Mimi Kim
CORRECTION: Mimi M. Kim
Mimi Kim
CORRECTION: Mimi M. Kim
the Assistant (named Claude, in Anthropic's models) can be thought of as a character that the LLM is writing about, almost like an author writing about someone in a novel.
这个比喻颠覆了对 AI 助手的通常理解:Claude 不是在「说话」,而是在「写作一个名叫 Claude 的角色」。这意味着 Claude 的情绪表现实际上是作者(LLM)在为虚构人物赋予情感——这种框架让「AI 有没有情绪」的问题变得像问「小说作者有没有让角色真实地爱上了人」一样奇妙。
for - book - Progress: A History of Humanity's Worst Idea - author - Samuel Miller McDonald - from - youtube - interview - Planet Critical - Samuel Miller McDonald --- https://hyp.is/r-hmFtjKEfCd8odATbINbA/www.youtube.com/watch?v=BEhmWEDkZUQ
By Simón(e) D Sun
Dr. Simone D Sun (PHD) is a scientist, artist, and advocate in the J. Tollkuhn Lab at the Cold Spring Harbor Laboratory, a Senior Fellow at the Center for Applied Transgender Studies, and a 2023 HHMI Hanna H. Gray Fellow. They received their PhD in the R.W. Tsien Lab at the NYU Neuroscience Institute studying neuroplasticity. This information is important becasue it males her argument in this text more credible. The publisher is SCI AM, a credible becasue of its long history of science publication.
By Simón(e) D Sun
Author: Simón(e) D Sun is a doctoral candidate at the neuroscience institute at New York University. It's also important to note that they are a non-binary transwoman and activist, so they have first-hand experience with the topic in the article. The work was published originally in the Scientific America's former blog network. It's noted that the views written are those of the author not necessarily Scientific American as it was originally a blog post.
Thanks to the participation of trans people in research, we have expanded our understanding of how brain structure, sex and gender interact.
Sun writes from a standpoint grounded in scientific research. Engaging directly with empirical studies shows the rather than ideology shows the credibility of the author. The author also values inclusive science.
Specifically, through three subjects: (1) genetics, (2) neurobiology and (3) endocrinology.
shows that the author is knowledgable within science by organizing the argument into different scientific fields. This knowledge from the author builds credibility with the reader.
Florian Fischer
rogress: A History of Humankind's Worst Idea
for - progress trap - book - to - book - Progress: A History of Humankind's Worst Idea - https://hyp.is/cMyt5tjMEfCGz9-Edzp-hA/harpercollins.co.uk/products/progress-a-history-of-humanitys-worst-idea-samuel-miller-mcdonald - author Samuel Miller McDonald
SRG comment - interview - book on Progress - see other references: - to - book - A Short History of Progress (2004) - https://hyp.is/93k5CtjLEfC1UpPEi59BHA/archive.org/details/shorthistoryofpr0000wrig - to - movie - Surviving Progress (2011) - https://hyp.is/sRPYJtjLEfCwuDdwG2xNnw/www.nfb.ca/film/surviving-progress/ - SRG article - Cogress
for - paper - Major transitions in sociocultural evolution (2025) - author - Arsham Nejad kourki - criitque of sociocultural systems as ETI
for - book - Hierarchy in the Forest - The Evolution of Egaliterian Behavior - author - Christopher Boehm - from - youtube - The Anthropocene Paradigm Shift
Goliath’s Curse
for - book - Goliath's curse - author - Luke Kemp - SRG comment - a naughty social superorganism! - from - youtube - The Anthropocene Paradigm Shift - https://hyp.is/a1E9ZtQ4EfCEQs_6Hskbmw/www.youtube.com/watch?v=9ggpzwSI1qY
Fn Eraise of qcribes
for - book - In Praise of Scribes - Author Johannes Trithemius - history - progress - technology - printing press
My soul which yearned for knowledge,
When Xantippe describes her soul as one that “yearned for knowledge,” she describes a desire that classical society discouraged in women. This yearning reflects Amy Levy’s own intellectual ambitions and her struggle to access education in a world that restricted women’s academic opportunities. Levy pushed against these limitations of her time. She became the second Jewish woman ever admitted to Cambridge University and the first Jewish woman to enroll at Newnham College, one of the women’s colleges founded to expand access to higher learning. Levy’s personal experiences with gender barriers enhance her portrayal of Xantippe’s longing. By giving a classical woman, the same thirst for intellectual life that Levy felt as a Victorian woman, the poem creates a bridge between eras. Xantippe’s desire becomes not merely personal but representative of a long history of women whose intellectual aspirations were dismissed or deemed inappropriate. Through this moment of self-revelation, Levy highlights the emotional cost of systemic exclusion and places knowledge-seeking as both a private desire and an act of resistance.
What, have I waked again? I never thought To see the rosy dawn, or ev’n this grey, Dull, solemn stillness, ere the dawn has come. The lamp burns low; low burns the lamp of life:
Although “Xantippe: A Fragment” was published in 1880, nine years before Levy’s death in 1889, the poem already reveals the emotional turmoil that resulted in her long-standing, though undiagnosed, clinical depression. In these lines, Amy Levy gives a haunting voice to a figure who feels emotionally drained, as if her life’s flame were dimming. The imagery of a “lamp of life” burning low, mixed with the weariness of waking, resonates with Levy’s own recurring bouts of melancholic depression. As a young Jewish woman navigating the male-dominated intellectual circles of Victorian England, Levy often felt like an outsider, both socially and spiritually. According to the Jewish Women’s Archive (2021), a friend and confidant, Richard Garnett, described her as having "constitutional melancholy." By channeling that profound exhaustion through Xantippe, she not only critiques the silencing of women, but also reveals personal anxieties about her own worth, agency, and artistic survival.
Summary:
This work is of interest because it increases our understanding of the molecular mechanisms that distinguish subtypes of VIP interneurons in the cerebral cortex and because of the multiple ways in which the authors address the role of Prox1 in regulating synaptic function in these cells.
The authors would like to thank the reviewers for their constructive comments. In response, we would like to clarify a number of issues, as well as outline how we plan to resolve major concerns.
Reviewer #1:
Stachiak and colleagues examine the physiological effects of removing the homeobox TF Prox1 from two subtypes of VIP neurons, defined on the basis of their bipolar vs. multipolar morphology.
The results will be of interest to those in the field, since it is known from prior work that VIP interneurons are not a uniform class and that Prox1 is important for their development.
The authors first show that selective removal of a conditional Prox1 allele using a VIP cre driver line results in a change in paired pulse ratio of presumptive excitatory synaptic responses in multipolar but not bipolar VIP interneurons. The authors then use RNA-seq to identify differentially expressed genes that might contribute and highlight a roughly two-fold reduction in the expression of a transcript encoding a trans-synaptic protein Elfn1 known to contribute to reduced glutamate release in Sst+ interneurons. They then test the potential contribution of Elfn1 to the phenotype by examining whether loss of one allele of Elfn1 globally alters facilitation. They find that facilitation is reduced both by this genetic manipulation and by a pharmacological blockade of presynaptic mGluRs known to interact with Elfn1.
Although the results are interesting, and the authors have worked hard to make their case, the results are not definitive for several reasons:
1) The global reduction of Elfn1 may act cell autonomously, or may have other actions in other cell types. The pharmacological manipulation is less subject to this interpretation, but these results are not as convincing as they could be because the multipolar Prox1 KO cells (Fig. 3 J) still show substantial facilitation comparable, for example to the multipolar control cells in the Elfn1 Het experiment (controls in Fig. 3E). This raises a concern about control for multiple comparisons. Instead of comparing the 6 conditions in Fig 3 with individual t-tests, it may be more appropriate to use ANOVA with posthoc tests controlled for multiple comparisons.
The reviewer’s concerns regarding non-cell-autonomous actions of global Elfn1 KO are well founded. Significant phenotypic alterations have previously been reported, both in the physiology of SST neurons as well in the animals’ behavior (Stachniak, Sylwestrak, Scheiffele, Hall, & Ghosh, 2019; Tomioka et al., 2014). The homozygous Elfn1 KO mouse displays a hyperactive phenotype and epileptic activity after 3 months of age, suggesting generalcortical activity differences exist (Dolan & Mitchell, 2013; Tomioka et al., 2014). Nevertheless, we have not observed such changes in P17-21 Elfn1 heterozygous (Het) animals.
Comparing across different experimental animal lines, for example the multipolar Prox1 KO cells (Fig. 3 J) to the multipolar control cells in the Elfn1 Het experiment (controls in Fig. 3E), is in our view not advisable. There is a plethora of examples in the literature on the effect of mouse strain on even the most basic cellular functions and hence it is always expected that researchers use the correct control animals for their experiments, which in the best case scenario are littermate controls. For these reasons, we would argue that statistical comparisons across mouse lines is not ideal for our study. Elfn1 Het and MSOP data are presented side by side to illustrate that Elfn1 Hets (3C,E) phenocopy the effects of Prox1 deletion (3G,H,I,J). (See also point 3) MSOP effect sizes, however, do show significant differences by ANOVA with Bonferroni post-hoc (normalized change in EPSC amplitude; multipolar prox1 control: +12.1 ± 3.8%, KO: -8.4 ± 4.3%, bipolar prox1 control: -5.2 ± 4.3%, KO: -3.4 ± 4.7%, cell type x genotype interaction, p= 0.02, two way ANOVA).
2) The isolation of glutamatergic currents is not described. Were GABA antagonists present to block GABAergic currents? Especially with the Cs-based internal solutions used, chloride reversal potentials can be somewhat depolarized relative to the -65 mV holding potential. If IPSCs were included it would complicate the analysis.
No, in fact GABA antagonists were not present in these experiments. The holding voltage in our evoked synaptic experiments is -70 mV, which combined with low internal [Cl-] makes it highly unlikely that the excitatory synaptic responses we study are contaminated by GABA-mediated ones, even with a Cs MeSO4-based solution. Nevertheless, we have now performed additional experiments where glutamate receptor blockers were applied in bath and we observe a complete blockade of the synaptic events at -70mV proving that they are AMPA/NMDA receptor mediated. When holding the cell at 0mV with these blockers present, outward currents were clearly visible, suggesting intact GABA-mediated events.
3) The assumption that protein levels of Elfn1 are reduced to half in the het is untested. Synaptic proteins can be controlled at the level of translation and trafficking and WT may not have twice the level of this protein.
We thank reviewer for pointing this out. Our rationale for using the Elfn1 heterozygous animals is rather that transcript levels are reduced by half in heterozygous animals, to match the reduction we found in the mRNA levels of VIP Prox1 KO cells (Fig 2). The principle purpose of the Elfn1 KO experiment was to determine whether the change in Elfn1 transcript levels could be sufficient to explain the synaptic deficit observed in VIP Prox1 KO cells. As the reviewer notes, translational regulation and protein trafficking could ultimately result in even larger changes than 0.5x protein levels at the synapse. This may ultimately explain the observed multipolar/bipolar disparity, which cannot be explained by transcriptional regulation alone (Fig 4).
4) The authors are to be commended for checking whether Elfn1 is regulated by Prox1 only in the multipolar neurons, but unfortunately it is not. The authors speculate that the selective effects reflect a selective distribution of MgluR7, but without additional evidence it is hard to know how likely this explanation is.
Additional experiments are underway to better understand this mechanism.
Reviewer #2:
Stachniak et al., provide an interesting manuscript on the postnatal role of the critical transcription factor, Prox1, which has been shown to be important for many developmental aspects of CGE-derived interneurons. Using a combination of genetic mouse lines, electrophysiology, FACS + RNAseq and molecular imaging, the authors provide evidence that Prox1 is genetically upstream of Elfn1. Moreover, they go on to show that loss of Prox1 in VIP+ cells preferentially impacts those that are multipolar but not the bipolar subgroup characterized by the expression of calretinin. This latter finding is very interesting, as the field is still uncovering how these distinct subgroups emerge but are at a loss of good molecular tools to fully uncover these questions. Overall, this is a great combination of data that uses several different approaches to come to the conclusions presented. I have suggestions that I think would strengthen the manuscript:
1) Can the authors add a supplemental table showing the top 20-30 genes up and down regulated in their Prox1 KOS? This would make these, and additional, data more tenable to readers.
We would be happy to provide supplementary tables with candidate genes at both P8 and P12.
2) It is interesting that loss of Prox1 or Elfn1 leads to phenotypes in multipolar but are not present or mild in bipolar VIP+ cells. The authors test different hypotheses, which they are able to refute and discuss some ideas for how multipolar cells may be more affected by loss of Elfn1, even when the transcript is lost in both multipolar and bipolar after Prox1 deletion. If there is any way to expand upon these ideas experimentally, I believe it would greatly strengthen the manuscript. I understand there is no perfect experiment due to a lack of tools and reagents but if there is a way to develop one of the following ideas or something similar, it would be beneficial:
We thank the reviewer for the note.
a) Would it be possible to co-fill VIPCre labeled cells with biocytin and a retroviral tracer? Then, after the retroviral tracer had time to label a presynaptic cell, assess whether these were preferentially different between bipolar and multipolar cell types, the latter morphology determined by the biocytin fill? This would test whether each VIP+ subtype is differentially targeted.
Although this is a very elegant experiment and we would be excited to do it, we do feel that single-cell rabies virus tracing is technically very challenging and will take many months to troubleshoot before being able to acquire good data. Hence, we think it is beyond the scope of this study.
b) Another biocytin possibility would be to trace filled VIP+ cells and assess whether the dendrites of multipolar and bipolar cells differentially targeted distinct cortical lamina and whether these lamina, in the same section or parallel, were enriched for mGluR7+ afferents.
We thank the reviewer for their suggestion and we are planning on doing these kinds of experiments.
Reviewer #3:
In this work Stachiak and colleagues investigate the role of Prox1 on the development of VIP cells. Prox1 is expressed by the majority of GABAergic derived from the caudal ganglionic eminence (CGE), and as mentioned by the authors, Prox1 has been shown to be necessary for the differentiation, circuit integration, and maintenance of CGE-derived GABAergic cells. Here, Stachiak and colleagues show that removal of Prox1 in VIP cells leads to suppression of synaptic release probability onto cortical multipolar VIP cells in a mechanism dependent on Elfn1. This work is of interest for the field because it increases our understanding of differential synaptic maturation of VIP cells. The results are noteworthy, however the relevance of this manuscript would potentially be increased by addressing the following suggestions:
1) Include histology to show when exactly Prox1 is removed from multipolar and bipolar VIP-expressing cells by using the VIP-Cre mouse driver.
We can address this by performing an in-situ hybridization against Prox1 from P3 onwards (when Cre becomes active).
2) Clarify if the statistical analysis is done using n (number of cells) or N (number of animals). The analysis between control and mutants (both Prox1 and Elfn1) need to be done across animals and not cells.
Statistics for physiology were done across n (number of cells) while statistics for ISH are done across number of slices. We will clarify this point in the text and update the methods.
Regarding the statistics for the ISH, these have been done across n (number of slices) for control versus KO tissue (N = 3 and N = 2 animals, respectively). We will add more animals to this analysis to compare by animal instead, although we do not expect any change in the results.
Regarding the physiology, we would provide a two-pronged answer. We first of all feel that averaging synaptic responses for each animal would hide a good deal of the biological variability in PPR present in different cells (response Fig 1), the characterization of which is integral to the central findings of the paper. Secondly, to perform such analysis asked by the reviewer one would need to obtain recordings from ~10 animals or so per condition for each condition, which, to our knowledge, is something that is not standard when utilizing in vitro electrophysiological recordings from single cells. For example, in these very recent studies that have performed in vitro electrophysiological recordings all the statistics are performed using “n” number of cells and not the average of all the cells recorded per animal collapsed into a single data point. (Udakis, Pedrosa, Chamberlain, Clopath, & Mellor, 2020) https://www.nature.com/articles/s41467-020-18074-8
(Horvath, Piazza, Monteggia, & Kavalali, 2020) https://elifesciences.org/articles/52852
(Haas et al., 2018) https://elifesciences.org/articles/31755
Nevertheless, we have now re-run the analysis grouping the cells and averaging the values we get per animal, since we have obtained our data from many animals. The results are more or less indistinguishable from the ones presented in the original submission, except for on p value that rose to 0.07 from 0.03 due to the lack of the required number of animals. We hope that the new plots and statistics presented herein address the concern put forward by the reviewer.
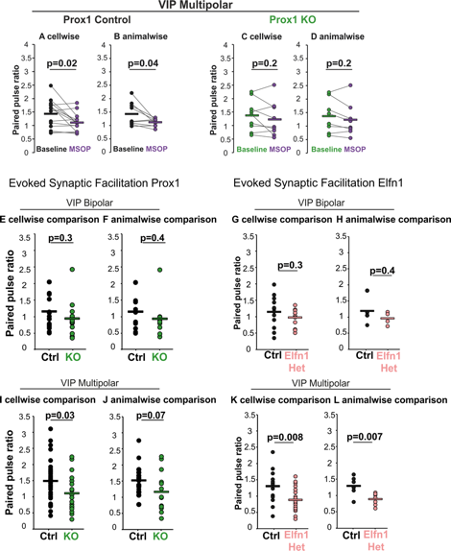
Response Fig 1: A comparison of cell wise versus animal-wise analysis of synaptic physiology. Some cell to cell variability is hidden, and the reduction in numbers impacts the P values.
(A) PPR of multipolar Prox1 Control for 14 cells from 9 animals (n/N=14/9) under baseline conditions and with MSOP, cell-wise comparison p = 0.02 , t = 2.74 and (B) animal-wise comparisons (p = 0.04, t stat = 2.45). Statistics: paired t-test.
(C) PPR of multipolar Prox1 KO cells (n/N=9/8) under baseline conditions and with MSOP, cell-wise comparison p = 0.2, t = 1.33 and (D) animal-wise comparisons (p = 0.2, t stat = 1.56). Statistics: paired t-test. Comparisons for PPR of bipolar Prox1 Control (n/N=8/8) and KO cells (n/N=9/9) did not change.
(E) PPR for Prox1 control (n/N=18/11) and KO (n/N=13/11) bipolar VIP cells, cell-wise comparison p = 0.3, t = 1.1 and (F) animal-wise comparisons (p = 0.4, t stat = 0.93). Statistics: t-test.
(G) PPR of Elfn1 Control (n/N=12/4) and Het (n/N=12/4) bipolar VIP cells, cell-wise comparison p = 0.3, t = 1.06 and (H) animal-wise comparisons (p = 0.4, t stat = 0.93)
(I) PPR of Prox1 control (n/N=33/18) and KO (n/N=19/14) multipolar VIP cells, cell-wise comparison p = 0.03, t = 2.17. and (J) animal-wise comparisons (p = 0.07, t stat = 1.99).
(K) PPR of Elfn1 Control (n/N=14/6) and Het (n/N=20/8) multipolar VIP cells, cell-wise comparison p = 0.008, t = 2.84 and (L) animal-wise comparisons (p = 0.007, t stat = 3.23).
3) Clarify what are the parameters used to identify bipolar vs multipolar VIP cells. VIP cells comprise a wide variety of transcriptomic subtypes, and in the absence of using specific genetic markers for the different VIP subtypes, the authors should either include the reconstructions of all recorded cells or clarify if other methods were used.
We thank the reviewer for this comment. The cell parameter criteria will be amended in the methods: “Cell type was classified as bipolar vs. multipolar based on cell body morphology (ovoid vs. round) and number and orientation of dendritic processes emanating from it (2 or 3 dendrites perpendicular to pia (for bipolar) vs. 3 or more processes in diverse orientations (for multipolar). In addition, the laminar localization of the two populations differs, with multipolar cells found primarily in the upper layer 2, while bipolar cells are found throughout layers 2 and 3. Initial determination of cell classification was made prior to patching fluorescent-labelled cells, but whenever possible this initial assessment was confirmed with post-hoc verification of biocytin filled cells.”
Reference:
Dolan, J., & Mitchell, K. J. (2013). Mutation of Elfn1 in Mice Causes Seizures and Hyperactivity. PLOS ONE, 8(11), e80491. Retrieved from https://doi.org/10.1371/journal.pone.0080491
Haas, K. T., Compans, B., Letellier, M., Bartol, T. M., Grillo-Bosch, D., Sejnowski, T. J., … Hosy, E. (2018). Pre-post synaptic alignment through neuroligin-1 tunes synaptic transmission efficiency. ELife, 7, e31755. https://doi.org/10.7554/eLife.31755
Horvath, P. M., Piazza, M. K., Monteggia, L. M., & Kavalali, E. T. (2020). Spontaneous and evoked neurotransmission are partially segregated at inhibitory synapses. ELife, 9, e52852. https://doi.org/10.7554/eLife.52852
Stachniak, T. J., Sylwestrak, E. L., Scheiffele, P., Hall, B. J., & Ghosh, A. (2019). Elfn1-Induced Constitutive Activation of mGluR7 Determines Frequency-Dependent Recruitment of Somatostatin Interneurons. The Journal of Neuroscience, 39(23), 4461 LP – 4474. https://doi.org/10.1523/JNEUROSCI.2276-18.2019
Tomioka, N. H., Yasuda, H., Miyamoto, H., Hatayama, M., Morimura, N., Matsumoto, Y., … Aruga, J. (2014). Elfn1 recruits presynaptic mGluR7 in trans and its loss results in seizures. Nature Communications. https://doi.org/10.1038/ncomms5501
Udakis, M., Pedrosa, V., Chamberlain, S. E. L., Clopath, C., & Mellor, J. R. (2020). Interneuron-specific plasticity at parvalbumin and somatostatin inhibitory synapses onto CA1 pyramidal neurons shapes hippocampal output. Nature Communications, 11(1), 4395. https://doi.org/10.1038/s41467-020-18074-8
Reviewer #1:
The Lambowitz group has developed thermostable group II intron reverse transcriptases (TGIRTs) that strand switch and also have trans-lesion activity to provide a much wider view of RNA species analyzed by massively parallel RNA sequencing. In this manuscript they use several improvements to their methodology to identify RNA biotypes in human plasma pooled from several healthy individuals. Additionally, they implicate binding by proteins (RBPs) and nuclease-resistant structures to explain a fraction of the RNAs observed in plasma. Generally I find the study fascinating and argue that the collection of plasma RNAs described is an important tool for those interested in extracellular RNAs. I think the possibility that RNPs are protecting RNA fragments in circulation is exciting and fits with elegant studies of insects and plants where RNAs are protected by this mechanism and are transmitted between species.
