and perhaps quantitative comparison of approaches is warranted.
Answer : See answer to reviewer 1 above, and more elaborate answers below. We provide now new RMSE analyses to quantify the accuracy of our demographic inference (Supplementary Tables 1,6,7,8,9,10). We also discuss the validity and usefulness of our approach when the epimutation rates are unknown. In short, the discussion was rewritten to further discuss the challenges of application to empirical methylation data. We clarify that in the case epimutations are well known and modelled (as much is known in A. thaliana for example), they can be integrated into a SMC framework to improve the accuracy of the method approach. When epimutations are not well understood and rates unknown, our approach can help understand the epimutational process through generations at the evolutionary time scale. Hence, whether makers are understood or not, our approach can be used to study the marker evolutionary processes through generations and/or to deepen our understanding of the population past history. We hope our discussion underlies better how our approach is designed and can be used.
The authors simulate methylated markers at 2% (and in some places up to 20%). In many plant genomes a large proportion of cytosines are methylated (e.g. 70% in maize: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8496265/). I don't know what % of these may be polymorphic, but this leads to an order of magnitude more methylated cytosines than there are SNPs. Couldn't this mean that any appreciable error in estimating methylation threatens to be of a similar order of magnitude to the SNP data? I would welcome the authors' thoughts here.
Answer : The reviewer is correct and this is an interesting question. First, studies show that heritable epimutations in plants are restricted to CG dinucleotides that are located well outside of the target regions of de novo methylation pathways in plants. Most of these CGs tend of fall within so-called gene body methylated regions. While it is true that plant species can differ substantially in their proportion of methylation at the genome-wide scale, the number of gene body methylated genes (i.e. genic CG methylation) is relatively similar, and at least well within the same order of magnitude (Takuno et al. Nature Plants 2016, review in Muyle et al. Genome Biol Evol 2022). Moreover, spontaneous CG epimutations in gene body methylated regions has been shown to be neutral (van Der Graaf et al. 2015, Vidali et al. 2016, Yao et al. 2023), which is an ideal property for phylogentic and demographic inference.
Second, CG methylation calls are sometimes affected by coverage or uncertainty. Stringent filtering for reliable SMP calls typically reduces the total proportion of CG sites that can be used as input for demographic inference. Here we only kept CG sites where the methylation information could be fully trusted after SMP calling (i.e. >99.9% posteriori certainty). Overall, this explains why the percentage of sites with methylation information is so small, and why we have decided to work on simulation with 2% of reliable methylated markers.
Nevertheless, for the sake of generality, it may be that in some species such as maize a higher percentage of polymorphic methylated sites can be used, and the number of SMPs could be higher than that of SNPs when the effective population size is very small (due to past demographic history and/or life history traits). In this case, any error in the epimutation rate and variance due to the finite site model estimation (and homoplasy) are not corrected by the lack of SNPs and can lead to mis-inference.
A few points of discussion about the biology of methylation might be worth including. For example, methylation can differ among cell types or cells within a tissue, yet sequencing approaches evaluate a pool of cells. This results in a reasonable fraction of sites having methylation rates not clearly 0 or 1. How does this variation affect the method? Similarly, while the authors cite literature about the stable inheritance of methylation, a sentence or so more about the time scale over which this occurs would be helpful.
Answer: We thank reviewer 3 for asking those very interesting questions, which we further developed below and mention in the discussion (lines 716-722).
For Arabidopsis thaliana:
Following up on our previous comment above, the majority of the CG sites that serve as input to our approach are located in body methylated genes. Previous work has shown that CG methylation in these regions shows essentially no tissue and cellular heterogeneity (e.g. Horvath et al. 2019). This means that bulk methylation measurements only show limited susceptibility to measurement error. That said, to guard against any spurious SMPs call that could arise from residual measurement variation, we applied stringent filtering of CG methylation. We have kept sites where the methylation percentage is close to either 0% or 100% (the rest being removed from the analysis). We have used similar filtering strategies in previous studies of epimutational processes in mutation accumulation lines and long-lived perennials (work of the Johannes lab). In these later studies we found that the SMP calls sufficiently accurate for inferences of phylogenetic parameters in experimental settings (Sharyhary et al. Genome Biology 2021, Yao et al. Science, 2023).
For other species:
It is true that currently, evaluating the methylation state of a site from a pool of cells may be problematic for some species for two main reasons: 1) it will add noise to the signal and SMP calling could be erroneous, and 2) the methylation state used in analysis might originate from different tissues at different location of the genome/methylome. Overall, this will lead to spurious SMPs and can render the inference inaccurate (see Sellinger et al 2021 for the effect of spurious SNPs). Hence, caution is advised when calling SMPs in other species and for different tissues.
Finally, in some species methylated cytosines have mutation rates an order of magnitude higher than other nucleotides. The authors mention they assume independence, but how would violation of this assumption affect their inference?
Answer: Indeed, we assume the mutation and epimutation process to be independent thus the probability for a SNP to occur does not depend on the local methylation state. If this was the case, the mutation rate use would indeed be wrong to a degree function of the dependency between the processes. We suggest that by ignoring this dependence, we are in the same situation as ignoring the variation of mutation rate along the genome. We have previously documented the effect of ignoring this biological feature of genomes in Strüt et al 2023 and Sellinger et al 2021. The variation in mutation rate along the genome if too extreme and not accounted for can lead to erroneous inference results. However, this problem could be easily solved (modelled) by adapting the emission matrix. To correctly model this dependency, additional knowledge is needed: either the mutation and epimutation rates must be known to quantify the dependency, or the dependency must be known to quantify the resulting rates. As far as we know, these data are at the moment not available, but could maybe be obtained using the MA lines of A. thaliana (used in Yao et al. 2023).
Recommendations for the authors:
All three reviewers liked this approach and found it a valuable contribution. I think it is important to address reviewer 1/3 concerns about quantifying the accuracy of inference (the TMRCA approach from reviewer 1 sounds pretty reasonable), and reviewer 1 also highlights an intriguing point about model accuracy being worse when the mutation rate is known. Additionally, I think some discussion is warranted about challenges dealing with empirical methylation data (points from Rev 2 and 3 as well as Rev 1's question about inferred vs published rates of epigenetic mutation).
Answer : We have added tables containing the root mean square error (RMSE) of every demographic inference in the manuscript to better quantify accuracy. We have below given the explanation on why accuracy in presence of site and region epimutations can in some cases decrease when real rates are known (because methylation state at the region level needs to be first inferred). We added evidence that accounting for methylation can improve the accuracy when recovering the TMRCA along the genome when the rates are known. We also have enhanced the discussion on the challenges of dealing with epimutations data for inference. As is suggested, we hope this study will generate an interest in tackling these challenges by applying the methods to various methylome datasets from different species.
Reviewer #1 (Recommendations For The Authors):
Major comments:
- For all of the simulated demographic inference results, only plots are presented. This allowsfor qualitative but not quantitative comparisons to be made across different methods. It is not easy to tell which result is actually better. For example, in Supp. Fig. 5, eSMC2 seems slightly better in the ancient past, and times the trough more effectively, while SMCm seems a bit better in the very recent past. For a more rigorous approach, it would be useful to have accompanying tables that measure e.g. mean-squared error (along with confidence intervals) for each of the different scenarios, similar to what is already done in Tables 1 and 2 for estimating $r$.
Answer : We understand the concern of reviewer #1 for a more quantitative approach to compare the inference results. We agree that plots are not sufficient to fully grasp a method performance. To provide better supports to quantity approaches performance, we added Sup tables 1,6,8,9 and 10 containing the RMSE (in log10 for visibility) for all Figures. The root mean-squared error is calculated as in Sellinger 2021 and a description of how the root mean-squared error is calculated and now found in the method section lines 886-893.
- 434: The discussion downplays the really odd result that inputting the true value of themutation rate, in some cases, produces much worse estimates than when they are learned from data (SFig. 6)! I can't think of any reason why this should happen other than some sort of mathematical error or software bug. I strongly encourage the authors to pin down the cause of this puzzling behaviour.
Answer : There are unfortunately no errors in this plot and those results are perfectly normal and coherent, but we understand they can be confusing at first.
As described in the method section and in the appendix, when accounting for regionlevel epimutations, our algorithm requires the regional methylation status which needs to be inferred as a first step from the data (real or simulated). Because region and single site epimutation events are occurring at similar rates in our simulated scenario, the methylation state of the region is very hard to correctly recover (e.g. there will be unmethylated site in methylated regions and methylated sites in unmethylated regions). In other words, the accuracy of the region estimation HMM procedure is decreased by the joint action of site and region epimutation processes.
When subsequently applying the HMM for inference, as described in the appendix, the probabilities of two CG site being in the same or different methylation state depends on the methlylation state of the "region". Hence the mislabelling of the region methylation state is (to some extent) equivalent to spurious SMPs (or inaccurate SMP calling).
If the true rates for site and region epimutations are given as input, the model forces the demography (and other inferred parameters) to fit the observed distribution of SMPs (given the inputted rates), resulting in the poor accuracy observed in the Figure (Now Supplementary Figure 7).
Note: The estimated rates from real data in A. thaliana suffer from the same issue as the region and site epimutation rates are independently estimated, and the existence of regions first quantified using an independent HMM method (Denkena et al. 2022).
However, when rates are freely inferred, they are inferred accordingly to the estimated methylation status of regions and SNPs. Therefore, even if the inferred rates are wrong, they are used by the SMC in a more consistent way.
Note: When methylation rates violate the infinite site assumption, such as here, we first estimate the tree sequence along the genome using SNPs (i.e. DNA mutations). The algorithm then infers the epimutations rates given the inferred coalescent times and the observed methylation diversity.
To summarise: when inputting rates to the model, if the model fails to correctly recover the region methylation status there will be conflicting information between SNPs and SMPs leading to accuracy loss. However if the rates are inferred this is realized with the help of SNPs, leading to less conflicting information and potentially smaller loss of accuracy. We apologize that the explanations were missing from the manuscript and have added them lines 449-460 and 702-716.
A further argument is that if region and site epimutations occur at rates of at least two orders of magnitude difference, the inference results are better (and accurate) when the true rates are given. The reason is that one epimutational process overrides the other (see Supplementary Table 2). In that case one epimutation process is almost negligible and we fall back to results from Figure 5 or Supplementary Figure 6.
- As noted at 580, all of the added power from integrating SMPs/DMRs should come fromimproved estimation of recent TMRCAs. So, another way to study how much improvement there is would be to look at the true vs. estimated/posterior TMRCAs. Although I agree that demographic inference is ultimately the most relevant task, comparing TMRCA inference would eliminate other sources of differences between the methods (different optimization schemes, algorithmic/numerical quirks, and so forth). This could be a useful addition, and may also give you more insight into why the augmented SMC methods do worse in some cases.
Answer : We fully agree with reviewer 1. We have added a comparison in TMRCA inference as proof of principle between using or not using methylation sites. The results are written in Supplementary Table 7 and methodology is inspired by Schiffels 2014 and described at the end of the method section (line 894-907). Those results demonstrate the potential gain in accuracy when using methylation polymorphic. However, TMRCA (or ARG) inference is a very vast and complex subject in its own right. Therefore, we are developing a complete TMRCA/ARG inference investigation and an improve methodology than the one presented in this manuscript. To do so we are currently working on a manuscript focusing on this topic specifically. We hence consider further investigations of TMRCA/ARG inference beyond the scope of this current study.
- A general remark on the derivations in Section 2 of the supplement: I checked theseformulas as best I could. But a cleaner, less tedious way of calculating these probabilities would be to express the mutation processes as continuous time Markov chains. Then all that is needed is to specify the rate matrices; computing the emission probabilities needed for the SMC methods reduces to manipulating the results of some matrix exponentials. In fact, because the processes are noninteracting, the rate matrix decomposes into a Kronecker sum of the individual rate matrices for each process, which is very easy to code up. And this structure can be exploited when computing the matrix exponential, if speed is an issue.
Answer: We thank the reviewer for this very interesting suggestion! Unfortunately, it is a bit late to re-implement the algorithm and reshape the manuscript according to this suggestion. Speed is not yet an issue but will most likely become one in the future when integrating many different rates or when using a more complex SMC model. Hence, we added reviewer #1 suggestions to the discussion (line 648) and hope to be using it in our future projects.
- Most (all?) of the SNP-only SMC methods allow for binning together consecutiveobservations to cut down on computation time. I did not see binning mentioned anywhere, did you consider it? If the method really processes every site, how long does it take to run?
Answer: This is a very good question. We do the binning exactly as described in Mailund 2013 & Terhorst 2017, and added this information in the method section (lines 801-809). However, as described in Terhorst 2017, one can only bin observation of the same "type" (to compute the Baum-Welch algorithm). Therefore, the computation time gain by binning is reduced when different markers spread along the genome in high proportion. This is the approach we used throughout the study when facing multiple markers as it had the best speed performance. As for example, when the proportion of site with methylated information is 1% or less, computation time is only slightly affected (i.e. same order of magnitude).
However, the binning method presented in Mailund 2013 can be extended to observation of different types, but parameters need to be estimated through a full likelihood approach (as presented in Figure 2). In our study this approach did not have the best speed performance. However, as our study is the first of its kind, it remains sub-optimal for now. Hence, we did not further investigate the performance of our approach in presence of many multiple different genomic marker (e.g. 5 different markers each representing ~20% of the genome each). Currently, with SMC approaches a high proportion of sites contain the information "No SNPs", making the Baum welch algorithm described in Terhorst 2017 very efficient. But when further developing our theoretical approach, we expect that most of the sites in a genome analysis will contain some "information", which could render the full likelihood approach computationally more tractable.
- 486: The assumed site and region (de)methylation rates listed here are several OOMdifferent from what your method estimated (Supp. Tables 5-6). Yet, on simulated data your method is usually correct to within an order of magnitude (Supp. Table 4). How are we to interpret this much larger difference between the published estimates and yours? If the published estimates are not reliable, doesn't that call into question your interpretation of the blue line in Fig. 7 at 533?
Answer: We thank the reviewer for asking this question. We believe answering this question is indeed the most interesting aspect of our study. Beyond demographic inference, our study has indeed unveiled a discrepancy between rates inferred through biological experiment and our study through the use of SNPs and branch length. There are several reasons which could explained the discrepancy between both approaches:
-
Firstly, our underlying HMM hypotheses are certainly violated. We ignoredpopulation structure, variation of mutations and recombination rate along the genome as well as the effect of selection. Hence, the branch lengths used for methylation rate estimations are to some extent inaccurate. We note that this is especially likely for the short branches of coalescent tree originating from background selection events in the coding regions and which are especially observable when using the methylation sites with a higher mutation rate than SNPs (Yao et al. 2023) at body methylated genes.
-
Secondly, calling single methylation site polymorphism is not 100 % reliable. If theerror rate is 0.1%, as the study was conducted on ~10 generations a minimum epimutation rate of 10-4 is to be expected. However, because our approach works at the evolutionary time scale, we expect that it suffers less from this bias as the proportion of diversity originating from actual epimutations, and not SMP calling error, should be greater.
-
Thirdly, as mentioned above, recovering the methylation status of a region is veryhard. Hence false region status inference could affect our inference accuracy as shown in Supplementary Figure 4.