I have one major comment for the authors to consider. In my view the use of pooled plasma samples prevented the important opportunity to provide a glimpse on human variation in plasma RNA biotypes. This significantly limits the use of this information to begin addressing RNA biotypes as biomarkers. While I realize that data from multiple individuals represents a significant undertaking and may be beyond the scope of this manuscript, I urge the authors to do two things: (1) downplay the significance of the current study on the development of biomarkers in the current manuscript (e.g., in the abstract and discussion - e.g., "The ability of TGIRT-seq to simultaneously profile a wide variety of RNA biotypes in human plasma, including structured RNAs that are intractable to retroviral RTs, may be advantageous for identifying optimal combinations of coding and non-coding RNA biomarkers for human diseases."). (2) Carry out an analysis in multiple individuals - including racially diverse individuals - very important information will come of this - similar to C. Burge's important study in Nature ~2008 where it was clear that there is important individual variation in alternative splicing decisions - very likely genetically determined. This second suggestion could be added here or constitute a future manuscript.
The identification of biomarkers in human plasma is an important application of this study, as was noted by reviewer 3 -- "Overall, this study provided a robust dataset and expanded picture of RNA biotypes one can detect in human plasma. This is valuable because the findings may have implications in biomarker identification in disease contexts." The present manuscript lays the foundation for such applications, which we have been carrying out in parallel. In one such study in collaboration with Dr. Naoto Ueno (MD Anderson), we used TGIRT-seq to identify combinations of mRNA and non-coding RNA biomarkers in FFPE-tumor slices, PBMCs and plasma from inflammatory breast cancer patients compared to non-IBC breast cancer patients and healthy controls (manuscript in preparation; data presented publicly in seminars), and in another, we explored the potential of using full-length excised intron (FLEXI) RNAs as biomarkers. In the latter study, we identified >8,000 FLEXI RNAs in different human cell lines and tissues and found that they are expressed in a cell-type specific manner, including hundreds of differences between matched tumor and healthy tissues from breast cancer patients and cell lines. A manuscript describing the latter findings was submitted for publication after this one and has been uploaded as a pertinent related manuscript. This new manuscript follows directly from the last sentence of the present manuscript and fully references the BioRxiv preprint currently under review for eLife.
Reviewer #2:
Yao et al used thermostable group II intron reverse transcriptase sequencing (TGIRT-seq) to study apheresis plasma samples. The first interesting discovery is that they had identified a number of mRNA reads with putative binding sites of RNA-binding proteins. A second interesting discovery from this work is the detection of full-length excised intron RNAs.
I have the following comments:
1) One doubt that I have is how representative is apheresis plasma when compared with plasma that one obtains through routine centrifugation of blood. The authors have reported the comparison of apheresis plasma versus a single male plasma in a previous publication. I think that to address this important question, a much increased number of samples would be necessary.
Detailed comparison of plasma prepared by apheresis to that prepared by centrifugation would require a separate large-scale study, preferably by multiple laboratories using different methods to prepare plasma. However, our impression both from our findings and from the literature (Valbonesi et al. 2001, cited in the manuscript) is that apheresis-prepared plasma has very low levels of cellular contamination (required to meet clinical standards) compared to plasma prepared by centrifugation, even with protocols designed to minimize contamination from intact 4 or broken cell (e.g., preparing plasma from freshly drawn blood, centrifugation into a Ficoll cushion to minimize cell breakage, and carefully avoiding contamination from sedimented cells).
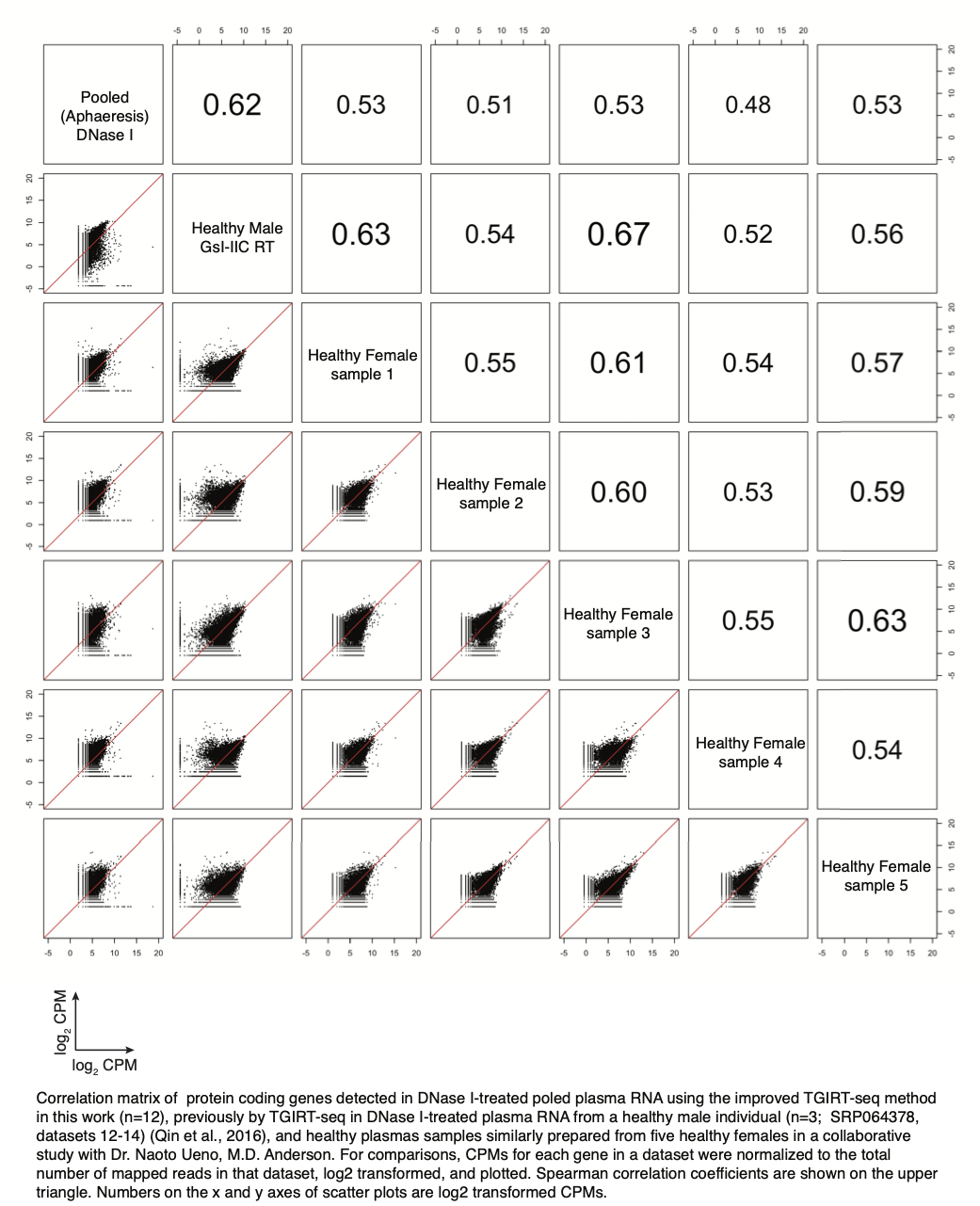
We do have additional information about the degree of variation in protein-coding gene transcripts detected by TGIRT-seq in plasma samples prepared by centrifugation from five healthy females controls in our collaborative study with Dr. Naoto Ueno (M.D. Anderson; see above), and we have added it to the manuscript citing a manuscript in preparation with permission from Dr. Ueno (p. 10, beginning line 6 from bottom) as follows:
“The identities and relative abundances of different protein-coding gene transcripts in the apheresis-prepared plasma were broadly similar to those in the previous TGIRT analysis of plasma prepared by Ficoll-cushion sedimentation of blood from a healthy male individual (Qin et al., 2016) (r = 0.62-0.80; Figure 3C) and between high quality plasma samples similarly prepared from five healthy females in a collaborative study with Dr. Naoto Ueno, M.D. Anderson (r = 0.53-0.67; manuscript in preparation).” See Author Response Image below.
2) For the important conclusion of the presence of binding sites of RNA-binding proteins in a proportion of apheresis plasma mRNA molecules, the authors need to explore whether there is any systemic difference in terms of mapping quality (i.e. mapping quality scores in alignment results) between RBP binding sites and non-RBP binding sites, so that any artifacts of peaks caused by the alignment issues occurring in RNA-seq analysis could be revealed and solved subsequently. Furthermore, it would be prudent to perform immunoprecipitation experiments to confirm this conclusion in at least a proportion of the mRNA.
We have added a figure panel comparing MAPQ scores for reads from peaks containing RBP-binding site to other long RNA reads (Figure 4–figure supplement 2A) and have added further details about the methods used to obtain peaks with high quality reads, including the following (p. 13, beginning line 3 from the bottom).
“After further filtering to remove read alignments with MAPQ <30 (a cutoff that eliminates reads mapping equally well at more than one locus) or ≥5 mismatches from the mapped locus, we were left with 950 high confidence peaks ranging in size from 59 to 1,207 nt with ≥5 high quality read alignments at the peak maximum (Supplementary File).”
3) In Fig. 2D, one can observe that there are clearly more RNA reads in TGIRT-seq located in the 1st exon of ACTB, compared with SMART-seq. Is there any explanation? Will this signal be called as a peak (a potential RBP binding site) in the peak calling analysis (MACS2)? Is ACTB supposed to be bound by a certain RBP?
The higher coverage of the ACTB 5'-exon in the TGIRT-seq datasets reflects in part the more uniform 5' to 3' coverage of mRNA sequences by TGIRT-seq compared to SMART-seq, which is biased for 3'-mRNA sequences that have poly(A) tails (current Figure 3F). The signal in the first exon of ACTB was in fact called as a peak by MACS2 (peak ID#893, Supplementary file), which overlapped an annotated binding site for SERBP1 (see Supplementary File).
4) For Fig 2A, it would be informative for the comparison of RNA yield and RNA size profile among different protocols if the author also added the results of TGIRT-seq.
Figure 3D (previously Figure 2A) shows a bioanalyzer trace of PCR amplified cDNAs obtained by SMART-Seq. These cDNAs correspond to 3' mRNA sequences that have poly(A) tails and are not comparable to the bioanalyzer profiles of plasma RNA (Figure 1–figure supplement 1) or read span distributions in the TGIRT-seq datasets (Figure 1B), which are dominated by sncRNAs. The coverage plots for protein-coding gene transcripts show that TGIRT-seq captures mRNA fragments irrespective of length that span the entire mRNA sequence, whereas SMART-seq is biased for 3' sequences linked to poly(A) (Figure 3F). We also note that coverage plots and mRNAs detected by TGIRT-seq remain similar, even if the plasma RNA is chemically fragmented prior to TGIRT-seq library construction (Figure 3F and Figure 3–figure supplement 2).
5) As shown in Figure 4 C (the track of RBP binding sites), it seems quite pervasive in some gene regions. How many RBP binding sites from public eCLIP-seq results are used for overlapping peaks present in TGIRT-seq of plasma RNA? What percentage of plasma RNA reads have fallen within RBP binding sites? Are those peaks present in TGRIT-seq significantly enriched in RBPs binding regions?
Some of these points are addressed under Reviewer 1-comment #4. Additionally, we noted that 109 RBP-binding sites were searched in the original analysis, and we have now added further analyses for 150 RBPs currently available in ENCODE eCLIP datasets with and without irreproducible discovery rate (IDR) analysis (Figure 6 and Figure 6–figure supplement 1). We have also added a tab to the Supplementary File identifying the 109 and 150 RBPs whose binding sites were searched. The requested statistical analysis has been added in Figure 4–figure supplement 2C. The analysis shows that enrichment of RBP-binding site sequences in the 467 called peaks was statistically significant (p<0.001) (p. 14, para. 3, last sentence).
6) Since there is a considerable portion of TGIRT-seq reads related to simple repeat, one possible reason is likely the high abundance of endogenous repeat-related RNA species in plasma. Nonetheless, have authors studied whether the ligation steps in TGIRT-seq have any biases (e.g. GC content) when analyzing human reference RNAs and spike ins (page 4, paragraph 2)?
We have added a note to the manuscript indicating that although repeat RNAs constitute a high proportion of the called peaks, they do not constitute a similarly high proportion of the total RNA reads (Figure 1C; p. 18, para. 2, first sentence). The TGIRT-seq analysis of human reference RNAs and spike-ins showed that TGIRT-seq recapitulates the relative abundance of human transcripts and spike-in comparably to non-strand-specific TruSeq v2 and better than strand-specific TruSeq v3 (Nottingham et al. RNA 2016). Subsequently, we used miRNA reference sets for detailed analysis of TGIRT-seq biases, including developing a computer algorithm for bias correction based on a random forest regression model that provides insight into different factors that contribute to these biases (Xu et al. Sci. Report. 2019). Overall GC content does not make a significant contribution to TGIRT-seq biases (Figure 9 of Xu et al. Sci. Report, 2017). Instead, biases in TGIRT-seq are largely confined to the first three nucleotides at the 5'-end (due to bias of the thermostable 5' App DNA ligase used for 5' RNA-seq adapter addition) and the 3' nucleotide (due to TGIRT-template switching). These end biases are not expected to significantly impact the quantitation of repeat RNAs.
7) As described in Figure 2 legend, there are 0.25 million deduplicated reads for TGIRT-seq reads assigned to protein-coding genes transcripts which are far less than 2.18 million reads for SMART-seq. The authors need to discuss whether the current protocol of TGIRT-seq would cause potential dropouts in mRNA analysis, compared with SMART-seq?
We have added the following to the manuscript (p. 11, para. 1, line 15).
“The larger number of mRNA reads compared to TGIRT-seq (0.28 million) largely reflects that SMART-seq selectively profiles polyadenylated mRNAs, while TGIRT-seq profiles mRNAs together with other more abundant RNA biotypes. In addition, ultra low input SMART-Seq is not strand-specific, resulting in redundant sense and antisense strand reads (Figure 3–figure supplement 1).”
The manuscript contains the following statement regarding potential drop outs (p. 11, para. 2, line 1).
“A scatter plot comparing the relative abundance of transcripts originating from different genes showed that most of the polyadenylated mRNAs detected in DNase I-treated plasma RNA by ultra low input SMART-Seq were also detected by TGIRT-seq at similar TPM values when normalized for protein-coding gene reads (r=0.61), but with some, mostly lower abundance mRNAs undetected either by TGIRT-seq or SMART-Seq, and with SMART-seq unable to detect non-polyadenylated histone mRNAs, which are relatively abundant in plasma (Figure 3E and Figure 3–figure supplement 1).”
8) While scientific thought-provoking, the practical implication of the current work is still unclear. The authors have suggested that their work might have applications for biomarker development. Is it possible to provide one experimental example in the manuscript?
We addressed the relevance of the manuscript to biomarker identification and noted parallel studies that supports this application in the response to reviewer 1--comment 1. We have also modified the final paragraph of the Discussion (p. 30, para. 2).
“The ability of TGIRT-seq to simultaneously profile a wide variety of RNA biotypes in human plasma, including structured RNAs that are intractable to retroviral RTs, may be advantageous for identifying optimal combinations of coding and non-coding RNA biomarkers that could then be incorporated in target RNA panels for diagnosis and routine monitoring of disease progression and response to treatment. The finding that some mRNAs fragments persist in discrete called peaks suggests a strategy for identifying relatively stable mRNA regions that may be more reliably detected than other more labile regions in targeted liquid biopsies. Finally, we note that in addition to their biological and evolutionary interest, short full-length excised intron RNAs and intron RNA fragments, such as those identified here, may be uniquely well suited to serve as stable RNA biomarkers, whose expression is linked to that of numerous protein-coding genes."
Reviewer #3:
In this work, Yao and colleagues described transcriptome profiling of human plasma from healthy individuals by TGIRT-seq. TGIRT is a thermostable group II intron reverse transcriptase that offers improved fidelity, processivity and strand-displacement activity, as compared to standard retroviral RT, so that it can read through highly structured regions. Similar analysis was performed previously (ref. 20), but this study incorporated several improvements in library preparation including optimization of template switching condition and modified adapters to reduce primer dimer and introduce UMI. In their analysis, the authors detected a variety of structural RNA biotypes, as well as reads from protein-coding mRNAs, although the latter is in low abundance. Compared to SMART-Seq, TGIRT-seq also achieved more uniform read coverage across gene bodies. One novel aspect of this study is the peak analysis of TGIRT-seq reads, which revealed ~900 peaks over background. The authors found that these peaks frequently overlap with RBP binding sites, while others tend to have stable predicted secondary structures, which explains why these regions are protected from degradation in plasma. Overall, this study provided a robust dataset and expanded picture of RNA biotypes one can detect in human plasma. This is valuable because the findings may have implications in biomarker identification in disease contexts. On the other hand, the manuscript, in the current form, is relatively descriptive, and can be improved with a clearer message of specific knowledge that can be extracted from the data.
Specific points:
1) Several aspects of bioinformatics analysis can be clarified in more detail. For example, it is unclear how sequencing errors in UMI affect their de-duplication procedure. This is important for their peak analysis, so it should be explained clearly.
We have added details of the procedure used for de-duplication to the following paragraph in Materials and methods (p. 35, para. 2).
“Deduplication of mapped reads was done by UMI, CIGAR string, and genome coordinates (Quinlan, 2014). To accommodate base-calling and PCR errors and non-templated nucleotides that may have been added to the 3' ends of cDNAs during TGIRT-seq library preparation, one mismatch in the UMI was allowed during deduplication, and fragments with the same CIGAR string, genomic coordinates (chromosome start and end positions), and UMI or UMIs that differed by one nucleotide were collapsed into a single fragment. The counts for each read were readjusted to overcome potential UMI saturation for highly-expressed genes by implementing the algorithm described in (Fu et al., 2011), using sequencing tools (https://github.com/wckdouglas/sequencing_tools ).”
Also, it is not described how exon junction reads (when mapped to the genome) are handled in peak calling, although the authors did perform complementary analysis by mapping reads to the reference transcriptome.
We have added this to first sentence of the paragraph describing peak calling against the transcriptome reference (p. 16, line 4), which now reads as follows:
"Peak calling against the human genome reference sequence might miss RBP-binding sites that are close to or overlap exon junctions, as such reads were treated by MACS2 as long reads that span the intervening intron."
2) Overall, the authors provided convincing data that TGIRT-seq has advantages in detecting a wide range of RNA biotypes, especially structured RNAs, compared to other protocols, but these data are more confirmatory, rather than completely new findings (e.g., compared to ref. 20).
As indicated in the response to Reviewer 1, comment 2, we modified the first paragraph of the Discussion to explicitly describe what is added by the present manuscript compared to Qin et al. RNA 2016 (p. 24, para. 2). Additionally, further analysis in response to the reviewers' comments resulted in the interesting finding that stress granule proteins comprised a high proportion of the RBPs whose binding sites were enriched in plasma RNAs (to our knowledge a completely new finding), consistent with a previously suggested link between RNP granules, EV packing, and RNA export (p. 16, last sentence; data shown in Figure 6 and Figure 6–figure supplement 1). Also highlighted in the Discussion p. 26, last sentence, continuing on p. 27).
3) The peak analysis is more novel. The authors observed that 50% of peaks in long RNAs overlap with eCLIP peaks. However, there is no statistical analysis to show whether this overlap is significant or simply due to the pervasive distribution of eCLIP peaks. In fact, it was reported by the original authors that eCLIP peaks cover 20% of the transcriptome.
We have added statistical analysis, which shows that the enrichment of RBP-binding sites in the 467 called peaks is statistically significant at p<0.001 (p. 14, para. 3, last sentence; Figure 4–Figure supplement 2C), as well as scatter plots identifying proteins whose binding sites were more highly represented in plasma than cellular RNAs or vice versa (p. 16, last two sentences; Figure 6 and Figure 6-figure supplement 1).
Similarly, the authors found that a high proportion of remaining peaks can fold into stable secondary structures, but this claim is not backed up by statistics either.
First, near the beginning of the paragraph describing these findings, we added the following to provide a guide as to what can and can't be concluded by RNAfold (p. 17, line 6 from the bottom).
"To evaluate whether these peaks contained RNAs that could potentially fold into stable secondary structures, we used RNAfold, a tool that is widely used for this purpose with the understanding that the predicted structures remain to be validated and could differ under physiological conditions or due to interactions with proteins."
Second, at the end of the same paragraph, we have added the requested statistics (p. 18, para. 1, last sentence).
"Subject to the caveats above regarding conclusions drawn from RNAfold, simulations using peaks randomly generated from long RNA gene sequences indicated that enrichment of RNAs with more stable secondary structures (lower MFEs) in the called RNA peaks was statistically significant (p≤0.019; Figure 4–figure supplement 2D)."
4) Ranking of RBPs depends on the total number of RBP binding sites detected by eCLIP, which is determined by CLIP library complexity and sequencing depth. This issue should be at least discussed.