-
Lastly and most importantly, the reason behind this discrepancy is the modelling ofepimutation and methylation between sites and regions. As we discuss, the current combination of rates and models is still limited to describe the observed diversity along the genome (as we intend in SMC methods). This is in contrast to the recent study by Yao et al. where very few regions of polymorphic SMPs are chosen, which implicitly avoids the influence of the methylation region effect. A study just published by Biffra et al. (Cell reports 2023) also uses a functional model of methylation modelling using a mix of region and site epimutation, albeit not tuned for evolutionary analyses. Thus we suggest, in line with functional studies, that epimutations are not independent from the local methylation context and may tend to stabilize the methylation state of a region. Therefore, the estimated methylation rates show a discrepancy to the previously measured ones. Indeed, the biological experiment would reveal a fast epimutation rate because epimutations can actually be tracked at sites which can mutate, while region mutation rate is much slower. However, because the methylation state of a region is rather stable through time it would reduce the methylation diversity over long time scale, and these rates would differ between methylated or unmethylated regions (i.e. the methylation rate is higher in methylated regions). Our results are thus in agreement with the observation by Biffra et al. that region methylation modelling is needed to explain patterns of methylation across the genome.
To solve the discrepancy, one would need to develop a theoretical region + site epimutation model capable of describing the observed diversity at the evolutionary time scale (possibly based on the Biffra et al. model within an underlying population evolution model), and then use this model to reanalyse the sequence data from the biological experiment (i.e. in de Graaf et al. 2015 & Denkena et al. 2022) to re-estimate the methylation region sizes and epimutation rates.
Minor comments:
- 189: "SMCtheo" first occurs here, but it's not mentioned until 247 that this is the newmethod being presented.
Answer : Fixed
- 199: Are the estimates in this section from a single diploid sequence? Or is it n=5 (diploid) as mentioned in the earlier section?
Answer : Yes, those results were obtained with 5 diploid individuals. We added it in the Table 1 description.
- 336: I'm confused by the wording: it sounds like the test rejects the null if there is positivecorrelation in the methylation status across sites. But then, shouldn't 339 read "if the test is significant" (not non-significant)?
Answer : We apologize for the confusion and rewrote the sentence line 339-348, the choice of word was indeed misleading .
- Fig. 6: for some reason fewer simulations were run for 10Mb (panels C nad D) than for100Mb (A and B). Since it's very difficult to tell what's happening on average in the 10Mb case, I suggest running the same number of simulations.
Answer : Yes we understand your concern. Actually, the same number of simulations were run but we plotted only the first 3 runs as it was less visually confusing. We now have added the missing lines to the plot C and D.
Typos:
-
104: "or or"
-
292: build => built
-
388: fulfil
-
683: sample => samples
Answer : Many thanks to reviewer 1 for pointing out the typos. They are all now fixed.
Reviewer #2 (Recommendations For The Authors):
The authors may find some valuable information in Pisupati et al (2023) "On the causes of gene-body methylation variation in Arabidopsis thaliana" on interpreting epimutation rates.
Answer: Many thanks for the recommended manuscript. We add it to the cited literature as it strongly supports our use of heritability or methylation. We also added the recent Biffra et al. paper.
Reviewer #3 (Recommendations For The Authors):
There are many places throughout the manuscript with minor grammatical errors. Please review these. A few noted below as I read:
L104: extra "or"
L123: built not build
L 160 "relies" instead of "do rely"
L161 "events"
L 336 "from methylation data"
L 378 "exists"
L 379 "regions are on average shorter" instead of "there are shorter"
L 338 "a regional-level"
L 349 "," instead of "but"
L 394 DMRs
Table 1 legend: parentheses not brackets?
Answer : Many thanks to reviewer #3 for finding those mistakes. They are all now fixed.
I think a paragraph in the discussion of considerations of when to use this approach might be helpful to readers. Comparison to e.g. increased sample size in MSMC2, while not necessary, might be helpful here. It may often be the case that doubling the number of haplotypes with SNP data may be easier and cheaper estimating methylation accurately.
Answer : We discuss (lines 691-698) that our approach is always useful by design, but cannot always be used for the same purpose. If the evolutionary properties of the used marker used are not understood, we suggest that our approach can be used to investigate the marker heritability process through generations. This could help to correctly design experiments aiming to study the marker heritability through lineages. And if the properties of the marker are well understood and modelled, it can be integrated into the SMC framework to improve inference accuracy.
Other minor notes:
L 486 "known" is a stretch. empirically estimated seems appropriate.
Answer : Fixed
L 573 ARG? You are not estimating the full ARG here.
Answer : We apologize for the wrong choice of word and have rephrased the sentence.
Fig. 2 is not super useful and could be supplemental.
Answer : We moved Figure 2 to the appendix (now sup fig 1)
-
Reviewer #1 (Public Review):
The authors developed an extension to the pairwise sequentially Markov coalecent model that allows to simultaneously analyze multiple types of polymorphism data. In this paper, they focus on SNPs and DNA methylation data. Since methylation markers mutate at a much faster rate than SNPs, this potentially gives the method better power to infer size history in the recent past. Additionally, they explored a model where there are both local and regional epimutational processes.
Integrating additional types of heritable markers into SMC is a nice idea which I like in principle. However, a major caveat to this approach seems to be a strong dependence on knowing the epimutation rate. In Fig. 6 it is seen that, when the epimutation rate is known, inferences do indeed look better; but this is not necessarily true when the rate is not known. (See also major comment #1 below about the interpretation of these plots.) A roughly similar pattern emerges in Supp. Figs. 4-7; in general, results when the rates have to be estimated don't seem that much better than when focusing on SNPs alone. This carries over to the real data analysis too: the interpretation in Fig. 7 appears to hinge on whether the rates are known or estimated, and the estimated rates differ by a large amount from earlier published ones.
Overall, this is an interesting research direction, and I think the method may hold more promise as we get more and better epigenetic data, and in particular better knowledge of the epigenetic mutational process. At the same time, I would be careful about placing too much emphasis on new findings that emerge solely by switching to SNP+SMP analysis.
Major comments:<br /> - For all of the simulated demographic inference results, only plots are presented. This allows for qualitative but not quantitative comparisons to be made across different methods. It is not easy to tell which result is actually better. For example, in Supp. Fig. 5, eSMC2 seems slightly better in the ancient past, and times the trough more effectively, while SMCm seems a bit better in the very recent past. For a more rigorous approach, it would be useful to have accompanying tables that measure e.g. mean-squared error (along with confidence intervals) for each of the different scenarios, similar to what is already done in Tables 1 and 2 for estimating $r$.
- 434: The discussion downplays the really odd result that inputting the true value of the mutation rate, in some cases, produces much worse estimates than when they are learned from data (SFig. 6)! I can't think of any reason why this should happen other than some sort of mathematical error or software bug. I strongly encourage the authors to pin down the cause of this puzzling behaviour. (Comment addressed in revision. Still, I find the explanation added at 449ff to be somewhat puzzling -- shouldn't the results of the regional HMM scan only improve if the true mutation rate is given?)
- As noted at 580, all of the added power from integrating SMPs/DMRs should come from improved estimation of recent TMRCAs. So, another way to study how much improvement there is would be to look at the true vs. estimated/posterior TMRCAs. Although I agree that demographic inference is ultimately the most relevant task, comparing TMRCA inference would eliminate other sources of differences between the methods (different optimization schemes, algorithmic/numerical quirks, and so forth). This could be a useful addition, and may also give you more insight into why the augmented SMC methods do worse in some cases. (Comment addressed in revision via Supp. Table 7.).
- A general remark on the derivations in Section 2 of the supplement: I checked these formulas as best I could. But a cleaner, less tedious way of calculating these probabilities would be to express the mutation processes as continuous time Markov chains. Then all that is needed is to specify the rate matrices; computing the emission probabilities needed for the SMC methods reduces to manipulating the results of some matrix exponentials. In fact, because the processes are noninteracting, the rate matrix decomposes into a Kronecker sum of the individual rate matrices for each process, which is very easy to code up. And this structure can be exploited when computing the matrix exponential, if speed is an issue.
- Most (all?) of the SNP-only SMC methods allow for binning together consecutive observations to cut down on computation time. I did not see binning mentioned anywhere, did you consider it? If the method really processes every site, how long does it take to run?
- 486: The assumed site and region (de)methylation rates listed here are several OOM different from what your method estimated (Supp. Tables 5-6). Yet, on simulated data your method is usually correct to within an order of magnitude (Supp. Table 4). How are we to interpret this much larger difference between the published estimates and yours? If the published estimates are not reliable, doesn't that call into question your interpretation of the blue line in Fig. 7 at 533? (Comment addressed in revision.)
-
-
www.biorxiv.org www.biorxiv.org
-
Author Response
The following is the authors’ response to the original reviews.
Reviewer #1 (Public Review):
Summary:
This study examines the role of host blood meal source, temperature, and photoperiod on the reproductive traits of Cx. quinquefasciatus, an important vector of numerous pathogens of medical importance. The host use pattern of Cx. quinquefasciatus is interesting in that it feeds on birds during spring and shifts to feeding on mammals towards fall. Various hypotheses have been proposed to explain the seasonal shift in host use in this species but have provided limited evidence. This study examines whether the shifting of host classes from birds to mammals towards autumn offers any reproductive advantages to Cx. quinquefasciatus in terms of enhanced fecundity, fertility, and hatchability of the offspring. The authors found no evidence of this, suggesting that alternate mechanisms may drive the seasonal shift in host use in Cx. quinquefasciatus.
Strengths:
Host blood meal source, temperature, and photoperiod were all examined together.
Weaknesses: The study was conducted in laboratory conditions with a local population of Cx. quinquefasciatus from Argentina. I'm not sure if there is any evidence for a seasonal shift in the host use pattern in Cx. quinquefasciatus populations from the southern latitudes.
We agree on the reviewers observation about the evidence on seasonal shift in the host use pattern in Cx. quinquefasciatus populations from southern latitudes. We include a paragraph in the Introduction section regarding this. Unfortunately, studies conducted in South America to understand host use by Culex mosquitoes are very limited, and there are virtually no studies on the seasonal feeding pattern. In Argentina, there is some evidence (Stein et al., 2013, Beranek, 2019) regarding the seasonal change in host use by Culex species, including Cx. quinquefasciatus, where the inclusion of mammals during the autumn has been observed. As part of a comprehensive study on characterising bridge vectors for SLE and WN viruses, our research group is currently working on the molecular identification of blood meals from engorged females to gain deeper insights into the seasonal feeding pattern of Culex mosquitoes. While the seasonal change in host use by Culex quinquefasciatus has not been reported in Argentina so far, there has been an observed increase in reported cases of SLE virus in humans between summer and fall (Spinsanti et al., 2008). It is based on this evidence that we hypothesise there is a seasonal change in host use by Cx. quinquefasciatus, similar to what occurs in the United States. This is also considering that both countries (Argentina and the United States) have regions with similar climatic conditions (temperate climates with thermal and hydrological seasonality). Since we work on the same species and in a similar temperate climate regimen, we assumed there is a seasonal shift in the host use by this mosquito species.
Reviewer #1 (Recommendations for the authors):
Abstract
Line 23: fed on two different hosts.
Accepted as suggested.
I think the concluding statement should be rewritten to say that immediate reproductive outcomes do not explain the shift in host use pattern of Cx. quinquefasciatus mosquitoes from birds to mammals towards autumn.
Accepted as suggested.
Introduction
No comments.
Materials and Methods
Please mention sample sizes in the text as well (n = ?) for each treatment.
Accepted as suggested.
Page 99: ......C. quinquefasciatus, since C. pipiens and its hybrids are present as well in Cordoba.
Accepted as suggested.
Results – Line 146: subsequently instead of posteriorly
Accepted all changes as suggested.
Line 148: were counted instead of was counted.
Accepted all changes as suggested.
Line 160: Subsequently instead of posteriorly
Accepted all changes as suggested.
Line 171: on fertility
Accepted all changes as suggested.
Line 174: there was an interaction effect on…
Accepted all changes as suggested.
Line 175: there were no differences in the number of eggs
Accepted all changes as suggested.
Discussion
I think the first paragraph in the discussion section is redundant and should be deleted.
The whole discussion was rewritten to be focused on our aims and results.
Line 282: this sentence needs to be rewritten.
Accepted as suggested.
Line 299: at 28{degree sign}C
Line 300: at 30{degree sign}C
Sorry, but we are not sure about your comment here. We checked. Temperatures are written as stated, 28°C and 30°C.
Line 363: I think the authors need to discuss more about the bigger question they were addressing. I think that the discussion section can be strengthened greatly by elaborating on whether there is evidence for a seasonal shift in host use pattern in Cx. quinquefasciatus in the southern latitudes. If yes, what alternate mechanisms they believe could be driving the seasonal change in host use in this species in the southern latitudes now that they show the 'deriving reproductive advantages' hypothesis to be not true for those populations.
Thanks for this observation. We agree and so the Discussion section was restructured to align it with our results, as suggested.
Reviewer #2 (Public Review):
Summary:
Conceptually, this study is interesting and is the first attempt to account for the potentially interactive effects of seasonality and blood source on mosquito fitness, which the authors frame as a possible explanation for previously observed host-switching of Culex quinquefasciatus from birds to mammals in the fall. The authors hypothesize that if changes in fitness by blood source change between seasons, higher fitness in birds in the summer and on mammals in the autumn could drive observed host switching. To test this, the authors fed individuals from a colony of Cx. quinquefasciatus on chickens (bird model) and mice (mammal model) and subjected each of these two groups to two different environmental conditions reflecting the high and low temperatures and photoperiod experienced in summer and autumn in Córdoba, Argentina (aka seasonality). They measured fecundity, fertility, and hatchability over two gonotrophic cycles. The authors then used a generalized linear mixed model to evaluate the impact of host species, seasonality, and gonotrophic cycle on fecundity and fertility and a null model analysis via data randomization for hatchability. The authors were trying to test their hypothesis by determining whether there was an interactive effect of season and host species on mosquito fitness. This is an interesting hypothesis; if it had been supported, it would provide support for a new mechanism driving host switching. While the authors did report an interactive impact of seasonality and host species, the directionality of the effect was the opposite of that hypothesized. While this finding is interesting and worth reporting, there are significant issues with the experimental design and the conclusions that are drawn from the results, which are described below. These issues should be addressed to make the findings trustworthy.
Strengths:
(1) Using a combination of laboratory feedings and incubators to simulate seasonal environmental conditions is a good, controlled way to assess the potentially interactive impact of host species and seasonality on the fitness of Culex quinquefasciatus in the lab.
(2) The driving hypothesis is an interesting and creative way to think about a potential driver of host switching observed in the field.
Weaknesses:
(1) There is no replication built into this study. Egg lay is a highly variable trait, even within treatments, so it is important to see replication of the effects of treatment across multiple discrete replicates. It is standard practice to replicate mosquito fitness experiments for this reason. Furthermore, the sample size was particularly small for some groups (e.g. 15 egg rafts for the second gonotrophic cycle of mice in the autumn, which was the only group for which a decrease in fecundity and fertility was detected between 1st and 2nd gonotrophic cycles). Replicates also allow investigators to change around other variables that might impact the results for unknown reasons; for example, the incubators used for fall/summer conditions can be swapped, ensuring that the observed effects are not artefacts of other differences between treatments. While most groups had robust sample sizes, I do not trust the replicability of the results without experimental replication within the study.