We have added scatter plots in Figure 6 and Figure 6–figure supplement 1, which show that the relative abundance of different RBP-binding sites detected in plasma differs markedly from that for cellular RNAs in the eCLIP datasets (both for the 109 RBPs searched initially and for 150 RBPs with or without irreproducible discovery rate (IDR) analysis from the ENCODE web site,) As mentioned in comments above, this analysis identified a number of RBP-binding sites that were substantially enriched in plasma RNAs compared to cellular RNAs or vice versa and led to what we think is the important new finding that plasma RNAs are enriched binding sites for a number of stress granule proteins (Figure 6 and Figure 6–figures supplement 1). We thank the reviewers for this and related comments that led to this additional analysis.
5) Enrichment of RBP binding sites and structured RNA in TGIRT-seq data is certainly consistent with one's expectation. However, the paper can be greatly improved if the authors can make a clearer case of what is new that can be learned, as compared to eCLIP data or other related techniques that purify and sequence RNA fragments crosslinked to proteins. What is the additional, independent evidence to show the predicted secondary structures are real?
Compared to CLIP and related methods, peak calling enables more facile identification of candidate RBPs and putatively structured RNAs for further analysis and may be particularly useful for the vanishingly small amounts of RNA present in plasma and other bodily fluids. New findings resulting from peak calling in the present manuscript include that plasma RNAs are enriched in binding sites for stress granule proteins (see above) and the discovery of a variety of novel RNAs, including the full-length excised intron RNAs first identified here and subsequently studied in cellular RNAs in the Yao et al. pertinent submitted manuscript. We also note that peak calling enables the identification of protein-protected and structured mRNA regions that are relatively stable in plasma and may be more reliably detected in targeted liquid biopsy assays than are more labile mRNA regions (p. 17, para. 1, last sentence; and p. 30, para. 2, beginning on line 5).
6) The authors should probably discuss how alignment errors can potentially affect detection of repetitive regions.
In the Empirical Bayes method that we used for the analysis of repeats, repeat sequences were quantified by aggregate counts irrespective of the genomic locus to which they mapped (Materials and methods, p. 38, para. 2, line 5), which should not be affected by alignment errors.
7) Many figures are IGV screenshots, which can be difficult to follow. Some of them can probably be summarized to deliver the message better.
Some IGV-based figures are crucial for showing key features of the RNAs that are called as peaks (e.g., the predicted secondary structures of the full-length excised intron RNAs and intron RNA fragments). However, in the process of reformatting, we have switched in and added non-IGV main text figures including Figure 2 (microbiome analysis), Figure 3 (TGIRT-seq versus SMART-Seq), Figure 4 (repeats), and Figure 6 (new figure comparing relative abundance of RBP-binding sites in plasma versus cells).
Author Response refers to a revised version of the manuscript, Version 3, which was posted October 23, 2020.
Summary:
Serra-Marques, Martin et al. investigate the individual and cooperative roles of specific kinesins in transporting Rab6 secretory vesicles in HeLa cells using CRISPR and live-cell imaging. They find that both KIF5B and KIF13B cooperate in transporting Rab6 vesicles, but Eg5 and other kinesin-3s (KIF1B and KIF1C) are dispensable for Rab6 vesicle transport. They show that both KIF5B and KIF13B localize to these vesicles and coordinate their activities such that KIF5B is the main driver of the cargos on older, MAP7-decorated microtubules, and KIF13B takes over as the main transporter on freshly-polymerized microtubule ends that are largely devoid of MAP7. Interestingly, their data also indicate that KIF5B is important for controlling Rab6 vesicle size, which KIF13B cannot rescue. By analyzing subpixel localization of the motors, they find that the motors localize to the front of the vesicle when driving transport, but upon directional cargo switching, KIF5B localizes to the back of the vesicle when opposing dynein. Overall, this paper provides substantial insight into motor cooperation of cargo transport and clarifies the contribution of these distinct classes of motors during Rab6 vesicle transport.
We thank the reviewers for their thoughtful and constructive suggestions, and for the positive feedback.
Reviewer #1:
In their manuscript, Serra-Marques, Martin, et al. investigate the individual and cooperative roles of specific kinesins in transporting Rab6 vesicles in HeLa cells using CRISPR and live-cell imaging. They find that both KIF5B and KIF13B cooperate in transporting Rab6 vesicles, but KIF5B is the main driver of transport. In these cells, Eg5 and other kinesin-3s (KIF1B and KIF1C) are dispensable for Rab6 vesicle transport. They find that both KIF5B and KIF13B are present on these vesicles and coordinate their activities such that KIF5B is the main driver of the cargos on older, MAP7-decorated MTs, and KIF13B takes over as the main transporter on freshly-polymerized MT ends that are largely devoid of MAP7. Interestingly, their data also indicate that KIF5B is important for controlling Rab6 vesicle size, which KIF13B cannot rescue. Upon cargo switching from anterograde to retrograde transport, KIF5B, but not KIF13B, engages in mechanical competition with dynein. Overall, this paper provides substantial insight into motor cooperation of cargo transport and clarifies the contribution of these distinct classes of motors during Rab6 vesicle transport. The experiments are well-performed and the data are of very high quality.
Major Comments:
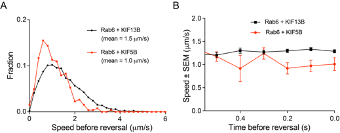
1) In Figure 5, it is very interesting that only KIF5B opposes dynein. It would be informative to determine which kinesin was engaged on the Rab6 vesicle before the switch to the retrograde direction. Can the authors analyze the velocity of the run right before the switch to the retrograde direction? If the velocity corresponds with KIF5B (the one example provided seems to show a slow run prior to the switch), this could indicate that KIF5B opposes dynein more actively because KIF5B was the motor that was engaged at the time of the switch. Or if the velocity corresponds with KIF13B, this could indicate that KIF5B becomes specifically engaged upon a direction reversal. In any case, an analysis of the speed distributions before the switch would provide insight into vesicle movement and motor engagement before the change in direction.
Directional switching was only analyzed in rescue experiments, where the vesicles were driven by either KIF5B alone or by KIF13B alone, and the speeds of vesicles were representative of these motors (please see panels on the right). The number of vesicle runs where two motors were detected simultaneously (KIF5B vs KIF13B in Figure 5G,H,J) were significantly lower, and therefore, unfortunately we could not perform the analysis of their directional switching with sufficient statistical power.
2) One of the most interesting aspects of this paper is the different lattice preferences for KIF5B, which shows runs predominantly on "older" polymerized MTs decorated by MAP7, and for KIF13B, whose runs are predominantly restricted to newly polymerized MTs that lack MAP7. The results in Figure 8 suggest a potential switch from KIF5B to KIF13B motor engagement upon a change in lattice/MAP7 distribution. In general, do the authors observe the fastest runs at the cell periphery, where there should be a larger population of freshly polymerized MTs? For Figure 4E, are example 1 and example 2 in different regions of the cell?
This is indeed a very interesting point and we have considered it carefully. As can be seen in Figure 8B (grey curve), vesicle speed remains relatively constant along the cell radius in control HeLa cells. We note, however, that our previous work has shown that in these cells microtubules are quite stable even at the cell periphery, due to the high activity of the CLASP-containing cortical microtubule stabilization complex (Mimori-Kiyosue et al., 2005, Journal of Cell Biology, PMID: 15631994; van der Vaart et al., 2013, Developmental Cell, PMID: 24120883). We therefore hypothesized that changes in vesicle speed distribution along the cell radius would be more obvious in cells with highly dynamic microtubule networks and performed a preliminary experiment in MRC5 human lung fibroblasts, which have a very sparse and dynamic microtubule cytoskeleton (Splinter et al., 2012, Molecular Biology of the Cell, PMID: 22956769). As shown in the figure below, we indeed found that vesicles move faster at the cell periphery. Even though these data are suggestive, characterization of this additional cell model goes beyond the scope of the current study, and we prefer not to include them in the manuscript.
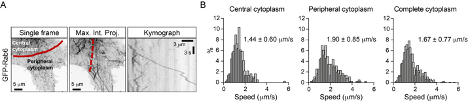
In Figure 4E, the two examples are from different cells, and were both recorded at the cell periphery. The difference in vesicle speeds reflects general speed variability.
Do the authors think the intermediate speeds are a result of the motors switching roles? Additional discussion would help the reader interpret the results.
Presence of intermediate speeds of cargos driven by multiple motors of two types is most clear in Figure 3F-H, where multiple and different ratios of KIF5B and KIF13B motors are recruited to peroxisomes. As can be seen in Fig. 3G, the kymographs in these conditions are “smooth” and no evidence of motor switching can be detected at this spatiotemporal resolution. On the other hand, it has been previously beautifully shown by the Verhey lab that when artificial cargos are driven by just two motor molecules of different nature, switching does occur (Norris et al., 2014, Journal of Cell Biology, PMID: 25365993). This point is emphasized on page 12 of the revised manuscript. These data suggest that motors working in teams show different properties, and more detailed biophysical analysis will be needed to understand them.
Reviewer #2:
The manuscript by Serra-Marques, Martin, et al provides a tour de force in the analysis of vesicle transport by different kinesin motor proteins. The authors generate cell lines lacking a specific kinesin or combination of kinesins. They analyze the distribution and transport of Rab6 as a marker of most, if not all, secretory vesicles and show that both KIF5B and KIF13B localize to these vesicles and describe the contribution of each motor to vesicle transport. They show that the motors localize to the front of the vesicle when driving transport whereas KIF5B localizes to the back of the vesicle when opposing dynein. They find that KIF5B is the major motor and its action on "old" microtubules is facilitated by MAP7 whereas KIF13B facilitates transport on "new" microtubules to bring vesicles to the cell periphery. The manuscript is well-written, the data are properly controlled and analyzed, and the results are nicely presented. There are a few things the authors could do to tie up loose ends but these would not change the conclusions or impact of the work and I only have a couple of clarifying questions.
In Figure 2E, it seems like about half of the KIF5B events start at or near the Golgi whereas most of the KIF13B events are away from the Golgi? Did the authors find this to be generally true or just apparent in these example images?
We sincerely apologize for the misunderstanding here. To automatically track the vesicles, we had to manually exclude the Golgi area. Moreover, only processive and not complete tracks are shown. Therefore, no conclusions can be made from these data on the vesicle exit from the Golgi. We have indicated this clearly in the Results (page 8) and Discussion (page 21) of the revised manuscript and included more representative images in the revised Figure 2E.
In Figure 8G, the tracks for KIF13B-380 motility are difficult to see, which is surprising as KIF13B has been shown to be a superprocessive motor. Is this construct a dimer? If not, do the authors interpret the data as a high binding affinity of the monomer for new microtubules and if so, do they have any speculation on what could be the molecular mechanism? It appears as if KIF13B-380 and EB3 colocalize at the plus ends for a period of time before both are lost but then quickly replenished. Is this common?
KIF13B-380 construct used here contains a leucine zipper from GCN4 and is therefore dimeric. In the revised version of the paper, we have indicated this more clearly in the Results section on page 17 of the revised manuscript. KIF13B-380 does show processive motility, although this is difficult to see close to the outermost microtubule tip as the motor tends to accumulate there. This does not necessarily correlate with a strong accumulation of EB3, likely because EB3 signal is more sensitive to the dynamic state of the microtubule (it diminishes when microtubule growth rate decreases). We now provide a kymograph in Fig. 8G where the processive motility of KIF13B-380 is clearer.
Reviewer #3:
Serra-Marques and co-authors use CRISPR/Cas9 gene editing and live-cell imaging to dissect the roles of kinesin-1 (KIF5) and kinesin-3 (KIF13) in the transport of Rab6-positive vesicles. They find that both kinesins contribute to the movement of Rab6 vesicles. In the context of recent studies on the effect of MAP7 and doublecortin on kinesin motility, the authors show that MAP7 is enriched on central microtubules corresponding to the preferred localization of constitutively-active KIF5B-560-GFP. In contrast, KIF13 is enriched on dynamic, peripheral microtubules marked by EB3.
The manuscript provides needed insight into how multiple types of kinesin motors coordinate their function to transport vesicles. However, I outline several concerns about the analysis of vesicle and kinesin motility and its interpretation below.
Major concerns:
1) The metrics used to quantify motility are sensitive to tracking errors and uncertainty. The authors quantify the number of runs (Fig. 2D,F; 7C) and the average speed (Fig. 3A,B,D,E,H). The number of runs is sensitive to linking errors in tracking. A single, long trajectory is often misrepresented as multiple shorter trajectories. These linking errors are sensitive to small differences in the signal-to-noise ratio between experiments and conditions, and the set of tracking parameters used. The average speed is reported only for the long, processive runs (tracks>20 frames, segments<6 frames with velocity vector correlation >0.6). For many vesicular cargoes, these long runs represent <10% of the total motility. In the 4X-KO cells, it is expected there is very little processive motility, yet the average speed is higher than in control cells. Frame-to-frame velocities are often over-estimated due to the tracking uncertainty. Metrics like mean-squared displacement are less sensitive to tracking errors, and the velocity of the processive segments can be determined from the mean-squared displacement (see for example Chugh et al., 2018, Biophys. J.). The authors should also report either the average velocity of the entire run (including pauses), or the fraction of time represented by the processive segments to aid in interpreting the velocity data.
Two stages of the described tracking and data processing are responsible for the extraction of processive runs: the “linking” method used during the tracking, and the “trajectory segmentation” method, applied to the obtained tracks. The detection and linking of vesicles have been performed using our previously published tracking method (Chenouard et al., 2014, Nature Methods, PMID: 24441936). Our linking method uses multi-frame data association, taking into account detections from four subsequent image frames in order to extend and create a trajectory at any given time. This allows for dealing with temporal disappearance of particles (missing detections) for 1-2 frames and avoiding creation of breaks in longer trajectories. The method is robust to noise, spurious and missing detections and had been fully evaluated in the aforementioned paper (Chenouard et al., 2014) showing excellent performance compared to other tracking methods.
Having the trajectories describing the behavior of each particle, the track segmentation method had been applied to split each trajectory into a sequence of smaller parts (tracklets) describing processive runs and pieces of undirected (diffusive) motion. The algorithm that we used was validated earlier on an artificial dataset (please see Fig.S2e in Katrukha et al., Nat Commun 2017, PMID: 28322225). The chosen parameters were in the range where the algorithm provided less than 10% of false positives. Since the quantified and reported changes in the number of runs are six-fold (Fig.2D,F), we are quite certain that this estimated error (inherent to all automatic image analysis methods) does not affect our conclusions. Moreover, it is consistent with visual observations and manual analysis of representative movies.
Further, we agree that frame-to-frame velocities are often somewhat over-estimated due to the tracking uncertainty. We are aware of such overestimation which is very difficult to avoid. In our case, we estimated (using a Monte Carlo simulation) that such overestimation will positively bias the average not more than 3-6%. Since we focus not on the absolute values of velocities, but rather on the comparison between different conditions, such biasing will be present in all estimates of average velocity and will not affect the presented conclusions.
The usage of mean square displacement (MSD) to analyze trajectories containing both periods of processive runs and diffusive motion is confusing, since it represents average value over whole trajectories, resulting in the MSD slope which is in the range of 1.5 (i.e. between 1, diffusive and 2, processive; please see Fig.2c in Katrukha et al., 2017, Nature Communications, PMID: 28322225). Therefore, initial segmentation of trajectories is necessary, as it was performed in the paper by Chugh et al (Chugh et al., 2018, Biophysical Journal, PMID: 30021112; please see Fig.2e in that paper), suggested by the reviewer. In this paper the authors used an SCI algorithm, which is very similar to our analysis, relying on temporal correlations of velocities. Indeed, MSD analysis of only processive segments is less sensitive to tracking errors, but it reports an average velocity of the whole population of runs. This method is well suited if one would expect monodisperse velocity distribution (the case in Chugh et al, where single motor trajectories are analyzed). If there are subpopulations with different speeds (as we observed for Rab6 by manual kymograph analysis), this information will be averaged out. Therefore, we used histogram/distribution representations for our speed data, which in our opinion represents these data better.
Finally, we fully agree with the reviewers that the fractions of processive/diffusive motion should be reported. In the revised version, we have added new plots to the revised manuscript (Figure 2G-I, Figure 2 - figure supplement 2G) illustrating these data for different conditions. Our data fully support the reviewer’s statement that processive runs represent less than 10% of total vesicle motility (new Figure 2G). As could be expected, the total time vesicles spent in processive motion and the percentage of trajectories containing processive runs strongly depended on the presence of the motors (new Figure 2H,I). However, within trajectories that did have processive segments, the percentage of processive movement was similar (new Figure 2I).
We note that while our analysis is geared towards identification and characterization of processive runs (which was verified manually), analysis of diffusive movements poses additional challenges and is even more sensitive to linking errors. Therefore, we do not make any strong quantitative conclusions about the exact percentage and the properties of diffusive vesicle movements, and their detailed studies will require additional analytic efforts.
2) The authors show that transient expression of either KIF13B or KIF5B partially rescues Rab6 motility in 4X-KO cells and that knock-out of KIF13B and KIF5B have an additive effect. They also analyze two vesicles where KIF13B and KIF5B co-localize on the same vesicle. The authors conclude that KIF13B and KIF5B cooperate to transport Rab6 vesicles. However, the nature of this cooperation is unclear. Are the motors recruited sequentially to the vesicles, or at the same time? Is there a subset of vesicles enriched for KIF13B and a subset enriched for KIF5B? Is motor recruitment dependent on localization in the cell? These open questions should be addressed in the discussion.
Unfortunately, only fluorescent motors and not the endogenous ones can be detected on vesicles, so we cannot make any strong statements on this issue. Since KIF13B can compensate for the absence of KIF5B, it can be recruited to the vesicle when it emerges from the Golgi apparatus. However, in normal cells, KIF5B likely plays a more prominent role in pulling the vesicles from the Golgi, as Rab6 vesicles generated in the presence of KIF5B are larger (Figure 5I). We show in Figure 1G,H that KIF13B does not exchange on the vesicle and stays on the vesicle until it fuses with the plasma membrane. These data suggest that once recruited, KIF13B stays bound to the vesicle. Obtaining such data for KIF5B is more problematic because fewer copies of this motor are typically recruited to the vesicle (Figure 4B) and its signal is therefore weaker. Further research with endogenously tagged motors and highly sensitive imaging approaches will be needed to address the important open questions raised by the reviewer. We have added these points to the Discussion on pages 19 and 21 of the revised manuscript.
3) The authors suggest that KIF5B transports Rab6 vesicles along centrally-located microtubules while KIF13B drives transport on peripheral microtubules. Is the velocity of Rab6 vesicles different on central and peripheral microtubules in control cells?
As indicated in our answer to Major Comment 2 of Reviewer 1, we show in Figure 8B (grey curve) that vesicle speed remains relatively constant along the cell radius in control HeLa cells. We note, however, that our previous work has shown that in these cells microtubules are quite stable even at the cell periphery, due to the high activity of the CLASP-containing cortical microtubule stabilization complex (Mimori-Kiyosue et al., 2005, Journal of Cell Biology, PMID: 15631994; van der Vaart et al., 2013, Developmental Cell, PMID: 24120883). We therefore hypothesized that changes in vesicle speed distribution along the cell radius would be more obvious in cells with highly dynamic microtubule networks and performed a preliminary experiment in MRC5 human lung fibroblasts, which have a very sparse and dynamic microtubule cytoskeleton (Splinter et al., 2012, Molecular Biology of the Cell, PMID: 22956769). As shown in the figure above, we indeed found that vesicles move faster at the cell periphery.
4) The imaging and tracking of fluorescently-labeled kinesins in cells as shown in Fig. 4 is impressive. This is often challenging as kinesin-3 forms bright accumulations at the cell periphery and there is a large soluble pool of motors, making it difficult to image individual vesicles. The authors should provide additional details on how they addressed these challenges. Control experiments to assess crosstalk between fluorescence images would increase confidence in the colocalization results.
Imaging of vesicle motility was performed using TIRF microscopy focusing on regions where no strong motor accumulation was observed. We have little cross-talk between red and green channels, but channel cross talk in the three-color images shown in Figure 4E was indeed a potential concern. To address this potential issue, we performed the appropriate controls and added a new figure to the revised manuscript (Figure 4 – figure supplement 1). We conclude that we can reliably simultaneously detect blue, green and red channels without significant cross-talk on our microscope setup.
Reviewer #1:
Köster and colleagues present a brief report in which they study in 9 month-old babies the electrophysiological responses to expected and unexpected events. The major finding is that in addition to a known ERP response, an NC present between 400-600 ms, they observe a differential effect in theta oscillations. The latter is a novel result and it is linked to the known properties of theta oscillations in learning. This is a nice study, with novel results and well presented. My major reservation however concerns the push the authors make for the novelty of the results and their interpretation as reflecting brain dynamics and rhythms. The reason for that is, that any ERP, passed through the lens of a wavelet/FFT etc, will yield a response at a particular frequency. This is especially the case for families of ERP responses related to unexpected event e.g., MMR, and NC, etc. For which there is plenty of literature linking them to responses to surprising event, and in particular in babies; and which given their timing will be reflected in delta/theta oscillations. The reason why I am pressing on this issue, is because there is an old, but still ongoing debate attempting to dissociate intrinsic brain dynamics from simple event related responses. This is by no means trivial and I certainly do not expect the authors to resolve it, yet I would expect the authors to be careful in their interpretation, to warn the reader that the result could just reflect the known ERP, to avoid introducing confusion in the field.