We agree egg lay is a variable trait and so we consider high numbers of mosquitoes and egg lay during experiments compared to our studies of the same topics. Evaluating variables such as fecundity, fertility, or other types of variables (collectively referred to as "life tables") is a challenging issue that depends on several intrinsic and extrinsic factors. Because all of this, in some experiments, sample sizes might not be very large, and in several articles, lower sample sizes could be found. For instance, in Richards et al. (2012), for Culex quinquefasciatus, during the second gonotrophic cycle, some experiments had 13 or even 6 egg rafts. For species like Aedes aegypti, the sample size for life table analysis is also usually small. As an example, Muttis et al. (2018) reported between 1 and 4 engorged females (without replicates). In addition, small sample size would be a problem if we would not have obtained any effect, which is not the case due to the fact that we were interested in finding an effect, regardless of the effect size. Because of this, we do find our sample sizes quite robust for our results.
Regarding the need to repeat the experiments in order to give more robustness to the study we also agree. However, after a review of the literature (articles cited in the original manuscript), it is apparent that similar experiments are not frequently repeated as such. Examples of this are the studies of Richards et al. (2012), Demirci et al. (2014) or Telang & Skinner (2019), which even they manipulate several cages at a time as “replicates”, they are not true replicates because they summarise and manipulate all data together, and do not repeat the experiment several times. We see these “replicates” as a way of getting a greater N.
As was stated by the reviewer, repetition is a resource and time-consuming activity that we are not able to do. Replicating the experiment poses a significant time and resources challenge. The original experiment took over three months to complete, and it is anticipated that a similar timeframe would be necessary for each replication (6 months in total considering two more replicates). Given our existing commitments and obligations, dedicating such an extensive period solely to this would impede progress on other crucial projects and responsibilities.
Given the limitations of resources and time and the infrequent use of experimental replication in this type of studies, we performed a simulation-based analysis via a Monte Carlo approach. This approach involved generating synthetic data that mimics the expected characteristics of the original experiment and subsequently subjecting it to the same analysis routine. The main goal of this simulation was to evaluate the potential spuriousness and randomness of the results that might arise due to the experimental conditions. So, evaluating the robustness and confidence of our results and data.
(2) Considering the hypothesis is driven by the host switching observed in the field, this phenomenon is discussed very little. I do not believe Cx. quinquefasciatus host switching has been observed in Argentina, only in the northern hemisphere, so it is possible that the species could have an entirely different ecology in Argentina. It would have been helpful to conduct a blood meal analysis prior to this experiment to determine whether using an Argentinian population was appropriate to assess this question. If the Argentinian populations don't experience host switching, then an Argentinian colony would not be the appropriate colony to use to assess this question. Given that this experiment has already been conducted with this population, this possibility should at least be acknowledged in the discussion. Or if a study showing host switching in Argentina has been conducted, it would be helpful to highlight this in the introduction and discussion.
Thanks for this observation. We agree. However, we conducted the experiment beside host use data from Argentina since we used the mosquito species, and the centre region of Argentina (Córdoba) has a similar temperate weather regimen that those observed in the east coast of US.
We are aware that few studies regarding host shifting in South America are available, some such that those conducted by Stein et al. (2013) and Beranek (2019) reported a moderate host switch for Culex quinquefasciatus in Argentina. We have already performed a study about seasonal host feeding patterns for this species. However, even though there are few studies regarding host shifting, our hypothesis is based mainly in the seasonality of human cases of WNV and SLEV, a pattern that has been demonstrated for our region, see for example the study of Spinsanti et al. (2008).
We include a new paragraph in the Introduction and Discussion sections. Please see answers Reviewer #1.
(3) The impacts of certain experimental design decisions are not acknowledged in the manuscript and warrant discussion. For example, the larvae were reared under the same conditions to ensure adults of similar sizes and development timing, but this also prevents mechanisms of action that could occur as a result of seasonality experienced by mothers, eggs, and larvae.
We understand the confusion that may have arisen due to a lack of further details in the methodology. If we are not mistaken, you are referring to our oversight regarding the consideration of carry-over effects of larvae rearing that could potentially impact reproductive traits. When investigating the effects of temperature or other environmental factors on reproductive traits, it is possible to acclimate either larvae or adults. This is due to the significant phenotypic plasticity that mosquitoes exhibit throughout their entire ontogenetic cycle. In our study, we followed an approach similar to that of other authors where the adults are exposed to experimental conditions (temperature and photoperiod). For a similar approach you can refer to the studies conducted by Ferguson et al. (2018) for Cx. pipiens, Garcia Garcia & Londoño Benavides (2007) for Cx. quinquefasciatus or Christiansen-Jucht et al. (2014, 2015) for Anopheles gambiae.
(4) There are aspects of the data analysis that are not fully explained and should be further clarified. For example, there is no explanation of how the levels of categorical variables were compared.
The methodology and statistical analysis were expanded for a better understanding.
(5) The results show the opposite trend as was predicted by the authors based on observed feeding switches from birds to mammals in the autumn. However, they only state this once at the end of the discussion and never address why they might have observed the opposite trend as was hypothesized.
The discussion was restructured to focus on our results and our model.
(6) Generally speaking, the discussion has information that isn't directly related to the results and/or is too detailed in certain parts. Meanwhile, it doesn't dig into the meaning of the results or the ways in which the experimental design could have influenced results.
As mentioned above, the discussion was restructured to reflect our findings. We also included the effect that our design might have influenced our results. However, as stated above we do not fully agree that the design is inadequate for our analysis, we performed standard protocols followed by other researchers and studies in this research field.
(7) Beyond the issue of lack of replication limiting trust in the conclusions in general, there is one conclusion reached at the end of the discussion that would not be supported, even if additional replicates are conducted. The results do not show that physiological changes in mosquitoes trigger the selection of new hosts. Host selection is never measured, so this claim cannot be made. The results don't even suggest that fitness might trigger selection because the results show that physiological changes are in the opposite direction as what would be hypothesized to produce observed host switches. Similarly, the last sentence of the abstract is not supported by the results.
We agree with this observation. However, we did not evaluate the impact of fitness on host selection in this study. Instead, we aimed to investigate the potential influence of seasonality on mosquito fitness as a potential trigger for a shift in host selection. We agree that we have incorrectly used the term “host selection” when we should actually be discussing “host use change”. Our results indicate a seasonal alteration in mosquito fitness in response to temperature and photoperiod changes. Building upon this observation, we re-discussed our hypothesis and theoretical model to explain this seasonal shift in host use.
(8) Throughout the manuscript, there are grammatical errors that make it difficult to understand certain sentences, especially for the results.
All English grammar and writing of the manuscript was revised and corrected to be easily understood.
This study is driven by an interesting question and has the potential to be a valuable contribution to the literature.
Reviewer #2 (Recommendations for The Authors):
I hope that the authors will consider the suggested revisions and experimental replication to improve the quality of the study and paper.
This study tests a very interesting hypothesis. I understand that additional replicates are difficult to conduct, but I do believe that fitness studies absolutely require experimental replicates. Unless you are able to replicate the observed effects, I personally would not trust the results of this study. I hope that you will consider conducting replicates so that this important question can be answered in a more robust manner. Below, I expand upon some additional points in the public review and also provide more specific suggestions. I provided some copy-editing feedback, but was not able to point out all grammatical mistakes. I suggest that you use ChatGPT to help you edit the English. For example, you can feed ChatGPT your MS and ask it to bold the grammatical errors or you can ask it to edit grammatical errors and bold the sections that were edited. I understand that writing in a second language is very difficult (from personal experience!), so I view ChatGPT as a great tool to help even the playing field for publishing. Below are line item suggestions. Apologies that wording is curt, I was trying to be efficient in writing.
20-21: I suggest that you emphasize that you are investigating the interactive effect.
Accepted as suggested.