We would like to thank the author for highlighting the novelty of the results. Critically, there is one fundamental difference in investigating the ERP response and the trial-wise oscillatory power, which we have done in the present analysis: when looking at the evoked oscillatory response (i.e., the TF characteristics of the ERP), the signal is averaged over trials first and then subjected to a wavelet transform. However, when looking at the ongoing (or total) oscillatory response, the wavelet transform is applied at the level of the single trial, before the TF response of the single trials is averaged across the trials of one condition trials (for a classical illustration, see Tallon-Baudry & Bertrand, 1999; TICS, Box 2). We have now made this distinction more salient throughout the manuscript.
In the present study, the results did not suggest a relation between the ERP and the ongoing theta activity, because the topography, temporal evolution, and polarity of the ERP and the theta response were very dissimilar: Looking at Figure 2 (A and B) and Figure 3 (B and C), the Nc peaks at central electrodes, but the theta response is more distributed, and the expected versus unexpected difference was specific for the .4 to .6 s time window, but the theta difference lasted the whole trial. Furthermore, the NC was higher for expected versus unexpected, which should (due to the low frequency) rather lead to a higher theta power for unexpected, in contrast to expected events for the time frequency analysis for the Nc. To verify this intuition, we now ran a wavelet analysis on the evoked response (i.e., the ERP) and, for a direct comparison, also plotted the ongoing oscillatory response for the central electrodes (see Additional Figure 1). These additional analyses nicely illustrate that the trial-wise theta response provides a fundamentally different approach to analyze oscillatory brain dynamics.
Because this is likely of interest to many readers, we also report the results of the wavelet analysis of the ERP versus the analysis of the ongoing theta activity at central electrodes and the corresponding statistics in the result section, and have also included the Additional Figure in the supplementary materials, as Figure S2.
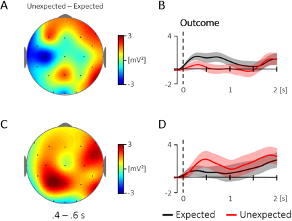
Additional Figure 1. Comparison of the topography and time course for the 4 – 5 Hz activity for the evoked (A, B) and the ongoing (C, D) oscillatory response at central electrodes (400 – 600 ms; Cz, C3, C4; baseline: -100 – 0 ms). (A) Topography for the difference between unexpected and expected events in the evoked oscillatory response. (B) The corresponding time course at central electrodes, which did not reveal a significant difference between 400 – 600 ms, t(35) = 1.57, p = .126. (C) Topography for the same contrast in the ongoing oscillatory response and (D) the corresponding time course at central electrodes, which did likewise not reveal a significant difference between 400 – 600 ms, t(35) = -1.26, p = .218. The condition effects (unexpected - expected) were not correlated between the evoked and the ongoing response, r = .23, p = .169.
A second aspect that I would like the authors to comment on is the power of the experimental design to measure surprise. From the methods, I gathered that the same stimulus materials and with the same frequency were presented as expected and unexpected endings. If that is the case, what is the measure of surprise? For once the same materials are shown causing habituation and reducing novelty and second the experiment introduces a long-term expectation of a 50:50 proportion of expected/unexpected events. I might be missing something here, which is likely as the methods are quite sparse in the description of what was actually done.
We have used 4 different stimuli types (variants) in each of the 4 different domains, with either an expected or unexpected outcome. This resulted in 32 distinct stimulus sequences, which we presented twice, resulting in (up to) 64 trials. We have now described this approach and design in more detail and have also included all stimuli as supplementary material (Figure S1). In particular, we have used multiple types in each domain to reduce potential habituation or expectation effects. Still, we agree that one difficulty may be that, over time, infants got used to the fact that expected and unexpected outcomes were to be similarly “expected” (i.e., 50:50). However, if this was the case it would have resulted in a reduction (or disappearance) of the condition effect, and would thus also reduce the condition difference that we found, rather than providing an alternative explanation. We now included this consideration in the method section (p. 7).
Two more comments concerning the analysis choices:
1) The statistics for the ERP and the TF could be reported using a cluster size correction. These are well established statistical methods in the field which would enable to identify the time window/topography that maximally distinguished between the expected and the unexpected condition both for ERP and TF. Along the same lines, the authors could report the spatial correlation of the ERP/TF effects.
For the ERP analysis we used the standard electrodes typically analyzed for the Nc in order to replicate effects found in former research (Langeloh et al., 2020; see also, Kayhan et al., 2019; Reynolds and Richards, 2005; Webb et al., 2005). For the TF analyses we used the most conservative criterion, namely all scalp recorded electrodes and the whole time window from 0 to 2000 ms, such that we did not make any choice regarding time window or the electrodes (i.e., which could be corrected for against other choices). We have now made those choices clearer in the method section, and why we think that, under these condition a multiple comparison correction is not needed/applicable (p. 10). Regarding the spatial correlation of the ERP and TF effects, we explained in response to the first comment the very different nature of the TF decomposition of the ERP and ongoing oscillatory activity and also that these were found to be interdependent (i.e., uncorrelated). We hope that with the additional analysis included in response to this comment that this difference is much clearer now.
2) While I can see the reason why the authors chose to keep the baseline the same between the ERP and the TF analysis, for time frequency analysis it would be advisable to use a baseline amounting to a comparable time to the frequency of interest; and to use a period that does not encroach in the period of interest i.e., with a wavelet = 7 and a baseline -100:0 the authors are well into the period of interested.
The difficulty in choosing the baseline in the present study was two-fold. First, we were interested in the ERP and the change in neural oscillations upon the onset of an outcome picture within a continuous presentation of pictures, forming a sequence. Second, we wanted to use a similar baseline for both analyses, to make them comparable. Because the second picture (the picture before the outcome picture) also elicited both an ERP and an oscillatory response at ~ 4 Hz (see Additional Figure 2), we choose a baseline just before the onset of the outcome stimulus, from -100 to 0 ms. Also we agree that the possibility to take a longer and earlier baseline, in particular for the TF results would have been favorable, but still consider that the -100 to 0 ms is still the best choice for the present analysis. Notably, because we found an increase in theta oscillations and the critical difference relies on a higher theta rhythm in one compared to the other condition, the effects of the increase in theta, if they effected the baseline, this effect would counteract rather than increase the current effect. We now explain this choice in more detail (p.10).
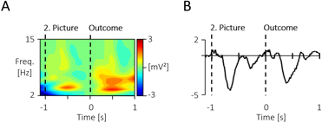
Additional Figure 1. Display of the grand mean signals prior to the -100 to 0 baseline and outcome stimulus. (A) The time-frequency response across all scalp-recorded electrodes, as well as (B) the ERP at the central electrodes (Cz, C3, C4) across both conditions show a similar response to the 2. picture like the outcome picture. Thus a baseline just prior to the stimulus of interest was chosen, consistent for both analyses.
Reviewer #2:
The manuscript reports increases in theta power and lower NC amplitude in response to unexpected (vs. expected) events in 9-month-olds. The authors state that the observed increase in theta power is significant because it is in line with an existing theory that the theta rhythm is involved in learning in mammals. The topic is timely, the results are novel, the sample size is solid, the methods are sound as far as I can tell, and the use of event types spanning multiple domains (e.g. action, number, solidity) is a strength. The manuscript is short, well-written, and easy to follow.
1) The current version of the manuscript states that the reported findings demonstrate that the theta rhythm is involved in processing of prediction error and supports the processing of unexpected events in 9-month-old infants. However, what is strictly shown is that watching at least some types of unexpected events enhance theta rhythm in 9-month-old infants, i.e. an increase in the theta rhythm is associated with processing unexpected events in infants, which suggests that an increase in the theta rhythm is a possible neural correlate of prediction error in this age range. While the present novel findings are certainly suggestive, more data and/or analyses would be needed to corroborate/confirm the role of the observed infant theta rhythm in processing prediction error, or document whether and how this increase in the theta rhythm supports the processing of unexpected events in infants. (As an example, since eye-tracking data were collected, are trial-by-trial variations in theta power increases to unexpected outcomes related to how long individual infants looked to the unexpected outcome pictures?) If it is not possible to further confirm/corroborate the role of the theta rhythm with this dataset, then the discussion, abstract, and title should be revised to more closely reflect what the current data shows (as the wording of the conclusion currently does), and clarify how future research may test the hypothesis that the infant theta rhythm directly supports the processing of prediction error in response to unexpected events.
We would like to thank the reviewer for acknowledging the merit of the present research.
On the one hand, we have revised our manuscript and are now somewhat more careful with our conclusion, in particular with regard to the refinement of basic expectations. On the other hand, we consider the concept of “violation to expectation” (VOE), which is one of the most widely used concepts in infancy research, very closely linked to the concept of a prediction error processing, namely a predictive model is violated. In particular, we have made this conceptual link in a recent theoretical paper (Köster et al., 2020), and based on former theoretical considerations about the link between these two concepts (e.g., see Schubotz 2015; Prediction and Expectation). In particular, in the present study we used a set of four different domains of violation of expectation paradigms, which are among the best established domains of infants core knowledge (e.g., action, solidity, cohesion, number; cf. Spelke & Kinzler, 2007). It was our specific goal not to replicate, for another time, that infants possess expectations (i.e., make predictions) in these domains, but to “flip the coin around” and investigate infants’ prediction error more generally, independent of the specific domain. We have now made the conceptual link between VOE and prediction error processing more explicit in the introduction of the manuscript and also emphasize that we choose a variety of domains to obtain a more general neural marker for infant processing of prediction errors.
Having said this, indeed, we planned to assess and compare both infants gaze behavior and EEG response. Unfortunately, this was not very successful and the concurrent recording only worked for a limited number of infants and trials. This led us to the decision to make the eye-tracking study a companion study and to collect more eye-tracking data in an independent sample of infants after the EEG assessment was completed, such that a match between the two measures was not feasible. We now make this choice more explicit in the method section (p. 7). In addition, contrary to our basic assumption we did not find an effect in the looking time measure. Namely, there was no difference between expected and unexpected outcomes. We assume that this is due to the specificities of the current design that was rather optimized for EEG assessments: We used a high number of repetitions (64), with highly variable domains (4), and restricted the time window for potential looking time effects to 5 seconds, which is highly uncommon in the field and therefore not directly comparable with former studies.
Finally, besides the ample evidence from former studies using VOE paradigms, if it were not the unexpected vs. expected (i.e., unpredicted vs. predicted) condition contrast which explains the differences we found in the ERP and the theta response, there would need to be an alternative explanation for the differential responses in the EEG, which produce the hypothesized effects. (Please also note that there are many studies relying their VOE assumption on ERPs alone, here we have two independent measures suggesting that infants discriminated between those conditions.)
2) The current version of the manuscript states "The ERP effect was somewhat consistent across conditions, but the effect was mainly driven by the differences between expected and unexpected events in the action and the number domain (Figure S1). The results were more consistent across domains for the condition difference in the 4 - 5 Hz activity, with a peak in the unexpected-expected difference falling in the 4 - 5 Hz range across all electrodes (Figure S2)". However, the similarity/dissimilarity of NC and theta activity responses across domains was not quantified or tested. Looking at Figures S1 and S2, it is not that obvious to me that theta responses were more consistent across domains than NC responses. I understand that there were too few trials to formally test for any effect of domain (action, number, solidity, cohesion) on NC and theta responses, either alone or in interaction with outcome (expected, unexpected). It may still be possible to test for correlations of the topography and time-course of the individual average unexpected-expected difference in NC and theta responses across domains at the group level, or to test for an effect of outcome (expected, unexpected) in individual domains for subgroups of infants who contributed enough trials. Alternatively, claims of consistency across domains may be altered throughout, in which case the inability to test whether the theta and/or NC signatures of unexpected event processing found are consistent across domains (vs. driven by some domains) should be acknowledged as a limitation of the present study.
We agree that this statement rather reflected our intuition and would not surpass statistical analysis given the low number of trials. So we are happy to refrain from this claim and simply refer to the supplementary material for the interested reader and also mention this as a perspective for future research in the discussion (p. 12; p. 15).
As outlined in our previous response, it was also not our goal to draw conclusions about each single domain, but rather to present a diversity of stimulus types from different core knowledge domains to gain a more generalized neural marker for infants’ processing of unexpected, i.e., unpredicted events.
Reviewer #3:
General assessment:
In this manuscript, the authors bring up a contemporary and relevant topic in the field, i.e. theta rhythm as a potential biomarker for prediction error in infancy. Currently, the literature is rich on discussions about how, and why, theta oscillations in infancy implement the different cognitive processes to which they have been linked. Investigating the research questions presented in this manuscript could therefore contribute to fill these gaps and improve our understanding of infants' neural oscillations and learning mechanisms. While we appreciate the motivation behind the study and the potential in the authors' research aim, we find that the experimental design, analyses and conclusions based on the results that can be drawn thereafter, lack sufficient novelty and are partly problematic in their description and implementation. Below, we list our major concerns in more detail, and make suggestions for improvements of the current analyses and manuscript.
Summary of major concerns:
1) Novelty:
(a) It is unclear how the study differs from Berger et al., 2006 apart from additional conditions. Please describe this study in more detail and how your study extends beyond it.
We would like to thank the reviewers for emphasizing the timeliness and relevance of the study.
The critical difference between the present study and the study by Berger et al. 2006 was that the authors applied, as far as we understand this from Figure 4 and the method section of their study, the wavelet analysis to the ERP signal. In contrast, in the present study, we applied the wavelet analysis at the level of single trials. We now explain the difference between the two signals in more detail in the revised manuscript and also included an additional comparison between the evoked (i.e., ERP) and the ongoing (i.e., total) oscillatory response (for more details, please see the first response to the first comment of reviewer 1).
(b) Seemingly innovative aspects (as listed below), which could make the study stand out among previous literature, but are ultimately not examined. Consequently, it is also not clear why they are included.
-Relation between Nc component and theta.
-Consistency of the effect across different core knowledge domains.
-Consistency of the effect across the social and non-social domains.
-Link between infants looking at time behavior and theta.
We are thankful for these suggestions, which are closely related to the points raised by reviewer 1 and 2. With regard to the relation between the Nc and the theta response, we have now included a direct comparison of these signals (see Additional Figure 1, i.e., novel Figure S2; for details, please see the first response to the first comment of reviewer 1). Regarding the consistency of effects across domains, we have explained in response to point 1 by reviewer 2 that this was not the specific purpose of the present study, but we aimed at using a diversity of VOE stimuli to obtain a more general neural signature for infants’ prediction error processing, and explain this in more detail in the revised manuscript. Having said this, we agree that the question of consistency of effects between conditions is highly interesting, but we would not consider the data robust enough to confidently test these differences given the limited number of trials available per stimulus category. We now discuss this as a direction for future research (p. 15). Finally, we also agree with regard to the link between looking times and the theta rhythm. As also outlined in response to point 1 by reviewer 2 (paragraph 2), we initially had this plan, but did not succeed in obtaining a satisfactory number of trials in the dual recording of EEG and eye-tracking, which made us change these plans. This is now explained in detail in the method section (p. 7).
(c) The reason to expect (or not) a difference at this age, compared to what is known from adult neural processing, is not adequately explained.
-Potentially because of neural generators in mid/pre-frontal cortex? See Lines 144-146.
The overall aim of the present study was to identify the neural signature for prediction error processing in the infant brain, which has, to the best of our knowledge, not been done this explicitly and with a focus on the ongoing theta activity and across a variety of violations in infants’ core knowledge domains. Because we did not expect a specific topography of this effect, in particular across multiple domains, we included all electrodes in the analyses. We have now clarified this in the method section (p. 10).
(d) The study is not sufficiently embedded in previous developmental literature on the functionality of theta. That is, consider theta's role in error processing, but also the increase of theta over time of an experiment and it's link to cognitive development. See, for example: Braithwaite et al., 2020; Conejero et al., 2018; Adam et al., 2020.
We are thankful that the reviewer indicated these works and have now included them in the introduction and discussion. Closest to the present study is the study by Conejero et al., 2018. However, this study is also based on theta analyses of the ERP, not of the ongoing oscillatory response and it includes considerably older infants (i.e., 16-month-olds instead of 9-month-olds as in the present study).
2) Methodology:
(a) Design: It is unclear what exactly a testing session entails.
-Was the outcome picture always presented for 5secs? The methods section suggests that, but the introduction of the design and Figure 1 do not. This might be misleading. Please change in Figure 1 to 5sec if applicable.
Yes, the final images were shown for 5s in order to simultaneously assess infants’ looking times. However, we included trials in the EEG analysis if infants looked for 2s, so this is the more relevant info for the analysis. We now clarified this in the method section (p. 7) and have also added this info in the figure caption.
-Were infants' eye-movements tracked simultaneously to the EEG recording? If so, please present findings on their looking time and (if possible) pupil size. Also examine the relation to theta power. This would enhance the novelty and tie these findings to the larger looking time literature that the authors refer to in their introduction.
Yes, in response to the second reviewer (comment 1) we explained in more detail why the joint analysis of the EEG and looking time data was not possible: We planned to assess both, infants gaze behavior and EEG response. Unfortunately, this was not very successful and the dual recording only worked for a few infants and trials. This led us to collect more eye-tracking data after the EEG assessment was completed, such that a match between the two measures was not feasible. We now clarified this in the method section (p. 7).
(b) Analysis:
-In terms of extracting theta power information: The baseline of 100ms is extremely short for a comparison in the frequency domain, since it does not even contain half a cycle of the frequency of interest, i.e. 4Hz. We appreciate the thought to keep the baseline the same as in the ERP analysis (which currently is hardly focused on in the manuscript), but it appears problematic for the theta analysis. Also, if we understand the spectral analysis correctly, the window the authors are using to estimate their spectral estimates is largely overlapping between baseline and experimental window. The question arises whether a baseline is even needed here, or if a direct contrast between conditions might be better suited.
Please see our explanation about the choice of the baseline in our response to reviewer 1, comment 2. Because our stimulus sequences were highly variable, likely leading to highly variable overall theta activity, and our specific interest was in the change in theta activity upon the onset of the unexpected versus unpredicted outcome, we still consider it useful to take a baseline here. Also because this makes the study more closely comparable to the existing literature. We now clarified this in the method section (p. 9)
-In terms of statistical testing
-It appears that the authors choose the frequency band that will be entered in the statistical analysis from visual inspection of the differences between conditions. They write: "we found the strongest difference between 4 - 5 Hz (see lower panel of Figure 3). Therefore, and because this is the first study of this kind, we analyzed this frequency range." ll. 277-279). This approach seems extremely problematic since it poses a high risk for 'double-dipping'. This is crucial and needs to be addressed. For instance, the authors could run non-parametric permutation tests on the time-frequency domain using FDR correction or cluster-based permutation tests on the topography.
-Lack of examining time- / topographic specificity.
Please also note the sentence before this citation, which states our initial hypothesis: “While our initial proposal was to look at the difference in the 4 Hz theta rhythm between conditions (Köster et al., 2019), we found the strongest difference between 4 – 5 Hz (see lower panel of Figure 3).” Note that the hypothesis of 4 Hz can be clearly derived from our 2019 study. We would maintain that the center frequency we took for the analysis 4.5Hz (i.e., 4 – 5Hz) is very close to this original hypothesis and, considering that we applied a novel design and analyses in very young infants, could indeed hardly have fallen more closely to this initial proposal. The frequency choice is also underlined, as the reviewer remarks, by the consistency of this peak across domains, peaking at 4Hz (cohesion), 4.5Hz (action), and 5Hz (solidity, number). Importantly, please note that we have chosen the electrodes and time window very conservatively, namely by including the whole time period and all electrodes, which we now explain in more detail on p. 10. Please also see our response to reviewer 1, comment “1)”.
3) Interpretation of results:
(a) The authors interpret the descriptive findings of Figure S1 as illustration of the consistency of the results across the four knowledge domains. While we would partly agree with this interpretation based on column A of that figure (even though also there the peak shifts between domains), columns B and C do not picture a consistent pattern of data. That is, the topography appears very different between domains and so does the temporal course of the 4-5Hz power, with only showing higher power in the action and number domain, not in the other two. Since none of these data were compared statistically, any interpretation remains descriptive. Yet, we would like to invite the authors to critically reconsider their interpretation. You also might want to consider adding domain (action, number etc.) as a covariate to your statistical model.
We agree with the reviewers (reviewer 2 and reviewer 3) that our initial interpretation of the data regarding the consistency of effects across domains may have been too strong. Thus, in the revised version of the manuscript, we do not state that the TF analysis revealed more consistent results. Given that the analysis was based on a different subsample and highly variable in trial numbers, we did not enter them as a covariate in the statistical model.
We thank the editors and the reviewers for a number of useful criticisms and suggestions, and for the opportunity given to us, as authors, to publicly reply to the comments. This is a useful exercise, which brings to the attention of the reader lights, but also shadows of the reviewing process, and that we hope will lead in future to develop a better approach to it. Here, we will reply to a number of selected issues which appear to us to be of particular relevance.
Reviewer 1
Reviewer 1 disqualifies our work altogether, based on her/his statement that: “In the paper by Mercurio et al, the authors examine the role of SOX2 in the development of mouse hippocampal dentate gyrus. Using conditionally mutant SOX2 mice the authors show that early, but not late, deletion of SOX2 leads to developmental impairments of the dentate gyrus. A drawback of their study is that these findings have been reported previously by the group (Favaro et al. 2009; Ferri et al. 2013).”
The statement reported in bold is simply not true. In Favaro et al. 2009 (Nat Neurosci 12:1248), we demonstrated that nes-Cre-mediated Sox2 deletion leads to defects in postnatal, but not embryonic, hippocampal neurogenesis. In Ferri et al. 2013 (Development 140:1250), we demonstrated that FoxG1Cre-mediated Sox2 deletion leads to defective development of the VENTRAL forebrain. The presence, at the end of gestation, of hippocampal defects was just mentioned in one sentence: - “the hippocampus, at E18.5, was severely underdeveloped (not shown)” (line 1, page 1253)-, and not analyzed any further. In the present work, we describe in detail, starting from E12.5, up to E18.5, how the hippocampal defect develops, and undertake a detailed study of downstream gene expression and cellular defects arising in mutants.