22: they weren't "reared" (from larvae) in different conditions, they were "maintained" as adults
Accepted as suggested.
26-27: increased/decreased is a bit misleading since you did not evaluate these groups sequentially in time. It might be more accurate to describe it as less than/greater than. Also, if you say increased/decreased or less than/greater than, you should always say what you are comparing to. The same applies throughout the MS.
Accepted as suggested.
29-30: "finding the" is not correct here; could be "with the lowest..."
Accepted as suggested.
34-36: I do not think that your results suggest this, even if you were to replicate the results of this experiment. You haven't shown metabolic changes.
We understand the point. Accepted as suggested.
42-44: "one of the main responsible" should be "one of the main species responsible..."
Accepted as suggested.
48: I think that "host preference" is better than selection here; -philic denotes preference
Accepted as suggested.
50: "Moreover" isn't the correct transition word here
Accepted as suggested.
57: "could" isn't correct here; consider saying "... species sometimes feed primarily on mammal hosts, including humans, in certain situations."
Accepted as suggested.
58: Different isn't correct word here
Accepted as suggested.
60: delete "feeding"
Accepted as suggested.
66-68: I am not familiar with any blood meal analysis studies in the southern hemisphere that show host switching for Culex species between summer and autumn. If this hasn't been shown, then this critique of the host migration hypothesis doesn't make sense.
There are some studies pointing this out (Stein et al., 2013, Beranek 2019), and unpublished data from us). However, our hypothesis has supported by epidemiological data observed in human population which indicate a seasonal activity pattern. It was explained in depth in the Introduction section.
68: ensures is not the right word; I suggest "suggests"
Accepted as suggested.
68-70: this explanation isn't clear to me; please revise
It will be revised. Accepted as suggested.
70: change cares to care
Accepted as suggested.
76-77: can you explain how they were not supported by the data for the benefit of those who are not familiar with these papers please?
Accepted as suggested.
87-89: I suggest the following wording: "In the autumn, we expect a greater number of eggs (fecundity) and larvae (fertility) in mosquitoes after feeding on a mammal host compared to an avian host, and the opposite relationship in the summer."
Accepted as suggested.
99: edit for grammar
Accepted as suggested.
102: suggest: "...offered a blood meal from a restrained chicken twice a month"
Accepted as suggested.
107: powder
Accepted as suggested.
108: inbred? Is this the term you meant to use?
Changed as suggested.
109: "several" cannot be used to describe 20 generations; suggest using "over twenty generations"; also, it would be good to acknowledge in your discussion that lab adaptation could force evolution, especially since mosquitoes are kept at constant temperatures and fed with certain hosts (with easy access) in the lab. Also, it would be good to know when the experiments were conducted to know the lapse of time between the creation of the colony and the experiments.
Accepted as suggested.
110-111: Does humidity vary between summer and fall in Córdoba? If so, I suggest acknowledging in the discussion that if humidity differences are involved in a potential interaction between host species and seasonality, then this would not have been captured by your experimental design.
Several variables change during seasons. We were interested in capturing the effects of temperature and photoperiod, since humidity is a variable difficult to control.
113-116: I suggest combining into one sentence to make more concise.
Accepted as suggested.
135: You might be obscuring the true impact of seasonality by rearing the larvae under the same conditions. There may be signals that mothers/eggs/larvae receive that influence their behavior (e.g. I believe this is the case for diapause), so this limitation should also be acknowledged. I understand why you decided to do this to control for development time and size, but it is something that should be considered in the discussion.
As it was explained above, Cx. quinquefasciatus do not suffer diapause in our country. Maintaining mosquitoes from adults was an approach selected by us based on other studies.
138: edit: "with cotton pads soaked in... on plastic..."; what is plastic glass? Do you mean plastic dishes?
Accepted as suggested.
141: here and throughout paragraph, full should be "fully"
Accepted as suggested.
144: located should be "placed"
Accepted as suggested.
147: suggest editing to "at which point, they were fixed with 1 mL of 96% ethanol and the number of L1 larvae per raft was counted."
Accepted as suggested.
154-155: edit for grammar
Accepted as suggested.
157: Your GLM explanation doesn't say anything about how you made pairwise comparisons between your levels; did you use emmeans?
This revised version includes a more detailed methodology and statistical analysis. Accepted as suggested.
158-160: I don't understand why you took this approach - it seems strange to me to use this analysis, but I am not familiar with it, so it might be that I lack the knowledge to be able to adequately evaluate. Please provide more explanation so that readers can better understand this analysis. A citation for this kind of application of the analysis would be helpful.
It was changed to be in accordance with the remaining analyses.
173: replace neither with either
Accepted as suggested.
174: this applies throughout; edit to : "An interaction effect was observed..."
Accepted as suggested.
175: "it was not found" is grammatically incorrect; instead : "We did not find ..." or "no differences in... were detected", etc
Accepted as suggested.
183: "it was detected" is grammatically incorrect
Accepted as suggested.
185-186: "being this treatment... in terms of fitness": I do not understand what this means. Please rephrase
Accepted as suggested.
170-199: you should provide the effect sizes and p values in text and/or in the figure for the pairwise comparisons
Accepted as suggested.
193-196. These two sentences are confusing and I am not sure what you mean, especially in the first sentence.
It was rewritten. Accepted as suggested.
Figure 1: This figure is great and easy to read and interpret! Thank you for the comment! 218-219: it is important to state which mosquito species you are referring to here.
Accepted as suggested.
226-227: you definitely should acknowledge the small sample size here.
Considered.
227: "it was observed" should be "We observed" or "A greater hatching rate.... was observed."
Accepted as suggested.
228-229: is the result really comparable even though you took very different approaches to the analysis for these outcomes?
Changed to be comparable.
230-278: the discussion of these hypotheses is too long and detailed, especially since the comparison of mouse vs chicken wasn't your main question; you really wanted to understand this in the context of seasonality. I suggest cutting this down a lot and making room to dig into your results more, and also to discuss the potential impacts of your experimental design/limitations on the results.
Discussion was changed to focus on our results and model. Accepted as suggested.
281: Hoffman is an old citation; I suggest you cite a modern review.
Accepted as suggested. We deleted it due to the re-writing of the manuscript.
282: "It can be recognise".. I am not sure what you are trying to say here
Accepted as suggested.
- After the first time you write a species name, you can abbreviate the genus in all future mentions unless it is at the beginning of a sentence.
Accepted as suggested.
303-305: Revise this sentence. E.g "Fewer studies are available regarding photoperiod and show mixed results; Mogi (1992) found that mid and long day lengths induced greater fecundity while Costanzo et al. (2015) did not find differences in fecundity by day length."
Accepted as suggested.
315-316: typically, unpublished data shouldn't be referenced; I'm not sure if eLife has a policy on this.
We will check this with eLife guidelines. However, since the lack of evidence on this pattern we consider important to include this unpublished data.
316: Aegypti should be lowercase
Accepted as suggested.
328-330: This sentence is redundant with the first sentence of the paragraph
Accepted as suggested.
321-336: You never reintroduced your hypothesis in your discussion. I suggest that you center your whole discussion more directly around the hypothesis that motivated the study. If you decide not to restructure your discussion, you should at least reintroduce your hypothesis here and discuss how your results do not support the hypothesis.
Accepted as suggested.
337-348: This paragraph is a bit confusing as you jump between fertility and hatchability
Accepted as suggested.
353: is viral transmission the right word to use here? I think you might mean bridge vector transmission to humans specifically?
Accepted as suggested.
357: you say "neither" but never define which traits you are referring to
Accepted as suggested.
361: I suggest "two variables previously analyzed separately..."
Accepted as suggested.
General: There is no statement about the availability of data; it is eLife policy to require all data to be publicly available. Also, it would be helpful to share your code to help understand how you conducted pairwise comparisons, etc.