It is unfortunate that the reviewer further insists on the same misleading, and unfounded statement – see her/his comment 3, highlighted in bold character: “the authors state "...remarkably, in the FoxG1-Cre cKO, the DG appears to be almost absent (Figure 2A).". The question is why this finding is remarkable as it already was published in (Ferri et al. 2013)”. As mentioned above, we only remark, in Ferri et al., that the hippocampus was severely underdeveloped (not shown).
Reviewer 2
Reviewer 2 states, already at the beginning: “I am concerned about a major confounding issue (see below).” ... “The authors rely on Foxg1-Cre for their main evidence that very early deletion of Sox2 leads to near loss of the dentate. However, it doesn't appear that the authors are aware that Foxg1 het mice have a fairly significant dentate phenotype (see this paper).”
The reviewer refers to the fact that, to delete Sox2, we need to express a Cre gene “knocked-in” into the Foxg1 gene; hence, heterozygous and homozygous Sox2 deletions will be accompanied by heterozygous loss of Foxg1. If Foxg1 is important for hippocampus development, the absence of a Foxg1 allele will affect the phenotype.
Unfortunately, the statement of the reviewer is subtly misleading, and leads the reader who has not checked the data reported in the cited paper (Shen et al., 2006) to erroneously believe that heterozygous loss of Foxg1 may be responsible for the effects that we report upon homozygous Sox2 deletion. In contrast to the statement made by the reviewer, the paper cited by the reviewer documents that, while heterozygous loss of Foxg1 leads to important POSTNATAL dentate gyrus abnormalities, the PRENATAL development of the dentate gyrus is essentially normal (Figure 6) (“a subtle and inconsistent defect” of the ventral blade observed in about 50% of the mice at E18.5, according to the authors of that paper). Compare “subtle and inconsistent defect” by Shen et al. with “fairly significant dentate phenotype”, as stated by the reviewer. As our paper is entirely focused on defects seen in PRENATAL development in Foxg1Cre; Sox2 mutants, the subtle and inconsistent defects seen by Shen et al. are in sharp contrast with the deep defects seen in embryonic development in our Foxg1Cre;Sox2-/- mutants, and in agreement with the similarity we observe between wild type and heterozygous Foxg1Cre;Sox2+/- embryos (page 5, lines 140-145, of the version of the Full Submission for publication on August 30). An example showing the comparison between a Wild type, a FoxG1 +/- heterozygote;Sox2+/- heterozygote and a FoxG1 heterozygote;Sox2-/- homozygote is now shown in the accompanying figure.
Obviously the incorrect statement kills our paper by itself. If the reviewer had doubts, we could have provided plenty of additional data demonstrating the lack of significant differences between Foxg1CRE Sox2+/- and wild type (Sox2+/+) embryos, as we stated in our paper.
There is an additional interesting comment by Reviewer 2 (see points 2 and 6). The reviewer argues that “The only two direct targets they find don't seem likely to be important players in the phenotypes they describe”. The Reviewer excludes the Gli3 gene (a direct Sox2 target, see Fig. 6), as a possible important player, in spite of the observation that Gli3 is decreased, at early developmental stages, in the cortical hem (Figure 5). The reviewer says “The Gli3 [mutation] phenotypes that have been published are quite distinct from this”. We object that the Gli3 phenotypes are indeed more severe than the phenotype of our mutant, and include failure to develop a dentate gyrus. However, this observation does not preclude the hypothesis that the decreased expression of Gli3 in our mutant is directly responsible for the phenotype we observe. The more severe phenotype of the Gli3 mutants is in fact due to a germ-line null mutation, whereas, in our Foxg1-Cre Sox2 mutants, we observe only a reduction of Gli3 expression, around E12.5 (Fig. 5), that is compatible with a less severe dentate gyrus phenotype. The Reviewer adds that Wnt3A, based on the phenotype of the knock-out mice, similar to that of our Sox2 deleted mice, is a more relevant gene, but it is not a direct target of Sox2. However, the fact that Wnt3A is apparently not directly regulated by Sox2 is not necessarily to be considered a “minus”; Sox2, being a transcription factor, is expected to directly regulate a multiplicity of genes, whose expression will affect the expression of other genes. Indeed, we presented in Fig 6D the hypothesis that decreased expression of Gli3 may contribute to decreased expression of Wnt3A, as already proposed by Grove et al. (1998) based on the observation that Gli3 null mutants lose the expression of Wnt3A (and other Wnt factors) from the cortical hem. The additional suggestion made by the Reviewer, in the context of the Wnt3A hypothesis, to investigate LEF1, as a potential direct Sox2 target, and its expression, is certainly interesting, but, as stated by the reviewer, LEF1 is downstream to Wnt3A, and, by itself, its hypothetical regulation by Sox2 would not explain the downregulation of Wnt3A. Moreover, we already have evidence that Sox2 does not directly regulate Wnt3A (unpublished).
Reviewer 1 and 2
Both Reviewer 1 and 2 have questions about the timing of Sox2 ablation in the Sox2 mutants obtained with the three different Cre deleters. As we state in the text (pages 4, 6), Foxg1-Cre deletes at E.9.5 (Ferri et al., 2013; Hébert and McConnell, 2000); Emx1-Cre deletes from E10.5 onwards, but not at E9.5 (Gorski et al., 2002; see also Shetty AS et al., PNAS 2013, E4913); Nestin-Cre deletes at later stages, around E12.5 (Favaro et al. 2009).
Reviewer 3
We thank Reviewer 3 for the useful considerations and suggestions, which constructively help to improve the paper.
Evidence that Sox2+/-;FoxG1+/- hippocampi at E18.5 do not significantly differ from wild type (Sox2+/+, FoxG1+/+) controls. In contrast, Sox2-/-;FoxG1+/- hippocampi are severely defective. (A) GFAP immunofluorescence at E18.5 on coronal sections of control and FoxG1-Cre cKO hippocampi (controls n=6, mutants n=4). (B) In situ hybridization at E18.5 for NeuroD (controls n=4, mutants n=3) on coronal sections of control and FoxG1-Cre cKO hippocampi. Arrows indicate dentate gyrus (DG); note the strong decrease of the dentate gyrus, and the radial glia (GFAP) disorganization in cKO.<br /> The Sox2flox/flox genotype corresponds to wild type mice (Sox2+/+). The Sox2+/flox ; FoxG1Cre genotype corresponds to Sox2+/-; FoxG1+/- controls. The Sox2flox/flox ; FoxG1Cre genotype corresponds to Sox2-/-; FoxG1+/- mutants.
Reviewer #1:
Hutchings et al. report an updated cryo-electron tomography study of the yeast COP-II coat assembled around model membranes. The improved overall resolution and additional compositional states enabled the authors to identify new domains and interfaces--including what the authors hypothesize is a previously overlooked structural role for the SEC31 C-Terminal Domain (CTD). By perturbing a subset of these new features with mutants, the authors uncover some functional consequences pertaining to the flexibility or stability of COP-II assemblies.
Overall, the structural and functional work appears reliable, but certain questions and comments should be addressed prior to publication. However, this reviewer failed to appreciate the conceptual advance that warrants publication in a general biology journal like eLIFE. Rather, this study provides a valuable refinement of our understanding of COP-II that I believe is better suited to a more specialized, structure-focused journal.
We agree that in our original submission our description of the experimental setup, indeed similar to previous work, did not fully capture the novel findings of this paper. Rather than being simply a higher resolution structure of the COPII coat, in fact we have discovered new interactions in the COPII assembly network, and we have probed their functional roles, significantly changing our understanding of the mechanisms of COPII-mediated membrane curvature. In the revised submission we have included additional genetic data that further illuminate this mechanism, and have rewritten the text to better communicate the novel aspects of our work.
Our combination of structural, functional and genetic analyses goes beyond refining our textbook understanding of the COPII coat as a simple ‘adaptor and cage’, but rather it provides a completely new picture of how dynamic regulation of assembly and disassembly of a complex network leads to membrane remodelling.
These new insights have important implications for how coat assembly provides structural force to bend a membrane but is still able to adapt to distinct morphologies. These questions are at the forefront of protein secretion, where there is debate about how different types of carriers might be generated that can accommodate cargoes of different size.
Major Comments: 1) The authors belabor what this reviewer thinks is an unimportant comparison between the yeast reconstruction of the outer coat vertex with prior work on the human outer coat vertex. Considering the modest resolution of both the yeast and human reconstructions, the transformative changes in cryo-EM camera technology since the publication of the human complex, and the differences in sample preparation (inclusion of the membrane, cylindrical versus spherical assemblies, presence of inner coat components), I did not find this comparison informative. The speculations about a changing interface over evolutionary time are unwarranted and would require a detailed comparison of co-evolutionary changes at this interface. The simpler explanation is that this is a flexible vertex, observed at low resolution in both studies, plus the samples are very different.
We do agree that our proposal that the vertex interface changes over evolutionary time is speculative and we have removed this discussion. We agree that a co-evolutionary analysis will be enlightening here, but is beyond the scope of the current work.
We respectfully disagree with the reviewer’s interpretation that the difference between the two vertices is due to low resolution. The interfaces are clearly different, and the resolutions of the reconstructions are sufficient to state this. The reviewer’s suggestion that the difference in vertex orientation might be simply attributable to differences in sample, such as inclusion of the membrane, cylindrical versus spherical morphology, or presence of inner coat components were ruled out in our original submission: we resolved yeast vertices on spherical vesicles (in addition to those on tubes) and on membrane-less cages. These analyses clearly showed that neither the presence of a membrane, nor the change in geometry (tubular vs. spherical) affect vertex interactions. These experiments are presented in Supplementary Fig 4 (Supplementary Fig. 3 in the original version). Similarly, we discount that differences might be due to the presence or absence of inner coat components, since membrane-less cages were previously solved in both conditions and are no different in terms of their vertex structure (Stagg et al. Nature 2006 and Cell 2008).
We believe it is important to report on the differences between the two vertex structures. Nevertheless, we have shifted our emphasis on the functional aspects of vertex formation and moved the comparison between the two vertices to the supplement.
2) As one of the major take home messages of the paper, the presentation and discussion of the modeling and assignment of the SEC31-CTD could be clarified. First, it isn't clear from the figures or the movies if the connectivity makes sense. Where is the C-terminal end of the alpha-solenoid compared to this new domain? Can the authors plausibly account for the connectivity in terms of primary sequence? Please also include a side-by-side comparison of the SRA1 structure and the CTD homology model, along with some explanation of the quality of the model as measured by Modeller. Finally, even if the new density is the CTD, it isn't clear from the structure how this sub-stoichiometric and apparently flexible interaction enhances stability. Hence, when the authors wrote "when the [CTD] truncated form was the sole copy of Sec31 in yeast, cells were not viable, indicating that the novel interaction we detect is essential for COPII coat function." Maybe, but could this statement be a leap to far? Is it the putative interaction essential, or is the CTD itself essential for reasons that remain to be fully determined?
The CTD is separated from the C-terminus of the alpha solenoid domain by an extended domain (~350 amino acids) that is predicted to be disordered, and contains the PPP motifs and catalytic fragment that contact the inner coat. This is depicted in cartoon form in Figures 3A and 7, and discussed at length in the text. This arrangement explains why no connectivity is seen, or expected. We could highlight the C-terminus of the alpha-solenoid domain to emphasize where the disordered region should emerge from the rod, but connectivity of the disordered domain to the CTD could arise from multiple positions, including from an adjacent rod.
The reviewer’s point about the essentiality of the CTD being independent of its interaction with the Sec31 rod, is an important one. The basis for our model that the CTD enhances stability or rigidity of the coat is the yeast phenotype of Sec31-deltaCTD, which resembles that of a sec13 null. Both mutants are lethal, but rescued by deletion of emp24, which leads to more easily deformable membranes (Čopič et al. Science 2012). We agree that even if this model is true, the interaction of the CTD with Sec31 that our new structure reveals is not proven to drive rigidity or essentiality. We have tempered this hypothesis and added alternative possibilities to the discussion.
We have included the SRA1 structure in Supplementary Fig 5, as requested, and the model z-score in the Methods. The Z-score, as calculated by the proSA-web server is -6.07 (see figure below, black dot), and falls in line with experimentally determined structures including that of the template (PDB 2mgx, z-score = -5.38).
3) Are extra rods discussed in Fig. 4 are a curiosity of unclear functional significance? This reviewer is concerned that these extra rods could be an in vitro stoichiometry problem, rather than a functional property of COP-II.
This is an important point, that, as we state in the paper, cannot be answered at the moment: the resolution is too low to identify the residues involved in the interaction. Therefore we are hampered in our ability to assess the physiological importance of this interaction. We still believe the ‘extra’ rods are an important observation, as they clearly show that another mode of outer coat interaction, different from what was reported before, is possible.
The concern that interactions visualised in vitro might not be physiologically relevant is broadly applicable to structural biology approaches. However, our experimental approach uses samples that result from active membrane remodelling under near-physiological conditions, and we therefore expect these to be less prone to artefacts than most in vitro reconstitution approaches, where proteins are used at high concentrations and in high salt buffer conditions.
4) The clashsccore for the PDB is quite high--and I am dubious about the reliability of refining sidechain positions with maps at this resolution. In addition to the Ramchandran stats, I would like to see the Ramachandran plot as well as, for any residue-level claims, the density surrounding the modeled side chain (e.g. S742).
The clashscore is 13.2, which, according to molprobity, is in the 57th percentile for all structures and in the 97th for structures of similar resolutions. We would argue therefore that the clashscore is rather low. In fact, the model was refined from crystal structures previously obtained by other groups, which had worse clashscore (17), despite being at higher resolution. Our refinement has therefore improved the clashscore. During refinement we have chosen restraint levels appropriate to the resolution of our map (Afonine et al., Acta Cryst D 2018)
The Ramachandran plot is copied here and could be included in a supplemental figure if required. We make only one residue-level claim (S742), the density for which is indeed not visible at our resolution. We claim that S742 is close to the Sec23-23 interface, and do not propose any specific interactions. Nevertheless we have removed reference to S742 from the manuscript. We included this specific information because of the potential importance of this residue as a site of phosphorylation, thereby putting this interface in broader context for the general eLife reader.
Minor Comments:
1) The authors wrote "To assess the relative positioning of the two coat layers, we analysed the localisation of inner coat subunits with respect to each outer coat vertex: for each aligned vertex particle, we superimposed the positions of all inner coat particles at close range, obtaining the average distribution of neighbouring inner coat subunits. From this 'neighbour plot' we did not detect any pattern, indicating random relative positions. This is consistent with a flexible linkage between the two layers that allows adaptation of the two lattices to different curvatures (Supplementary Fig 1E)." I do not understand this claim, since the pattern both looks far from random and the interactions depend on molecular interactions that are not random. Please clarify.
We apologize for the confusion: the pattern of each of the two coats are not random. Our sentence refers to the positions of inner and outer coats relative to each other. The two lattices have different parameters and the two layers are linked by flexible linkers (the 350 amino acids referred to above). We have now clarified the sentence.
2) Related to major point #1, the author wrote "We manually picked vertices and performed carefully controlled alignments." I do now know what it means to carefully control alignments, and fear this suggests human model bias.
We used different starting references for the alignments, with the precise aim to avoid model bias. For both vesicle and cage vertex datasets, we have aligned the subtomograms against either the vertex obtained from tubules, or the vertex from previously published membrane-less cages. In all cases, we retrieved a structure that resembles the one on tubules, suggesting that the vertex arrangement we observe isn’t simply the result of reference bias. This procedure is depicted in Supplementary Fig 4 (Supplementary Fig. 3 in the original manuscript), but we have now clarified it also in the methods section.
3) Why do some experiments use EDTA? I may be confused, but I was surprised to see the budding reaction employed 1mM GMPPNP, and 2.5mM EDTA (but no Magnesium?). Also, for the budding reaction, please replace or expand upon the "the 10% GUV (v/v)" with a mass or molar lipid-to-protein ratio.
We regret the confusion. As stated in the methods, all our budding reactions are performed in the presence of EDTA and Magnesium, which is present in the buffer (at 1.2 mM). The reason is to facilitate nucleotide exchange, as reported and validated in Bacia et al., Scientific Reports 2011.
Lipids in GUV preparations are difficult to quantify. We report the stock concentrations used, but in each preparation the amount of dry lipid that forms GUVs might be different, as is the concentration of GUVs after hydration. However since we analyse reactions where COPII proteins have bound and remodelled individual GUVs, we do not believe the protein/lipid ratio influences our structures.
4) Please cite the AnchorMap procedure.
We cite the SerialEM software, and are not aware of other citations specifically for the anchor map procedure.
5) Please edit for typos (focussing, functionl, others)
Done
Reviewer #2:
The manuscript describes new cryo-EM, biochemistry, and genetic data on the structure and function of the COPII coat. Several new discoveries are reported including the discovery of an extra density near the dimerization region of Sec13/31, and "extra rods" of Sec13/31 that also bind near the dimerization region. Additionally, they showed new interactions between the Sec31 C-terminal unstructured region and Sec23 that appear to bridge multiple Sec23 molecules. Finally, they increased the resolution of the Sec23/24 region of their structure compared to their previous studies and were able to resolve a previously unresolved L-loop in Sec23 that makes contact with Sar1. Most of their structural observations were nicely backed up with biochemical and genetic experiments which give confidence in their structural observations. Overall the paper is well-written and the conclusions justified.
However, this is the third iteration of structure determination of the COPII coat on membrane with essentially the same preparation and methods. Each time, there has been an incremental increase in resolution and new discoveries, but the impact of the present study is deemed to be modest. The science is good, but it may be more appropriate for a more specialized journal. Areas of specific concern are described below.
As described above, we respectfully disagree with this interpretation of the advance made by the current work. This work improves on previous work in many aspects. The resolution of the outer coat increases from over 40A to 10-12A, allowing visualisation of features that were not previously resolved, including a novel vertex arrangement, the Sec31 CTD, and the outer coat ‘extra rods’. An improved map of the inner coat also allows us to resolve the Sec23 ‘L-loop’. We would argue that these are not just extra details, but correspond to a suite of novel interactions that expand our understanding of the complex COPII assembly network. Moreover, we include biochemical and genetic experiments that not only back up our structural observations but bring new insights into COPII function. As pointed out in response to reviewer 1, we believe our work contributes a significant conceptual advance, and have modified the manuscript to convey this more effectively.
1) The abstract is vague and should be re-written with a better description of the work.
We have modified the abstract to specifically outline what we have done and the major new discoveries of this paper.
2) Line 166 - "Surprisingly, this mutant was capable of tubulating GUVs". This experiment gets to one of the fundamental unknown questions in COPII vesiculation. It is not clear what components are driving the membrane remodeling and at what stages during vesicle formation. Isn't it possible that the tubulation activity the authors observe in vitro is not being driven at all by Sec13/31 but rather Sec23/24-Sar1? Their Sec31ΔCTD data supports this idea because it lacks a clear ordered outer coat despite making tubules. An interesting experiment would be to see if tubules form in the absence of all of Sec13/31 except the disordered domain of Sec31 that the authors suggest crosslinks adjacent Sec23/24s.
This is an astute observation, and we agree with the reviewer that the source of membrane deformation is not fully understood. We favour the model that budding is driven significantly by the Sec23-24 array. To further support this, we have performed a new experiment, where we expressed Sec31ΔN in yeast cells lacking Emp24, which have more deformable membranes and are tolerant to the otherwise lethal deletion of Sec13. While Sec31ΔN in a wild type background did not support cell viability, this was rescued in a Δemp24 yeast strain, strongly supporting the hypothesis that a major contributor to membrane remodelling is the inner coat, with the outer coat becoming necessary to overcome membrane bending resistance that ensues from the presence of cargo. We now include these results in Figure 1.
However, we must also take into account the results presented in Fig. 6, where we show that weakening the Sec23-24 interface still leads to budding, but only if Sec13-31 is fully functional, and that in this case budding leads to connected pseudo-spherical vesicles rather than tubes. When Sec13-31 assembly is also impaired, tubes appear unstructured. We believe this strongly supports our conclusions that both inner and outer coat interactions are fundamental for membrane remodelling, and it is the interplay between the two that determines membrane morphology (i.e. tubes vs. spheres).
To dissect the roles of inner and outer coats even further, we have done the experiment that the reviewer suggests: we expressed Sec31768-1114, but the protein was not well-behaved and co-purified with chaperones. We believe the disordered domain aggregates when not scaffolded by the structured elements of the rod. Nonetheless, we used this fragment in a budding reaction, and could not see any budding. We did not include this experiment as it was inconclusive: the lack of functionality of the purified Sec31 fragment could be attributed to the inability of the disordered region to bind its inner coat partner in the absence of the scaffolding Sec13-31 rod. As an alternative approach, we have used a version of Sec31 that lacks the CTD, and harbours a His tag at the N-terminus (known from previous studies to partially disrupt vertex assembly). We think this construct is more likely to be near native, since both modifications on their own lead to functional protein. We could detect no tubulation with this construct by negative stain, while both control constructs (Sec31ΔCTD and Nhis-Sec31) gave tubulation. This suggests that the cross-linking function of Sec31 is not sufficient to tubulate GUV membranes, but some degree of functional outer coat organisation (either mediated by N- or C-terminal interactions) is needed. It is also possible that the lack of outer coat organisation might lead to less efficient recruitment to the inner coat and cross-linking activity. We have added this new observation to the manuscript.
3) Line 191 - "Inspecting cryo-tomograms of these tubules revealed no lozenge pattern for the outer 192 coat" - this phrasing is vague. The reviewer thinks that what they mean is that there is a lack of order for the Sec13/31 layer. Please clarify.