In the submission it was not mentioned anything about data availability. However, all data and scripts will be uploaded with the VOR if it is required.
Recommendations for the authors:
I found your study interesting and potentially promising. However, there are some fundamental problems with the study design and the hypothesis, including:
<(1) Seasonality simulation - Seasonality is strongly associated with time, so it is unusual to simulate seasonal factors without accounting for time. The actual factors associated with seasonal change in reproductive output may be neither a difference in host blood meal nor temperature and photoperiod. It is therefore, odd to reduce seasonality to a difference in photoperiod and temperature in summer and autumn without even mentioning the time of year when the experiment was carried (except for the mention of February as the time the stock samples were collected from the wild).
The temperature and photoperiod settings are established according to a representative day in both autumn and summer. To determine these settings, we utilized climate data spanning a 3-year period (2020-2022), encompassing the most frequently occurring temperatures and day lengths. The weather conditions remained notably consistent throughout this time frame, which is why the specific year was not mentioned. Moreover, including the year in laboratory experiment details is uncommon, as evident in various papers. This practice can be corroborated by referring to multiple sources (cited in the original manuscript). We mention this in the new version.
(2) Hypothesis - While the hypothesis alludes to the 'reason' for seasonal host shift, the prediction is on the outcome of the interaction between blood meal type and season.
It might be nicer to frame your hypothesis to be consistent with the aim, which is, testing the partial contributions of blood meal type, versus photoperiod and temperature to seasonal change in the reproductive output of Culex quinquefasciatus. A hypothesis like that can be accompanied by alternative predictions according to the expected individual and interactive effects of both factors.
It was rewritten in the revised version to be consistent with our predictions and findings.
Blood meal type, temperature, and photoperiod are all components of seasonality, so the strength of the study is its potential to decouple the effect of blood meal type from that of temperature and photoperiod on the seasonal reproductive output of Culex quinquefasciatus by comparing the two blood meal types under simulated summer and winter conditions. Ideally, this should have been over a natural summer and winter because a natural time difference captures the effect of other seasonal factors other than temperature and photoperiod.
Furthermore, the hypothesis stemmed from field observations, while the study itself was conducted under laboratory conditions using a local population of Culex quinquefasciatus from Argentina. It remains uncertain whether there is supporting evidence for a seasonal shift in host usage in Culex quinquefasciatus from the stock population. Discussing the field observations within the stock population would provide valuable insights.
It was considered in the new version.
-
-
openclassrooms.com openclassrooms.com
-
des exercices qui vous aideront à mettre en place votre code
Je ne trouve pas ces exercices.. Quelqu'un pourrait m'aider à les trouver ?
-
-
openai.com openai.com
-
The success of the LLM paradigm is enabled in part by the use of tokens that elegantly unify diverse modalities of text—code, math and various natural languages.
tokenizer的重要性。
-
-
www.biorxiv.org www.biorxiv.org
-
ABSTRACTAs genomic sequencing technology continues to advance, it becomes increasingly important to perform joint analyses of multiple datasets of transcriptomics. However, batch effect presents challenges for dataset integration, such as sequencing data measured on different platforms, and datasets collected at different times. Here, we report the development of BatchEval Pipeline, a batch effect workflow used to evaluate batch effect on dataset integration. The BatchEval Pipeline generates a comprehensive report, which consists of a series of HTML pages for assessment findings, including a main page, a raw dataset evaluation page, and several built-in methods evaluation pages. The main page exhibits basic information of the integrated datasets, a comprehensive score of batch effect, and the most recommended method for removing batch effect from the current datasets. The remaining pages exhibit evaluation details for the raw dataset, and evaluation results from the built-in batch effect removal methods after removing batch effect. This comprehensive report enables researchers to accurately identify and remove batch effects, resulting in more reliable and meaningful biological insights from integrated datasets. In summary, the BatchEval Pipeline represents a significant advancement in batch effect evaluation, and is a valuable tool to improve the accuracy and reliability of the experimental results.
This work has been published in GigaByte Journal under a CC-BY 4.0 license (https://doi.org/10.46471/gigabyte.108) as part of our Spatial Omics Methods and Applications series (https://doi.org/10.46471/GIGABYTE_SERIES_0005), and has published the reviews under the same license as follows:
**Reviewer 1. Chunquan Li **
- Page 1, Lines 14-16. The authors indicate that “it is crucial to thoroughly investigate the batch effects in the dataset before integrating and processing the data”. The term “thoroughly” may be not accurate enough. The current method can alleviate the batch effects, but it can’t thoroughly solve the related problems. In addition, this work proposes a batch evaluation tool, such “reasonably evaluate the batch effects” may be more accurate than “thoroughly investigate the batch effects”.
- In Figure 1, does the first box is “integrated datasets”?
- Page 5, Line 168, and Page 6, Lines 169-175, the content of these two paragraphs is similar, with some redundant descriptions. It is recommended to organize and write them into one paragraph.
- There is Table 1 in the table list, but Table 1 is missing in the main text.
- Page 8, Discussion section, it is better to discuss the differences between the proposed tool and a similar tool “batchQC”, especially the advantages of the proposed tool.
- Some other minor issues: Page 1, Line 22, “to do so” should be “to do it”. Page 3, Line 100, Ref. [13] should be cited when it first appears on Line 97. Page 4, Line 114 and Page 5, Line 146, “UMAP” should be given its full name when it first appears and abbreviated directly in the following text. The variable should be in italics, such as “p” on Page 4, Line 119, “H” on Page 6, Line 184.
Reviewer 2. W. Evan Johnson and Howard Fan
Is the source code available, and has an appropriate Open Source Initiative license (https://opensource.org/licenses) been assigned to the code?
Yes. However, the code could use substantial improvements.
Is installation/deployment sufficiently outlined in the paper and documentation, and does it proceed as outlined?
No. The manuscript is missing a section describing the software and its implementation.
Is there enough clear information in the documentation to install, run and test this tool, including information on where to seek help if required?
Yes. But it took a while to get it installed.
Have any claims of performance been sufficiently tested and compared to other commonly-used packages?
No. I think the most glaring deficiency in the paper is the lack of comparison with other methods. For example, there is no comparison of the tools available in BatchEval compared to other methods, such as BatchQC. Also, they mention that BatchQC might not work on larger datasets, but they perform no performance evaluation for BatchEval, and no comparison with BatchQC to demonstrate improved performance.
Are there (ideally real world) examples demonstrating use of the software?
Yes. Missed opportunity--I think the most exciting thing I observed from the paper was that the example data were from spatial transcriptomics data! To my knowledge, existing batch effect methods are not directly adapted to manage these data (although they did mention tools like BatchQC cannot handle large datasets, which may be true). But they don’t mention anything about batch adjustment/evaluation in spatial data in the manuscript. I feel that if the authors address this niche it would increase the value/impact of their work!
Additional Comments:
This review was conducted and written by Evan Johnson, who developed the competing BatchQC software.
The authors provide an interesting toolkit for assessing batch effects in genomics data. The paper was clear and well-written, albeit I had a few concerns (see below). We were also able to download the associated software and test it out (comments below as well).
I think the most exciting thing I observed from the paper was that the example data were from spatial transcriptomics data! To my knowledge, existing batch effect methods are not directly adapted to manage these data (although they did mention tools like BatchQC cannot handle large datasets, which may be true). But they don’t mention anything about batch adjustment/evaluation in spatial data in the manuscript. I feel that if the authors address this niche it would increase the value/impact of their work!
In addition, this toolkit is written in Python, while BatchQC and other tools are written in R, so this is an advantage of the method as well—it addresses an audience that uses Python for gene expression analysis (not as big as the R community, but substantial). Their Python toolkit might also be more accessible to implementation in a pipeline workflow (for a core or large project) than R-based tools like BatchQC—this might be important to mention this as well.