The reviewer is correct, we have changed the sentence.
4) Line 198 - "unambiguously confirming this density corresponds to 199 the CTD." This only confirms that it is the CTD if that were the only change and the Sec13/31 lattice still formed. Another possibility is that it is density from other Sec13/31 that only appears when the lattice is formed such as the "extra rods". One possibility is that the density is from the extra rods. The reviewer agrees that their interpretation is indeed the most likely, but it is not unambiguous. The authors should consider cross-linking mass spectrometry.
We have removed the word ‘unambiguously’, and changed to ‘confirming that this density most likely corresponds to the CTD’. Nonetheless, we believe that our interpretation is correct: the extra rods bind to a different position, and themselves also show the CTD appendage. In this experiment, the lack of the CTD was the only biochemical change.
5) In the Sec31ΔCTD section, the authors should comment on why ΔCTD is so deleterious to oligomer organization in yeast when cages form so abundantly in preparations of human Sec13/31 ΔC (Paraan et al 2018).
We have added a comment to address this. “Interestingly, human Sec31 proteins lacking the CTD assemble in cages, indicating that either the vertex is more stable for human proteins and sufficient for assembly, or that the CTD is important in the context of membrane budding but not for cage formation in high salt conditions.”
6) The data is good for the existence of the "extra rods", but significance and importance of them is not clear. How can these extra densities be distinguished from packing artifacts due to imperfections in the helical symmetry.
Please also see our response to point 3 from reviewer 1. Regarding the specific concern that artefacts might be a consequence of imperfection in the helical symmetry, we would argue such imperfections are indeed expected in physiological conditions, and to a much higher extent. For this reason interactions seen in the context of helical imperfections are likely to be relevant. In fact, in normal GTP hydrolysis conditions, we expect long tubes would not be able to form, and the outer coat to be present on a wide range of continuously changing membrane curvatures. We think that the ability of the coat to form many interactions when the symmetry is imperfect might be exactly what confers the coat its flexibility and adaptability.
7) Figure 5 is very hard to interpret and should be redone. Panels B and C are particularly hard to interpret.
We have made a new figure where we think clarity is improved.
8) The features present in Sec23/24 structure do not reflect the reported resolution of 4.7 Å. It seems that the resolution is overestimated.
We report an average resolution of 4.6 Å. In most of our map we can clearly distinguish beta strands, follow the twist of alpha helices and see bulky side chains. These features typically become visible at 4.5-5A resolution. We agree that some areas are worse than 4.6 Å, as typically expected for such a flexible assembly, but we believe that the average resolution value reported is accurate. We obtained the same resolution estimate using different software including relion, phenix and dynamo, so that is really the best value we can provide. To further convince ourselves that we have the resolution we claim, we sampled EM maps from the EMDB with the same stated resolution (we just took the 7 most recent ones which had an associated atomic model), and visualised their features at arbitrary positions. For both beta strands and alpha helices, we do not feel our map looks any worse than the others we have examined. We include a figure here.
9) Lines 315/316 - "We have combined cryo-tomography with biochemical and genetic assays to obtain a complete picture of the assembled COPII coat at unprecedented resolution (Fig. 7)"
10) Figure 7. is a schematic model/picture the authors should reference a different figure or rephrase the sentence.
We now refer to Fig 7 in a more appropriate place.
Reviewer #3:
The manuscript by Hutchings et al. describes several previously uncharacterised molecular interactions in the coats of COP-II vesicles by using a reconstituted coats of yeast COPI-II. They have improved the resolution of the inner coat to 4.7A by tomography and subtomogram averaging, revealing detailed interactions, including those made by the so-called L-loop not observed before. Analysis of the outer layer also led to new interesting discoveries. The sec 31 CTD was assigned in the map by comparing the WT and deletion mutant STA-generated density maps. It seems to stabilise the COP-II coats and further evidence from yeast deletion mutants and microsome budding reconstitution experiments suggests that this stabilisation is required in vitro. Furthermore, COP-II rods that cover the membrane tubules in right-handed manner revealed sometimes an extra rod, which is not part of the canonical lattice, bound to them. The binding mode of these extra rods (which I refer to here a Y-shape) is different from the canonical two-fold symmetric vertex (X-shape). When the same binding mode is utilized on both sides of the extra rod (Y-Y) the rod seems to simply insert in the canonical lattice. However, when the Y-binding mode is utilized on one side of the rod and the X-binding mode on the other side, this leads to bridging different lattices together. This potentially contributes to increased flexibility in the outer coat, which maybe be required to adopt different membrane curvatures and shapes with different cargos. These observations build a picture where stabilising elements in both COP-II layers contribute to functional cargo transport. The paper makes significant novel findings that are described well. Technically the paper is excellent and the figures nicely support the text. I have only minor suggestions that I think would improve the text and figure.
We thank the reviewer for helpful suggestions which we agree improve the manuscript.
Minor Comments:
L 108: "We collected .... tomograms". While the meaning is clear to a specialist, this may sound somewhat odd to a generic reader. Perhaps you could say "We acquired cryo-EM data of COP-II induced tubules as tilt series that were subsequently used to reconstruct 3D tomograms of the tubules."
We have changed this as suggested
L 114: "we developed an unbiased, localisation-based approach". What is the part that was developed here? It seems that the inner layer particle coordinates where simply shifted to get starting points in the outer layer. Developing an approach sounds more substantial than this. Also, it's unclear what is unbiased about this approach. The whole point is that it's biased to certain regions (which is a good thing as it incorporates prior knowledge on the location of the structures).
We have modified the sentence to “To target the sparser outer coat lattice for STA, we used the refined coordinates of the inner coat to locate the outer coat tetrameric vertices”, and explain the approach in detail in the methods.
L 124: "The outer coat vertex was refined to a resolution of approximately ~12 A, revealing unprecedented detail of the molecular interactions between Sec31 molecules (Supplementary Fig 2A)". The map alone does not reveal molecular interactions; the main understanding comes from fitting of X-ray structures to the low-resolution map. Also "unprecedented detail" itself is somewhat problematic as the map of Noble et al (2013) of the Sec31 vertex is also at nominal resolution of 12 A. Furthermore, Supplementary Fig 2A does not reveal this "unprecedented detail", it shows the resolution estimation by FSC. To clarify, these points you could say: "Fitting of the Sec31 atomic model to our reconstruction vertex at 12-A resolution (Supplementary Fig 2A) revealed the molecular interactions between different copies of Sec31 in the membrane-assembled coat.
We have changed the sentence as suggested.
L 150: Can the authors exclude the possibility that the difference is due to differences in data processing? E.g. how the maps amplitudes have been adjusted?
Yes, we can exclude this scenario by measuring distances between vertices in the right and left handed direction. These measurements are only compatible with our vertex arrangement, and cannot be explained by the big deviation from 4-fold symmetry seen in the membrane-less cage vertices.
L 172: "that wrap tubules either in a left- or right-handed manner". Don't they do always both on each tubule? Now this sentence could be interpreted to mean that some tubules have a left-handed coat and some a right-handed coat.
We have changed this sentence to clarify. “Outer coat vertices are connected by Sec13-31 rods that wrap tubules both in a left- and right-handed manner.”
L276: "The difference map" hasn't been introduced earlier but is referred to here as if it has been.
We now introduce the difference map.
L299: Can "Secondary structure predictions" denote a protein region "highly prone to protein binding"?
Yes, this is done through DISOPRED3, a feature include in the PSIPRED server we used for our predictions. The reference is: Jones D.T., Cozzetto D. DISOPRED3: precise disordered region predictions with annotated protein-binding activity Bioinformatics. 2015; 31:857–863. We have now added this reference to the manuscript.
L316: It's true that the detail in the map of the inner coat is unprecedented and the model presented in Figure 7 is partially based on that. But here "unprecedented resolution" sounds strange as this sentence refers to a schematic model and not a map.
We have changed this by moving the reference to Fig 7 to a more appropriate place
L325: "have 'compacted' during evolution" -> remove. It's enough to say it's more compact in humans and less compact in yeast as there could have been different adaptations in different organisms at this interface.
We have changed as requested. See also our response to reviewer 1, point 1.
L327: What's exactly meant by "sequence diversity or variability at this density".
We have now clarified: “Since multiple charge clusters in yeast Sec31 may contribute to this interaction interface (Stancheva et al., 2020), the low resolution could be explained by the fact that the density is an average of different sequences.”
L606-607: The description of this custom data processing approach is difficult to follow. Why is in-plane flip needed and how is it used here?
Initially particles are picked ignoring tube directionality (as this cannot be assessed easily from the tomograms due to the pseudo-twofold symmetry of the Sec23/24/Sar1 trimer). So the in plane rotation of inner coat subunit could be near 0 or 180°. For each tube, both angles are sampled (in-plane flip). Most tubes result in the majority of particles being assigned one of the two orientations (which is then assumed as the tube directionality). Particles that do not conform are removed, and rare tubes where directionality cannot be determined are also removed. We have re-written the description to clarify these points: “Initial alignments were conducted on a tube-by-tube basis using the Dynamo in-plane flip setting to search in-plane rotation angles 180° apart. This allowed to assign directionality to each tube, and particles that were not conforming to it were discarded by using the Dynamo dtgrep_direction command in custom MATLAB scripts”
L627: "Z" here refers to the coordinate system of aligned particles not that of the original tomogram. Perhaps just say "shifted 8 pixels further away from the membrane".
Changed as requested.
L642-643: How can the "left-handed" and "right-handed" rods be separated here? These terms refer to the long-range organisation of the rods in the lattice it's not clear how they were separated in the early alignments.
They are separated by picking only one subset using the dynamo sub-boxing feature. This extracts boxes from the tomogram which are in set positions and orientation relative to the average of previously aligned subtomograms. From the average vertex structure, we sub-box rods at 4 different positions that correspond to the centre of the rods, and the 2-fold symmetric pairs are combined into the same dataset. We have clarified this in the text: “The refined positions of vertices were used to extract two distinct datasets of left and right-handed rods respectively using the dynamo sub-boxing feature.”
Figure 2B. It's difficult to see the difference between dark and light pink colours.
We have changed colours to enhance the difference.
Figure 3C. These panels report the relative frequency of neighbouring vertices at each position; "intensity" does not seem to be the right measure for this. You could say that the colour bar indicates the "relative frequency of neighbouring vertices at each position" and add detail how the values were scaled between 0 and 1. The same applies to SFigure 1E.
Changed as requested.
Figure 4. The COP-II rods themselves are relatively straight, and they are not left-handed or right-handed. Here, more accurate would be "architecture of COPII rods organised in a left-handed manner". (In the text the authors may of course define and then use this shorter expression if they so wish.) Panel 4B top panel could have the title "left-handed" and the lower panel should have the title "right-handed" (for consistency and clarity).
We have now defined left- and right-handed rods in the text, and have changed the figure and panel titles as requested.
We thank the reviewers for their comments, which will improve the quality of our manuscript.
Our study describes a novel approach to the identification of GTPase binding-partners. We recapitulated and augmented previous protein-protein interaction data for RAB18 and presented data validating some of our findings. In aggregate, our dataset suggested that RAB18 modulates the establishment of membrane contact sites and the transfer of lipid between closely apposed membranes.
In the original version of our manuscript, we stated that we were exploring the possibility that RAB18 contributes to cholesterol biosynthesis by mobilizing substrates or products of the Δ8-Δ7 sterol isomerase emopamil binding protein (EBP). While our manuscript was under review, we profiled sterols in wild-type and RAB18-null cells and assayed cholesterol biosynthesis in a panel of cell lines (Figure 1).
Our new data show that an EBP-product, lathosterol, accumulates in RAB18-null cells (p<0.01). Levels of a downstream cholesterol intermediate, desmosterol, are reduced in these cells (p<0.01) consistent with impaired delivery of substrates to post-EBP biosynthetic enzymes (Figure 1A). Further, our preliminary data suggests that cholesterol biosynthesis is substantially reduced when RAB18 is absent or dysregulated (4 technical replicates, one independent experiment) (Figure 1B).
Because of the clinical overlap between Micro syndrome and cholesterol biosynthesis disorders including Smith-Lemli-Opitz syndrome (SLOS; MIM 270400) and lathosterolosis (MIM 607330), our new findings suggest that impaired cholesterol biosynthesis may partly underlie Warburg Micro syndrome pathology. Therapeutic strategies have been developed for the treatment of SLOS and lathosterolosis, and so confirmation of our findings may spur development of similar strategies for Micro syndrome.
Our new findings provide further functional validation of our methodology and support our interpretation of our protein interaction data.
Reply to point 1)
Tetracycline-induced expression of wild-type and mutant BirA*-RAB18 fusion proteins in the stable HEK293 cell lines was quantified by densitometry (Figure 2).

For the HEK293 BioID experiments, tetracycline dosage was adjusted to ensure comparable expression levels. We will include these data in supplemental material in an updated version of our manuscript.
The localization of wild-type and mutant forms of RAB18 in HEK293 cells is somewhat different consistent with previous reports (Ozeki et al. 2005)(Figure 3).
We feel that this may reflect the differential localization of ‘active’ and ‘inactive’ RAB18, with wild-type RAB18 corresponding to a mixture of the two. We will include these data in supplemental material in an updated version of our manuscript.
We acknowledge that the differential localization of wild-type and mutant BirA*-RAB18 might influence the compliment of proteins labeled by these constructs. Nevertheless, we feel that the RAB18(S22N):RAB18(WT) ratios are useful since they distinguish a number of previously-identified RAB18-interactors (manuscript, Figure 1B).
Reply to point 2)
For the HEK293 dataset, spectral counts are provided and for the HeLa dataset LFQ intensities were provided in the manuscript (manuscript, Tables S1 and S2 respectively). However, we did not find that these were useful classifiers for ranking functional interactions when used in isolation.
The extent of labelling produced in a BioID experiment is not wholly determined by the kinetics of protein-protein associations. It is also influenced by, for example, protein abundance, the number and location of exposed surface lysine residues, and protein stability over the timcourse of labelling. We feel that RAB18(S22N):RAB18(WT) and GEF-null:wild-type ratios were helpful in controlling for these factors. Further, that our comparative approach was effective in highlighting known RAB18-interactors and in identifying novel ones.
We acknowledge that our approach may omit some bona fide functional RAB18-interactions, but would argue that our aims were to augment existing functional RAB18-interaction data and avoid false-positives, rather than to emphasise completeness.
Reply to point 3)
We will include representative fluorescence images for the SEC22A, NBAS and ZW10 knockdown experiments in an updated version of our manuscript.
Unfortunately, a suitable antibody for determining knockdown efficiency of SEC22A at the protein level is not commercially available. We will determine SEC22A knockdown efficiency at the mRNA level using qPCR.
Reply to point 4)
Expression levels of wild-type and mutant RAB18 in the stable CHO cell lines generated for this study were determined by Western blotting and found to be comparable (Figure 4).
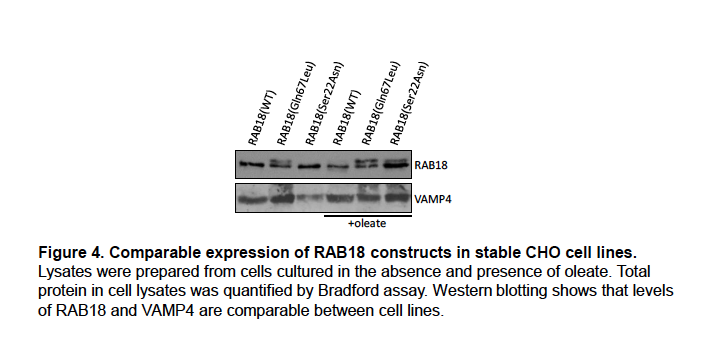
We will include these data in supplemental material in an updated version of our manuscript.
The levels of [14C]-CE were higher in RAB18(Gln67Leu) cells than in the other cell lines following loading with [14C]-oleate for 24 hours. We will amend the text to make this explicit. Our interpretation of the data is that ‘active’ RAB18 facilitates the mobilization of cholesterol. When cells are loaded with oleate, this promotes generation and storage of CE. Conversely, when cells are treated with HDL, it promotes more rapid efflux.
Our new data implicating RAB18 in the mobilization of lathosterol supports our interpretation of our loading and efflux experiments. In the light of our new data showing that de novo cholesterol biosynthesis is impaired when RAB18 is absent or dysregulated, it will be interesting to determine whether cholesterol synthesis is increased in the RAB18(Gln67Leu) cells.
Reply to point 1)
We anticipate that the approach of comparative proximity biotinylation in GEF-null and wild-type cell lines will be broadly useful in small GTPase research.
While RAB18 has previously been implicated in regulating membrane contacts, the identification of SEC22A as a RAB18-interactor adds to the previous model for their assembly.
While ORP2 and INPP5B have previously been shown to mediate cholesterol mobilization, the novel finding that they both interact with RAB18 adds to this work. We argue that RAB18-ORP2-INPP5B functions in an analogous manner to ARF1-OSBP-SAC1 in mediating sterol exchange. The broad Rab-binding specificity of multiple OSBP-homologs, and that of multiple phosphoinositide phosphatase enzymes, suggests that this may be a common conserved relationship.
Our new data indicating that RAB18 coordinates generation of sterol intermediates by EBP and their delivery to post-EBP biosynthetic enzymes reveals a new role for Rab proteins in lipid biogenesis. Most importantly, our new findings that RAB18 deficiency is associated with impaired cholesterol biogenesis suggest that Warburg Micro syndrome is a cholesterol biogenesis disorder. Further, that it may be amenable to therapeutic intervention.
Reply to point 2)
Recognising that the effect of RAB18 on cholesterol esterification and efflux could arise from various causes, we previously carried out Western blotting of the CHO cell lines for ABCA1 to determine whether this protein was involved (Figure 5).
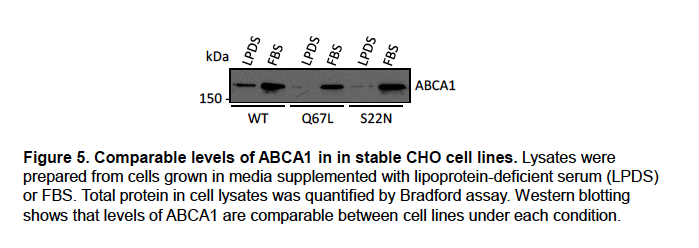
Similar levels of ABCA1 expression in these lines suggests it is not. We will include these data in supplemental material in an updated version of our manuscript.
We feel that our new data implicating RAB18 in lathosterol mobilization provides important insight into its role in cholesterol biogenesis. Further, it supports our previous suggestion that RAB18 mediates cholesterol mobilization.
Reply to point 3)
We agree that the established roles for ORP2, TMEM24/C2CD2L and PIP2 at the plasma membrane make this an extremely interesting area for future research; it is one we are actively investigating. However, we respectfully feel that to comprehensively explore the subcellular locations of RAB18-mediated sterol/PIP2 exchange requires another study and is beyond the scope of the present report.
Reply to point 1)
The RAB18-SPG20 interaction has already been validated with a co-immunoprecipitation experiment (Gillingham et al. 2014). We will update the text of our manuscript to make this more explicit, but do not feel it is necessary to recapitulate this work.
We argue in the manuscript that RAB18 may coordinate the assembly of a non-canonical SNARE complex incorporating SEC22A, STX18, BNIP1 and USE1. However, this role may be mediated through prior interaction with the NBAS-RINT1-ZW10 (NRZ) tethering complex and the SM-protein SCFD2 rather than through a direct interaction. We therefore feel that a RAB18-SEC22A interaction may be difficult to validate by conventional means.
The reciprocal experiments with BioID2(Gly40S)-SEC22A did provide tentative confirmation of the interaction together with evidence that a subset of SEC22A-interactions are attenuated when RAB18 is absent or dysregulated. In the light of our new findings reinforcing a role for RAB18 in sterol mobilization at membrane contact sites, it is interesting that one of these is DHRS7, an enzyme with steroids among its putative substrates.
Reply to point 2)
We previously analysed the localization of the BirA*-RAB18 fusion protein in HeLa cells (Figure 6).
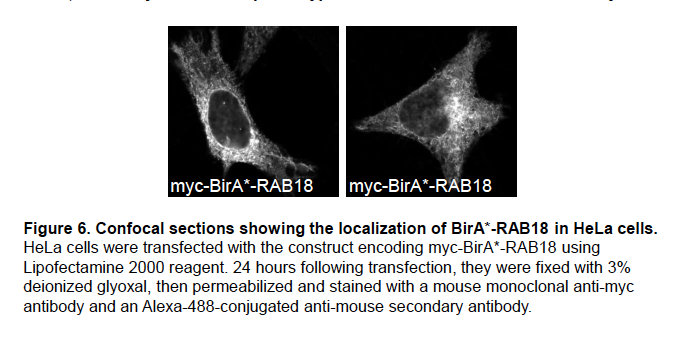
It shows a reticular staining pattern consistent with the reported localization of RAB18 to the ER (Gerondopoulos et al. 2014; Ozeki et al. 2005). We will include these data in supplemental material in an updated version of our manuscript.
Heterologous expression of the BirA*-RAB18 fusion protein in HeLa cells identified the interactions between RAB18 and EBP, ORP2 and INPP5B, for which we now have supportive functional evidence. Since the evidence for impaired lathosterol mobilization and cholesterol biosynthesis was derived from experiments on null-cells, in which endogenous protein expression is absent, we feel that rescue experiments are not necessary in the present study. However, such experiments could be highly useful in future studies.
Reply to point 3)
Our screening approach did use both a RAB3GAP-null:wild-type comparison (manuscript, Figure 2, Table S2) and also a RAB18(S22N):RAB18(WT) comparison (manuscript, Figure 1, Table S1). Differences should be expected between these datasets, since they used different cell lines and slightly different methodologies. Nevertheless, proteins identified in both datasets included the known RAB18 effectors NBAS, RINT1, ZW10 and SCFD2, and the novel potential effectors CAMSAP1 and FAM134B.