I think the most glaring deficiency in the paper is the lack of comparison with other methods. For example, there is no comparison of the tools available in BatchEval compared to other methods, such as BatchQC. Also, they mention that BatchQC might not work on larger datasets, but they perform no performance evaluation for BatchEval, and no comparison with BatchQC to demonstrate improved performance.
Similarly, the authors claim: “Manimaran [10] has developed user-friendly software for evaluating batch effects. However, the software does not take into account nonlinear batch effects and may not be able to provide objective conclusions.” I don’t understand what the authors mean by “may not be able to provide objective conclusions” – BatchQC provides – several visual and numerical evaluations of batch effect – more so than even the proposed BatchEval does. Did the authors mean something else, maybe that the lack of non-linear correction may lead to less accurate conclusions?
A related concern: does BatchEval provide non-linear adjustments? I may have missed this, but it seems that BatchEval is not providing non-linear adjustments either. Also, regarding non-linear adjustments, the authors should show in an example the problems with a lack non-linear adjustments and show that pre-transforming the data before using BatchQC does not perform as well as the non-linear BatchEval adjustments.
In Equation 10, should “batchScore” be BatchEvalScore?
Also, in the bottom of Figure on page 15, should the “BatchQCScore” also be BatchEvalScore??
The manuscript is missing a section describing the software and its implementation.
I asked my research scientist, who recently graduated with his PhD in Bioinformatics, to assess the software and examples. First of all, much of the software is named “BatchQC”. I think this is confusing, since the method is really named BatchEval and it will be confused with BatchQC which is another existing/competing software. Furthmore, it took him a significant effort to install the BatchEval software and get is working on our cluster. I would recommend the authors make their software more accessible and easier to install.
The output of the software was a nice .html report diagnosing the batch effects in the data—very useful (attached is a combined .pdfs of the .htmls that we generated). We were also able to generate a report for the harmony adjusted example using their code. One major disadvantage was that these reports are separate files, and this could get very complicated comparing cases using multiple batch effect methods that will all be in separate reports (refer to a recent single cell batch comparison that compared more than a dozen methods – Tran et al. Genome Biology, 2020 – it would be hard to use BatchEval for this comparison).
Also, it seems that the user is required to conduct the batch correction themselves, BatchEval does not help with the correction except for their example code for Harmony.
Finally, on comparing the raw and Harmony adjusted datasets, inspection of the visual assessments (e.g. PCA) show some improvement—although not a perfect correction. But must of the numerical assessments are still the sample. The BatchEvalScore in both cases leads to the conclusion “Need to do batch effect removal”. What’s missing is the difference or improvement that Harmony makes on its correction. Maybe this is just because Harmony doesn’t fully remove the batch effects? Or is there something not working in the code? Might be good to see another example where the batch effect correction improves the BatchEvalScore significantly.
Additional Files: https://gigabyte-review.rivervalleytechnologies.com/journal/gx/download-files?YXJ0aWNsZT00NDImZmlsZT0xNzEmdHlwZT1nZW5lcmljJnZpZXc9dHJ1ZQ~~
Re-review:
I find this paper to be much improved in this version. The authors have clearly worked hard to address my concerns and have addressed them in a satisfactory manner. I fully support the publication of this paper, and I believe their tools are a nice addition to the field.
-
-
www.biorxiv.org www.biorxiv.org
-
Reviewer #3 (Public Review):
Summary: The paper "Unveiling the signaling network of FLT3-ITD AML improves drug sensitivity prediction" reports the combination of prior knowledge signaling networks, multiparametric cell-based data on the activation status of 14 crucial proteins emblematic of the cell state downstream of FLT3 obtained under a variety of perturbation conditions and Boolean logic modeling, to gain mechanistic insight into drug resistance in acute myeloid leukemia patients carrying the internal tandem duplication in the FLT3 receptor tyrosine kinase and predict drug combinations that may reverse pharmacoresistant phenotypes. Interestingly, the utility of the approach was validated in vitro and using real-world data.
Strengths:
The model predictions have been validated in vitro and using external data.
This is a complex study, but readability is enhanced by the inclusion of a section that summarizes the study design, plus relevant figures. The availability of data as supplementary material and the availability of code in GitHub are also high points.
Weaknesses:
There are some apparent discrepancies between predicted and observed data that have been seemingly overlooked.
-
-
pressbooks.library.torontomu.ca pressbooks.library.torontomu.ca
-
Two-factor authentication, or two-step authentication, is a login process where the user is asked to provide two authentication points, such as a password and a code shared through a text message. Two-factor authentication enhances login security.
While two-factor authentication is a great method for authorizing identity, it does have it's drawbacks. One example is if you use your cell phone number and you change phone numbers. It can be very difficult to access your account and you may have to go through lots of red tape! Speaking from personal experience.
-
-
www.nytimes.com www.nytimes.com
-
Regardless of the native tongue of the subjects, or whether they were baboons, college students or robots, the results were the same. When individuals passed the code on to one another, the code became simpler but also less precise.
this is strange because baboons and humans are from the same evolution
-
-
openclassrooms.com openclassrooms.com
-
Vous pouvez retrouver le code du screencast dans le repository GitHub de cette partie du cours : le fichier HTML et le fichier JS.
le lien ne correspond pas aux bon fichiers, en plus de ça le code ne correspond pas du tout à celui de la vidéo, c'est vraiment inacceptable sachant que le cours est présenté pour des débutants qu'ils ont besoins de comprendre étapes par étape
-
-
www.biorxiv.org www.biorxiv.org
-
eLife assessment
The fMRI study is important because it investigates fundamental questions about the neural basis of multimodal binding using an innovative multi-day learning approach. The results provide solid evidence for learning-related changes in the anterior temporal lobe, however, the interpretation of these changes is not straightforward, and the study does not (yet) provide direct evidence for an integrative code. This paper is of potential interest to a broad audience of neuroscientists.
-
Reviewer #1 (Public Review):
This study used a multi-day learning paradigm combined with fMRI to reveal neural changes reflecting the learning of new (arbitrary) shape-sound associations. In the scanner, the shapes and sounds are presented separately and together, both before and after learning. When they are presented together, they can be either consistent or inconsistent with the learned associations. The analyses focus on auditory and visual cortices, as well as the object-selective cortex (LOC) and anterior temporal lobe regions (temporal pole (TP) and perirhinal cortex (PRC)). Results revealed several learning-induced changes, particularly in the anterior temporal lobe regions. First, the LOC and PRC showed a reduced bias to shapes vs sounds (presented separately) after learning. Second, the TP responded more strongly to incongruent than congruent shape-sound pairs after learning. Third, the similarity of TP activity patterns to sounds and shapes (presented separately) was increased for non-matching shape-sound comparisons after learning. Fourth, when comparing the pattern similarity of individual features to combined shape-sound stimuli, the PRC showed a reduced bias towards visual features after learning. Finally, comparing patterns to combined shape-sound stimuli before and after learning revealed a reduced (and negative) similarity for incongruent combinations in PRC. These results are all interpreted as evidence for an explicit integrative code of newly learned multimodal objects, in which the whole is different from the sum of the parts.
The study has many strengths. It addresses a fundamental question that is of broad interest, the learning paradigm is well-designed and controlled, and the stimuli are real 3D stimuli that participants interact with. The manuscript is well written and the figures are very informative, clearly illustrating the analyses performed.
There are also some weaknesses. The sample size (N=17) is small for detecting the subtle effects of learning. Most of the statistical analyses are not corrected for multiple comparisons (ROIs), and the specificity of the key results to specific regions is also not tested. Furthermore, the evidence for an integrative representation is rather indirect, and alternative interpretations for these results are not considered.
-





