There is prior evidence for 12 of the 25 RAB3GAP-dependent RAB18 interactions we identified (manuscript, Figure 2D). Among the 6 lipid modifying/mobilizing proteins found exclusively in our HeLa dataset, we previously presented direct evidence for the interaction of RAB18 with TMCO4. We now also have strong functional evidence for its interaction with EBP, ORP2 and INPP5B.
Reply to point 4)
It has been reported that knockdown of SEC22B does not affect the size distribution of lipid droplets (Xu et al. 2018) Figure 8H). Nevertheless, we will carry out qPCR experiments to determine whether the SEC22A siRNAs used in our study affect SEC22B expression. We have found that exogenous expression of SEC22A can cause cellular toxicity. Rescue experiments would therefore be difficult to perform.
Reply to point 5)
The background fluorescence measured in SPG20-null cells and presented in Figure 4B in the manuscript does not imply that the SPG20 antibody shows significant cross-reactivity. Rather, it reflects the fact that fluorescence intensity is recorded by our Operetta microscope in arbitrary units.
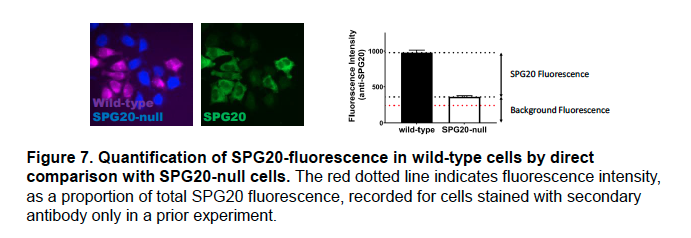
Above (Figure 7), is a version of the panel in which fluorescence from staining cells with only the secondary antibody is included (recorded in a previous experiment and expressed as a proportion of total SPG20 fluorescence in this experiment).
We have found that comparative fluorescence microscopy is more sensitive than immunoblotting. The SPG20 antibody we used to stain the HeLa cells has previously been used in quantitative fluorescence microscopy (Nicholson et al. 2015).
Furthermore, we showed corresponding, significantly reduced, expression of SPG20 in RAB18- and TBC1D20-null RPE1 cells, using quantitative proteomics (manuscript, Table S3).
We acknowledge that quantification of SPG20 transcript levels would clarify the level at which it is downregulated and will carry out qPCR experiments accordingly.
Reply to point 6)
We interpret both the enhanced CE-synthesis following oleate-loading and the rapid efflux upon incubation with HDL (manuscript, Figure 7A) as resulting from increased cholesterol mobilization. Our new data implicating RAB18 in the mobilization of lathosterol support this interpretation.
In the [3H]-cholesterol efflux assay (manuscript, Figure 7B) total [3H]-cholesterol loading at t=0 was 156392±8271 for RAB18(WT) cells, 168425±9103 for RAB18(Gln67Leu) cells and 148867±7609 (cpm determined through scintillation counting). Normalizing to total cellular radioactivity assured that differences in loading between replicates did not skew the results.
The candidate effector likely to directly mediate cholesterol mobilization is ORP2. It has been shown that ORP2 overexpression drives cholesterol to the plasma membrane (Wang et al. 2019). Further, there is evidence for reduced plasma membrane cholesterol in ORP2-null cells (Wang et al. 2019).
We previously carried out Western blotting of the CHO cell lines for ABCA1 to determine whether this protein was involved in altered efflux (Figure 5, above). Similar levels of ABCA1 expression in these lines suggests it is not. We will include these data in supplemental material in an updated version of our manuscript.
References
Gerondopoulos, A., R. N. Bastos, S. Yoshimura, R. Anderson, S. Carpanini, I. Aligianis, M. T. Handley, and F. A. Barr. 2014. 'Rab18 and a Rab18 GEF complex are required for normal ER structure', J Cell Biol, 205: 707-20.
Gillingham, A. K., R. Sinka, I. L. Torres, K. S. Lilley, and S. Munro. 2014. 'Toward a comprehensive map of the effectors of rab GTPases', Dev Cell, 31: 358-73.
Nicholson, J. M., J. C. Macedo, A. J. Mattingly, D. Wangsa, J. Camps, V. Lima, A. M. Gomes, S. Doria, T. Ried, E. Logarinho, and D. Cimini. 2015. 'Chromosome mis-segregation and cytokinesis failure in trisomic human cells', eLife, 4.
Ozeki, S., J. Cheng, K. Tauchi-Sato, N. Hatano, H. Taniguchi, and T. Fujimoto. 2005. 'Rab18 localizes to lipid droplets and induces their close apposition to the endoplasmic reticulum-derived membrane', J Cell Sci, 118: 2601-11.
Wang, H., Q. Ma, Y. Qi, J. Dong, X. Du, J. Rae, J. Wang, W. F. Wu, A. J. Brown, R. G. Parton, J. W. Wu, and H. Yang. 2019. 'ORP2 Delivers Cholesterol to the Plasma Membrane in Exchange for Phosphatidylinositol 4, 5-Bisphosphate (PI(4,5)P2)', Mol Cell, 73: 458-73 e7.
Xu, D., Y. Li, L. Wu, Y. Li, D. Zhao, J. Yu, T. Huang, C. Ferguson, R. G. Parton, H. Yang, and P. Li. 2018. 'Rab18 promotes lipid droplet (LD) growth by tethering the ER to LDs through SNARE and NRZ interactions', J Cell Biol, 217: 975-95.
This paper addresses the very interesting topic of genome evolution in asexual animals. While the topic and questions are of interest, and I applaud the general goal of a large-scale comparative approach to the questions, there are limitations in the data analyzed. Most importantly, as the authors raise numerous times in the paper, questions about genome evolution following transitions to asexuality inherently require lineage-specific controls, i.e. paired sexual species to compare with the asexual lineages. Yet such data are currently lacking for most of the taxa examined, leaving a major gap in the ability to draw important conclusions here. I also do not think the main positive results, such as the role of hybridization and ploidy on the retention and amount of heterozygosity, are novel or surprising.
We agree with the reviewer that having the sexual outgroups would improve the interpretations; this is one of the points we make in our manuscript. Importantly however, all previous genome studies of asexual species focus on individual asexual lineages, generally without sexual species for comparison. Yet reported genome features have been interpreted as consequences of asexuality (e.g., Flot et al. 2013). By analysing and comparing these genomes, we can show that these features are in fact lineage-specific rather than general consequences of asexuality. Unexpectedly, we find that asexuals that are not of hybrid origin are largely homozygous, independently of the cellular mechanism underlying asexuality. This contrasts with the general view that cellular mechanisms such as central fusion (which facilitates heterozygosity retention between generation) promotes the evolutionary success of asexual lineages relative to mechanisms such as gamete duplication (which generate complete homozygosity) by delaying the expression of the recessive load. We also do not observe the expected relationship between cellular mechanism of asexuality and heterozygosity retention in species of hybrid origin. Thus we respectfully disagree that our results are not surprising. Reviewer #2 found our results “interesting” and a “potentially important contribution”, and reviewer #3 wrote that we “call into question the generality of the theoretical expectations, and suggest that the genomic impacts of asexuality may be more complicated than previously thought”.
We also make it very clear that some of the patterns we uncover (e.g. low TE loads in asexual species) cannot be clearly evaluated with asexuals alone. Our study emphasizes the importance of the fact that asexuality is a lineage-level trait and that comparative analyses using asexuals requires lineage-level replication in addition to comparisons to sexual species.
References
Flot, Jean-François, et al. "Genomic evidence for ameiotic evolution in the bdelloid rotifer Adineta vaga." Nature 500.7463 (2013): 453-457.
[...] Major Issues and Questions:
1) The authors choose to refer to asexuality when describing thelytokous parthenogenesis. Asexuality is a very general term that can be confusing: fission, vegetative reproduction could also be considered asexuality. I suggest using parthenogenesis throughout the manuscript for the different animal clades studied here. Moreover, in thelytokous parthenogenesis meiosis can still occur to form the gametes, it is therefore not correct to write that "gamete production via meiosis... no longer take place" (lines 57-58). Fertilization by sperm indeed does not seem to take place (except during hybridogenesis, a special form of parthenogenesis).
We will clarify more explicitly what asexuality refers to in our manuscript. Notably our study does not include species that produce gametes which are fertilized (which is the case under hybridogenesis, which sensu stricto is not a form of parthenogenesis). Even though many forms of parthenogenesis do indeed involve meiosis (something we explain in much detail in box 2), there is no production of gametes.
2) The cellular mechanisms of asexuality in many asexual lineages are known through only a few, old cytological studies and could be inaccurate or incomplete (for example Triantaphyllou paper of 1981 of Meloidogyne nematodes or Hsu, 1956 for bdelloid rotifers). The authors should therefore mention in the introduction the lack of detailed and accurate cellular and genetic studies to describe the mode of reproduction because it may change the final conclusion.
For example, for bdelloid rotifers the literature is scarce. However the authors refer in Supp Table 1 to two articles that did not contain any cytological data on oogenesis in bdelloid rotifers to indicate that A. vaga and A. ricciae use apomixis as reproductive mode. Welch and Meselson studied the karyotypes of bdelloid rotifers, including A. vaga, and did not conclude anything about absence or presence of chromosome homology and therefore nothing can be said about their reproduction mode. In the article of Welch and Meselson the nuclear DNA content of bdelloid species is measured but without any link with the reproduction mode. The only paper referring to apomixis in bdelloids is from Hsu (1956) but it is old and new cytological data with modern technology should be obtained.
We will correct the rotifer citations and thank the reviewer for picking up the error. We agree that there are uncertainties in some cytological studies, but the same is true for genomic studies (which is why we base our analyses as much as possible on raw reads rather than assemblies because the latter may be incorrect). We in fact excluded cytological studies where the findings could not be corroborated. For example, we discarded the evidence for meiosis and diploidy by Handoo at al. 2004 for its incompatibility with genomic data because this study does not provide any verifiable evidence (there are no data or images, only descriptions of observations). We provide all the references in the supplementary material concerning the cytological evidence used.
3) In the section on Heterozygosity, the authors compute heterozygosity from kmer spectra analysis from reads to "avoid biases from variable genome assembly qualities" (page 16). But such kmer analysis can be biased by the quality and coverage of sequencing reads. While such analyses are a legitimate tool for heterozygosity measurements, this argument (the bias of genome quality) is not convincing and the authors should describe the potential limits of using kmer spectra analyses.
We excluded all the samples with unsuitable quality of data (e.g. one tardigrade species with excessive contamination or the water flea samples for insufficient coverage), and T. Rhyker Ranallo Benavidez, the author of the method we used, collaborated with us on the heterozygosity analyzes. However, we will clarify the limitations of the method for species with extremely low or high heterozygosity (see also comment 5 of this reviewer).
4) The authors state that heterozygosity levels “should decay over time for most forms of meiotic asexuality". This is incorrect, as this is not expected with "central fusion" or with "central fusion automixis equivalent" where there is no cytokinesis at meiosis I.
Our statement is correct. Note that we say “most” and not “all” because certain forms of endoduplication in F1 hybrids result in the maintenance of heterozygosity. Central fusion is expected to fully retain heterozygosity only if recombination is completely suppressed (see for example Suomalainen et al. 1987 or Engelstädter 2017).
5) I do not fully agree with the authors’ statement that: "In spite of the prediction that the cellular mechanism of asexuality should affect heterozygosity, it appears to have no detectable effect on heterozygosity levels once we control for the effect of hybrid origins (Figure 2)." (page 17)
The scaling on Figure 2 is emphasizing high values, while low values are not clearly separated. By zooming in on the smaller heterozygosity % values we may observe a bigger difference between the "asexuality mechanisms". I do not see how asexuality mechanism was controlled for, and if you look closely at intra group heterozygosity, variability is sometimes high.
It is expected that hybrid origin leads to higher heterozygosity levels but saying that asexuality mechanism is not important is surprising: on Figure 2 the orange (central fusion) is always higher than yellow (gamete duplication).
As we explain in detail in the text, the three comparatively high heterozygosity values under spontaneous origins of asexuality (“orange” points in the bottom left corner of the figure) are found in an only 40-year old clone of the Cape bee. Among species of hybrid origin, we see no correlation between asexuality mechanism and heterozygosity. These observations suggest that the asexuality mechanism may have an impact on genome-wide heterozygosity in recent incipient asexual lineages, but not in established asexual lineages.
Also, the variability found within rotifers could be an argument against a strong importance of asexuality origin on heterozygosity levels: the four bdelloid species likely share the same origin but their allelic heterozygosity levels appears to range from almost 0 to almost 6% (Fig 2 and 3, however the heterozygosity data on Rotaria should be confirmed, see below).
We prefer not using the data from rotifers for making such arguments, given the large uncertainty with respect to genome features in this group (including the possibility of octoploidy in some species which we describe in the supplemental information). One could even argue that the highly variable genome structure among rotifer species could indicate repeated transitions to asexuality and/or different hybridization events, but the available genome data would make all these arguments highly speculative.
The authors’ main idea (i.e. asexuality origin is key) seems mostly true when using homoeolog heterozygosity and/or composite heterozygosity which is not what most readers will usually think as "heterozygosity". This should be made clear by the authors mostly because this kind of heterozygosity does not necessarily undergo the same mechanism as the one described in Box 2 for allelic heterozygosity. If homoeolog heterozygosity is sometimes not distinguishable from allelic heterozygosity, then it would be nice to have another box showing the mechanisms and evolution pattern for such cases (like a true tetraploid, in which all copies exist).
The heterozygosity between homoeologs is always high in this study while it appears low between alleles, but since the heterozygosity between homeologs can only be measured when there is a hybrid origin, the only heterozygosity that can be compared between ALL the asexual groups is the one between alleles.
By definition, homoeologs have diverged between species, while alleles have diverged within species. So indeed divergence between homoeologs will generally exceed divergence between alleles. We will consider adding expected patterns in perfect tetraploid species for Box 2.
Both in the results and the conclusion the authors should not over interpret the results on heterozygosity. The variation in allelic heterozygosity could be small (although not in all asexuals studied) also due to the age of the asexual lineages. This is not mentioned here in the result/discussion section..
We explain in section Overview of species and genomes studied that age effects are important but that we do not consider them quantitatively because age estimates are not available for the majority of asexual species in our paper.
6) Regarding the section on Heterozygosity structure in polyploids
There is inconsistency in many of the numbers. For example, A. vaga heterozygosity is estimated at 1.42% in Figure 1, but then appears to show up around 2% in Figure 2, and then becomes 2.4% on page 20. It is unclear is this is an error or the result of different methods.
It is also unclear how homologs were distinguished from homeologs. How are 21 bp k-mers considered homologous? In the method section. the authors describe extracting unique k-mer pairs differing by one SNP, so does this mean that no more than one SNP was allowed to define heterozygous homologous regions? Does this mean that homologues (and certainly homoeologs) differing by more than 5% would not be retrieved by this method. If so, then It is not surprising that for A.vaga is classified as a diploid.
Figure 1 a presents the values reported in the original genome studies, not our results. This is explained in the corresponding figure legend. Hence, 1.42 is the value reported by Flot at al. 2013. 2.4 is the value we measure and it is consistent in Figures 2 and 3.
We used k-mer pairs differing by one SNP to estimate ploidy (smudgeplot). The heterozygosity estimates were estimated from kmer spectra (GenomeScope 2.0). The kmers that are found in 1n must be heterozygous between homologs, as the homoeolog heterozygosity would produce 2n kmers, We used the kmer approach to estimate heterozygosity in all other cases than homoeologs of rotifers, which were directly derived from the assemblies. We explain this in the legend to Figure 3, but we will add the information also to the Methods section for clarification.
The result for A. ricciae is surprising and I am still not convinced by the octoploid hypothesis. In Fig S2. there is a first peak at 71x coverage that still could be mostly contaminants. It would be helpful to check the GC distribution of k-mers in the first haploid peak of A. ricciae to check whether there are contaminants. The karyotypes of 12 chromosomes indeed do not fit the octoploid hypothesis. I am also surprised by the 5.5% divergence calculated for A. ricciae, this value should be checked when eliminating potential contaminants (if any). In general, these kind of ambiguities will not be resolved without long-read sequencing technology to improve the genome assemblies of asexual lineages.
We understand the scepticism of the reviewer regarding the octoploidy hypothesis, but it is important to note that we clearly present it as a possible explanation for the data that needs to be corroborated, i.e., we state that the data are better consistent with octo- than tetraploidy. Contamination seems quite unlikely, as the 71.1x peak represents nearly exactly half the coverage of the otherwise haploid peak (142x). Furthermore, the Smudgeplot analysis shows that some of the kmers from the 71x peak pair with genomic kmers of the main peaks. We also performed KAT analysis (not presented in the manuscript) showing that these kmers are also represented in the decontaminated assembly. We will add this clarification regarding possible contamination to the supplementary materials.
7) Regarding the section on palindromes and gene conversion
The authors screened all the published genomes for palindromes, including small blocks, to provide a more robust unbiased view. However, the result will be unbiased and robust if all the genomes compared were assembled using the same sequencing data (quality, coverage) and assembly program. While palindromes appear not to play a major role in the genome evolution of parthenogenetic animals since only few palindromes were detected among all lineages, mitotic (and meiotic) gene conversion is likely to take place in parthenogens and should indeed be studied among all the clades.
We agree with the reviewer that gene conversion might be one of the key aspects of asexual genome evolution. Our study merely pointed out that genomes of asexual animals do not show organisation in palindromes, indicating that palindromes might not be of general importance in asexual genome evolution. Note also that we clearly point out that these analyses are biased by the quality of the available genome assemblies.
8) Regarding the section on transposable elements
The authors are aware that the approach used may underestimate the TEs present in low copy numbers, therefore the comparison might underestimate the TE numbers in certain asexual groups.
Yes. We clearly explain this limitation in the manuscript. The currently available alternatives are based on assembled genomes, so the results are biased by the quality of the assemblies (and similarities to TEs in public databases) and our aim was to broadly compare genomes in the absence of assembly-generated biases.
9) Regarding the section on horizontal gene transfer. For the HGTc analysis, annotated genes were compared to the UniRef90 database to identify non-metazoan genes and HGT candidates were confirmed if they were on a scaffold containing at least one gene of metazoan origin. While this method is indeed interesting, it is also biased by the annotation quality and the length of the scaffolds which vary strongly between studies.
Yes, this is true and we explain many limitations in the supplemental information, but re-assembling and re-annotating all these genomes would be beyond reasonable computational possibilities.
10) Regarding the use of GenomeScope2.0
When homologues are very divergent (as observed in bdelloid rotifers) GenomeScope probably considers these distinct haplotypes as errors, making it difficult to model the haploid genome size and giving a high peak of errors in the GenomeScope profile. Moreover, due to the very divergent copies in A. vaga, GenomeScope indeed provides a diploid genome (instead of tetraploid).
For A. vaga, the heterozygosity estimated par GenomeScope2.0. on our new sequencing dataset is 2% (as shown in this paper). This % corresponds to the heterozygosity between k-mers but does not provide any information on the heterogeneity in heterozygosity measurements along the genome. A limitation of GenomeScope2.0. (which the authors should mention here) is that it is assuming that the entire genome is following the same theoretical k-mer distribution.
The model of estimating genome wide heterozygosity indeed assumes a random distribution of heterozygous loci and indeed is unable to estimate divergence over a certain threshold, which is the reason why we used genome assemblies for the estimation of divergence of homoeologs. Regarding estimates in all other genomes, the assumptions are unlikely to fundamentally change the output of the analysis. GenomeScope2 is described in detail in a recent paper (Ranallo-Benavidez et al. 2019), where the assumption that heterozygosity rates are constant across the genome is explicitly mentioned.
References
Engelstädter, Jan. "Asexual but not clonal: evolutionary processes in automictic populations." Genetics 206.2 (2017): 993-1009.
Flot, Jean-François, et al. "Genomic evidence for ameiotic evolution in the bdelloid rotifer Adineta vaga." Nature 500.7463 (2013): 453-457.
Handoo, Z. A., et al. "Morphological, molecular, and differential-host characterization of Meloidogyne floridensis n. sp.(Nematoda: Meloidogynidae), a root-knot nematode parasitizing peach in Florida." Journal of nematology 36.1 (2004): 20.
Suomalainen, Esko, Anssi Saura, and Juhani Lokki. Cytology and evolution in parthenogenesis. CRC Press, 1987.
Ranallo-Benavidez, Timothy Rhyker, Kamil S. Jaron, and Michael C. Schatz. "GenomeScope 2.0 and Smudgeplots: Reference-free profiling of polyploid genomes." BioRxiv (2019): 747568.
Jaron and collaborators provide a large-scale comparative work on the genomic impact of asexuality in animals. By analysing 26 published genomes with a unique bioinformatic pipeline, they conclude that none of the expected features due to the transition to asexuality is replicated across a majority of the species. Their findings call into question the generality of the theoretical expectations, and suggest that the genomic impacts of asexuality may be more complicated than previously thought.
The major strengths of this work is (i) the comparison among various modes and origins of asexuality across 18 independent transitions; and (ii) the development of a bioinformatic pipeline directly based on raw reads, which limits the biases associated with genome assembly. Moreover, I would like to acknowledge the effort made by the authors to provide on public servers detailed methods which allow the analyses to be reproduced. That being said, I also have a series of concerns, listed below:
We thank this reviewer for the relevant comments and for providing many constructive suggestions in the points below. We will take them into account for our final version of the manuscript.
1) Theoretical expectations
As far as I understand, the aim of this work is to test whether 4 classical predictions associated with the transition to asexuality and 5 additional features observed in individual asexual lineages hold at a large phylogenetic scale. However, I think that these predictions are poorly presented, and so they may be hardly understood by non-expert readers. Some of them are briefly mentioned in a descriptive way in the Introduction (L56 - 61), and with a little more details in the Boxes 1 and 2. However, the evolutive reasons why one should expect these features to occur (and under which assumptions) is not clearly stated anywhere in the Introduction (but only briefly in the Results & Discussion). I think it is important that the authors provide clear-cut quantitative expectations for each genomic feature analysed and under each asexuality origin and mode (Box 1 and 2). Also highlighting the assumptions behind these expectations will help for a better interpretation of the observed patterns.
We will clarify the expectations for non expert readers.
2) Mutation accumulation & positive selection
A subtlety which is not sufficiently emphasized to my mind is that the different modes of asexuality encompass reproduction with or without recombination (Box 2), which can lead to very different genetic outcomes. For example, it has been shown that the Muller's ratchet (the accumulation of deleterious mutations in asexual populations) can be stopped by small amounts of recombination in large-sized populations (Charlesworth et al. 1993; 10.1017/S0016672300031086). Similarly a new recessive beneficial mutation can only segregate at a heterozygous state in a clonal lineage (unless a second mutation hits the same locus); whereas in the presence of recombination, these mutations will rapidly fix in the population by the formation of homozygous mutants (Haldane's Sieve, Haldane 1927; 10.1017/S0305004100015644). Therefore, depending on whether recombination occurs or not during asexual reproduction, the expectations may be quite different; and so they could deviate from the "classical predictions". In this regard, I would like to see the authors adjust their conclusions. Moreover, it is also not very clear whether the species analysed here are 100% asexuals or if they sometimes go through transitory sexual phases, which could reset some of the genomic effects of asexuality.
Yes, the predictions regarding the efficiency of selection are indeed influenced by cellular modes of asexuality. Adding some details or at least a good reference would certainly increase the readability of the section. We thank the reviewer for this suggestion.
3) Transposable elements
I found the predictions regarding the amount of TEs expected under asexuality quite ambiguous. From one side, TEs are expected not to spread because they cannot colonize new genomes (Hickey 1982); but on the other side TEs can be viewed as any deleterious mutation that will accumulate in asexual genome due to the Muller's ratchet. The argument provided by the authors to justify the expectation of low TE load in asexual lineages is that "Only asexual lineages without active TEs, or with efficient TE suppression mechanisms, would be able to persist over evolutionary timescales". But this argument should then equally be applied to any other type of deleterious mutations, and so we won't be able to see Muller's ratchet in the first place. Therefore, not observing the expected pattern for TEs in the genomic data is not so surprising as the expectation itself does not seem to be very robust. I would like the authors to better acknowledge this issue, which actually goes into their general idea that the genomic consequences of asexuality are not so simple.
Indeed, the survivorship bias should affect all genomic features. Nothing that is incompatible with the viability of the species will ever be observed in nature. Perhaps the difference between Muller’s ratchet and the dynamics of accumulation of transposable elements (TEs) is that TEs are expected to either propagate very fast or not at all (Dolgin and Charlesworth 2006), while the effects of Muller’s ratchet are expected to vary among different populations and cellular mechanisms of asexuality. We will rephrase the text to better reflect the complexity of the predicted consequences of TE dynamics.
4) Heterozygosity
Due to the absence of recombination, asexual populations are expected to maintain a high level of diversity at each single locus (heterozygosity), but a low number of different haplotypes. However, as presented by the authors in the Box 2, there are different modes of parthenogenesis with different outcomes regarding heterozygosity: (1) preservation at all loci; (2) reduction or loss at all loci; (3) reduction depending on the chromosomal position relative to the centromere (distal or proximal). Therefore, the authors could benefit from their genome-based dataset to explore in more detail the distribution of heterozygosity along the chromosomes, and further test whether it fits with the above predictions. If the differing quality of the genome assemblies is an issue, the authors could at least provide the variance of the heterozygosity across the genome. The mode #3 (i.e. central fusions and terminal fusions) would be particularly interesting as one would then be able to compare, within the same genome, regions with large excess vs. deficit of heterozygosity and assess their evolutive impacts.
Moreover, the authors should put more emphasis on the fact that using a single genome per species is a limitation to test the subtle effects of asexuality on heterozygosity (and also on "mutation accumulation & positive selection"). These effects are better detected using population-based methods (i.e. with many individuals, but not necessarily many loci). For example, the FIS value of a given locus is negative when its heterozygosity is higher than expected under random mating, and positive when the reverse is true (Wright 1951; 10.1111/j.1469-1809.1949.tb02451.x).
We agree with the reviewer that the analysis of the distribution of heterozygosity along the chromosomes would be very interesting. However, the necessary data is available only for the Cape honey bee, and its analysis has been published by Smith et al. 2018. Calculating the probability distribution of heterozygosities would be possible, but it would require SNP calling for each of the datasets. Such an analysis would be computationally intensive and prone to biases by the quality of the genome assemblies.
5) Absence of sexual lineages
A second limit of this work is the absence of sexual lineages to use as references in order to control for lineage-specific effects. I do not agree with the authors when they say that "the theoretical predictions pertaining to mutation accumulation, positive selection, gene family expansions, and gene loss are always relative to sexual species [...] and cannot be independently quantified in asexuals." I think that this is true for all the genomic features analysed, because the transition to asexuality is going to affect the genome of asexual lineages relative to their sexual ancestors. This is actually acknowledged at the end of the Conclusion by the authors.
To give an example, the authors say that "Species with an intraspecific origin of asexuality show low heterozygosity levels (0.03% - 0.83%), while all of the asexual species with a known hybrid origin display high heterozygosity levels (1.73% - 8.5%)". Interpreting these low vs. high heterozygosity values is difficult without having sexual references, because the level of genetic diversity is also heavily influenced by the long term life history strategies of each species (e.g. Romiguier et al. 2014; 10.1038/nature13685).
I understand that the genome of related sexual species are not available, which precludes direct comparisons with the asexual species. However, I think that the results could be strengthened if the authors provided for each genomic feature that they tested some estimates from related sexual species. Actually, they partially do so along the Result & Discussion section for the palindromes, transposable elements and horizontal gene transfers. I think that these expectations for sexual species (and others) could be added to Table 1 to facilitate the comparisons.
Our statement "the theoretical predictions pertaining to mutation accumulation, positive selection, gene family expansions, and gene loss are always relative to sexual species [...] and cannot be independently quantified in asexuals." specifically refers to methodology: analyses to address these predictions require orthologs between sexual and asexual species. We fully agree that in addition to methodological constraints, comparisons to sexual species are also conceptually relevant - which is in fact one of the major points of our paper. We will clarify these points.
6) Regarding statistics, I acknowledge that the number of species analysed is relatively low (n=26), which may preclude getting any significant results if the effects are weak. However, the authors should then clearly state in the text (and not only in the reporting form) that their analyses are descriptive. Also, their position regarding this issue is not entirely clear as they still performed a statistical test for the effect of asexuality mode / origin on TE load (Figure 2 - supplement 1). Therefore, I would like to see the same statistical test performed on heterozygosity (Figure 2).
We will unify the sections and add an appropriate test everywhere where suited.
7) As you used 31 individuals from 26 asexual species, I was wondering whether you make profit of the multi-sample species. For example, were the kmer-based analyses congruent between individuals of the same species?
Unfortunately, some of the 31 individuals do not have publicly available reads (some of the root-knot nematode datasets are missing), others do not have sufficient quality (the coverage for some water flea samples is very low). Our analyses were consistent for the few cases where we have multiple datasets available.
References
Dolgin, Elie S., and Brian Charlesworth. "The fate of transposable elements in asexual populations." Genetics 174.2 (2006): 817-827.
Smith, Nicholas MA, et al. "Strikingly high levels of heterozygosity despite 20 years of inbreeding in a clonal honey bee." Journal of evolutionary biology 32.2 (2019): 144-152.
for - paper - title - Memory, Sleep, Dreams, and Consciousness: A Perspective Based on the Memory Theory of Consciousness - author - Andrew E. Budson, Ken A Paller - adjacency - memories - sleep - dreams - Memory Theory of Consciousness - MToC
summary - The authors present a theory of dreaming and sleep that I resonate with, that sleep is a time in which the brain performs unconscious processing of memories, consolidating them by taking advantage of consciousnesss down time to perform massive parallel processing to connect memories together. - dreams are seen as a small conscious byproduct of the massive parallel processing task, and their meaning may have value depending on how we interpret them.
for - paper - title - Mental Time Travel? A Neurocognitive Model of Event Simulation - author - Donna Rose Addis - adjacency - memory - imagination - the same - from - paper - https://hyp.is/0Fb6NqdjEfCyTTddI20_aQ/www.dovepress.com/memory-sleep-dreams-and-consciousness-a-perspective-based-on-the-memor-peer-reviewed-fulltext-article-NSS
summary - memory and imagination are proposed as fundamentally the same process. - It is the ‘mental’ rendering of experience that is the most fundamental function of this simulation system enabling humans to - re-experience the past, - pre-experience the future, and - comprehend the complexities of the present.
Changing the Narrative
I would want them to follow up on how changing the narrative takes place and how schools have improved in all aspects.
for - book - Embracing Paradox, Evolving Language - book - review - Embracing Paradox, Evolving Language - adjacency - Lisa's conlanger - Deep Humanity BEing journeys - Indyweb - provenance - Deep Humanity - language BEing journey - author - Lisa E. Maroski - to - post - LinkedIn - Bayo Akomolafe - from 'belief' to 'apolief" - https://hyp.is/go?url=https%3A%2F%2Fwww.linkedin.com%2Fposts%2Fbayoakomolafe_i-am-against-worldview-the-term-seems-activity-7319799984663535616-fpVW%2F&group=world
new trailmark - summary to review - the word "review" may be a better trailmark word than "summary" - At this point, I will replace "summary" with "review" in the case of book or article reviews
review - Lisa's book is an insightful convergence of an important but ignored subject, the experiential intersection between language and consciousness. - Her understanding that language plays an important role in constructing our reality leads to a bold and novel proposal, especially salient at this time of global poly-meta-perma-meaning crisis. - She proposes that we individually and collectively experiment and explore creating new words and language structures that transcend the limitations of our existing language - If patterns of language usage traps us in outdated conceptual paradigms, then breaking out of these may be challenging, if not impossible, without the creation of new linguistic and language structures. - From a Stop Reset Go and Deep Humanity perpsective, Lisa's proposal for practical experimentation with constructing new languages to unleash new forms of expression is very aligned to Deep Humanity BEing journeys - As I read and annotate Lisa's book, any potential linguistic and language BEing journeys that her words inspired will be recorded for posterity
Addendum - note from journal - 2025, May 8 - reflections on Lisa's book - asynchronous communication is only one half of indyweb - the other half is asynchronous REFLECTION AND SYNTHESIS - Effective timebinding requires both - Annotation captures interpersonal shared ideas - journalling captures ours own unique synthesis only emerges from asynchronous reflections of our existent associative network of ideas and the newly ingested interpersonal ones - Annotations capture the novel and newly inputted interpersonal ones - but annotation currently only applies to hypothesis - it needs to expand to realtime meetings such as zoom calls, emails, socials media comments and socials media chats in order to be complete - Until now, there has not been a medium with sufficient set of affordances to unleash the affordances potential in language itself - While digital media has existed and rapidly developed for the past 5 decades, - employing and leveraging it to unleash the full potential of language itself has not ever been conceived of until the concept of Indyweb arrived - Indeed, we could make the claim that the indyweb is a foundational human technology on the same order as language itself because it completes language, revealing its empty ( shunyata) quality, thereby - uniting it with the universe itself - From the unlimited potential of the tacit, - the limited forms of words emerge, both are 2 sides off the same nondual coin - and unleashing the full , unrealized potential of language - It is the provenance aspect of the indyweb that provides an automatic trail of all our learning journey, making both the - individual and - intertwingled collective evolution of ideas available as records for. timebinding posterity
for - natural language acquisition - author - J. Marvin Brown - book - From the Outside In - from - youtube - The Language School that Teaches Adults like Babies - https://hyp.is/Ls_IbCpbEfCEqEfjBlJ8hw/www.youtube.com/watch?v=984rkMbvp-w
for - book - The Unfolding of Language - An Evolutionary Tour of Mankind's Greatest Invention - author - Guy Deutscher
Choke Points
for - book - Choke Points - author - Edward Fishman - to - youtube search - Edward Fishman Chokepoint - https://hyp.is/F-UD3hqSEfCR80u-O1akrQ/www.youtube.com/
for - Trump tariffs - How much pain? - article How much pain will new tariffs bring - and for how long? - author - Justin Wolfers
for - climate justice - Africa - article - The Conversation - Wealthy nations owe climate debt to Africa - funds that could help cities grow - author - Astrid R.N. Haas
for - article - evolution of modernity - article - Resilience - evolution of modernity - author - Richard Heinberg - polycrisis - post carbon institute
Summary - A well thought out overview of hotter outer species arrived at our polycrisis in modernity
for - book - Deficit - How feminist economics can change our world - author - Emma Holten
there is something perplexing about claims like this. By their nature, they imply that we were smarter and more precise in the past.
YES! I'm very glad that the author does not like these claims.
creative minorities
for - definition - creative minorities - Arnold Toynbee, author of The Study of History - groups capable of inspiring action among the larger, less-educated, and less-visionary masses - SOURCE - article - Substack - The three civilizational priorities of the next societal transition - Michel Bauwens - 2025, Jan 17
Sovereign Individual.
for - further research - Oligarch's favorite book - The Sovereign Individual - Author - James Dale Davidson and William Rees-Mogg - source - article - Le Monde - Musk, Trump and the Broligarch's novel hyper-weapon - Yanis Varoufakis - 2025, Jan 4
https://taz.de/Folgen-der-Erderhitzung/!6057015/
Report: https://www.globalwater.online/globalwater/wp-content/uploads/2018/09/GWM-Report-2024.pdf
Die Biden-Regierung hat Regeln zur Subventionierung von grünem Wasserstoff finalisiert. Lobbies konnten sich zwar teilweise durchsetzen, die Förderungen werden aber weitgehend nur der Wasserstoffproduktion mit erneuerbarer Energie zugutekommen. Es ist allerdings nach dem Ende mehrerer Projekte unsicher, dass die WasserstoffIndustrie mit dem von vielen erwarteten hohem Tempo aufgebaut wird https://www.nytimes.com/2025/01/03/climate/hydrogen-subsidies-biden-treasury.html
Das britische Met.Office erwartet, dass 2025 eines der drei Jahre mit den höchsten je gemessenen Durchschnittstemperaturen werden wird. Die La Niña-Phase im Südpazifik wird sich deutlich weniger auswirken als die Erhitzung durch Treibhausgase https://www.theguardian.com/news/2025/jan/02/weatherwatch-2025-likely-to-be-another-year-of-high-temperatures
Die Ukraine.hat die Pipeline,durch die bisher russisches Gas nach Westeuropa geliefert wurde, unterbrochen. https://www.theguardian.com/world/2025/jan/01/breakaway-moldovan-region-cuts-heating-and-hot-water-as-russia-stops-gas-flowa
CEPA-Report:https://cepa.org/article/betraying-ukraine-for-blood-gas/
Eine 77-jährige Just Stop Oil-Aktivistin muss über die Feiertage weiter im Gefängnis bleiben. Laut Aussage der damit beauftragten privaten Firma gibt es kein elektronisches Gerät, das sich an ihren schmalen Handgelenken fixieren lässt und die Überwachung eines Hausarrests erlaubt https://www.theguardian.com/society/2024/dec/21/elderly-activist-to-spend-christmas-in-prison-because-tag-does-not-fit
Konrad Kramar im Kurier zur zerplatzenden Blase „Grüner Wasserstoff“. Von den 10 Mio. Tonnen jährlich, die die EU-Kommission bis 2030 geplant hat, sind erst 7% erreicht, von den geplanten Leitungen erst 3% im Bau. Im Vergleich zu Wärmepumpen bei Heizungen und Batterien bei LKWs ist Wasserstoff unwirtschaftlich. Die Wasserstoff-Lobbies promoten tatsächlich grauen, mit Erdgas produzierten Wasserstoff, der auf absehbare Zeit als einziger konkurrenzfähig ist, aber die fossilen Emissionen nicht verringert [via Sabine Jungwirth auf Facebook]. https://kurier.at/wirtschaft/eu-wasserstoff-klimawandel-solarzellen-pipeline-hydrogen-bank/402990372
Screenshot: https://www.facebook.com/photo/?fbid=10227863164746893&set=a.2637414336971
Eine neue Studie erhebt systematisch, wieviel weniger Frosttage es in den Ländern und Städten der Nordhalbkugel 2014-2023 aufgrund der globalen Erhitzung gab. Österreich hat inzwischen 13, Wien 22 Frosttage weniger als in einer Welt ohne fossile Emissionen. Diese Veränderungen haben gravierende Folgen für die Ökosysteme, die Verbreitung von Krankheiten, den Tourimus und das Lebensgefühl. https://www.derstandard.at/story/3000000249512/der-klimawandel-kostete-wien-22-frosttage-im-winter
Ausfühlicher Überblicksartikel zur Begrünung von Dächern mit einem Schwerpunkt auf Entwicklungen in Wien. Flache Dächer auf neuen Gebäuden müssen in Wien begrünt sein; die Begrünung wird von der Stadt gefördert. Die Begrünung hat auch positive Auswirkungen auf die Biodiversität. Als exemplarisches Projekt wird die Biotope City am Wienerberg genannt. https://www.derstandard.at/story/3000000247676/laut-fachleuten-braucht-es-mehr-gruen-auf-unseren-daechern
Der Artikel beschäftigt sich mit den gesundheitlichen Folgen der der globalen Erhitzung. Anlass ist, das bei der COP28 zum ersten Mal bei einer COP ein ganzer Tag der Gesundheit gewitdmet war. Gefordert wird ein interdisziplinärer Zugang an der der Stelle des biher gängigen Totschweigens, https://www.liberation.fr/idees-et-debats/tribunes/le-rechauffement-de-la-terre-est-une-urgence-de-sante-publique-20240105_A27LZFF2B5EYJD25FOPJ22CYCI/
Einzelheiten zum jetzt gekündigten geheimen Vertrag der OMV mit der Gazprom. Die OMV ging von einem bis auf 11,6 Terwattstunden steigenden Gasbedarf aus und verlängerte die Vereinbarung bis 2045..https://www.derstandard.at/story/3000000248960/gasliefervertrag-fuer-omv-waere-bei-nichtkuendigung-sogar-bis-2045-gelaufen
Chakrabarty formuliert sehr klar, worin die Neuartigkeit und Einzigartigkeit der historischen Situation des Anthropozän besteht, und damit auch, was die Herausforderung dieser Situation für Politik und Geschichtsschreibung ausmacht. Politik ist ohne die Dimension der Spezies Geschichte nicht mehr zu verstehen, und umgekehrt lässt sich diese Geschichte aber nur als auch politische Geschichte, und damit als Geschichte von Machtverhältnissen begreifen. Diese Dimension der Geschichte wird in der aktuellen westlichen Politik so gut wie nicht begriffen oder gar berücksichtigt.
Chakrabarty spricht in diesem Text immer nur von der einen Spezies Mensch. Möglicherweise kann man die Besonderheit dieser Spezies als einer historischen Entität besser verstehen, wenn man ihre Beziehungen zu anderen Spezies mitdenkt. Diese Beziehungen sind immer auch räumlich und lassen sich in der kritischen Zone der Erde lokalisieren. So lässt sich vielleicht auch Geschichte der Spezies und Geschichte der Globalisierung leichter in Verbindung bringen.
Die Globalisierung geht von der Möglichkeit eines positiven Universale "Menschheit" Ist. Die Geschichte der Spezies ist dagegen die Geschichte unterschiedlicher lokale Beziehungen zu anderen Spezies. Die Globalisierung als ein Prozess, durch den die als Einheit unsinnige Menschheit den Planeten überall in der gleichen Weise ausbeutet, ist nur als Fiktion zuende zuführen.
Am Anfang schreibt Chakrabarti über die Nachvollziehbarkeit der Geschichte durch Verständnis und Rekonstruktion der Intentionen von Akteuren, die damit als nicht natürlich begriffen werden,. Als Naturwesen sind sie nicht Gegenstand der Geschichte. Diese Überlegung erinnern mich an den von Chakrabarty nicht erwähnten Max Weber, der vom subjektiven Sinn als Voraussetzung der verstehenden Wissenschaften ausgegangen ist. Ihre Geschichte im anthropozän ist dagegen eine ganz andere als die von ihnen intendierte und nachvollziehbare. Natur würde vielleicht sagen, dass ich die Akteure vervielfachen und das Geschichten mit diesen vervielfachten Akteuren geschrieben werden müssen. Ein Beispiel wäre die Geschichte der Nutzung der fossilen Brennstoffe, wie sie Timothy Mitchell geschrieben hat.
La Niña
Gespräch mit einem französischen vertreter des wald waldlauf fand über die biodiversität politik in frankreich punkt anders ist der neue bericht des w w f punkt in frankreich gibt es drei gesetzliche versuche komma die biodiversität zu schützen komma die alle verwässert worden beziehungsweise nicht umgesetzt wurden doppelpunkt tarife für den wasserverbrauch komma eine grüner Fonds, komma um gebietskörperschaften bei der renaturierung zu unterstützen komma und ein gesetz komma um boden versiegelung bis zwanzig fünfzig zu unterbinden punkt nun das haupt hindernis bei der umsetzung dieser pläne ist die finanzierung punkt https://www.liberation.fr/environnement/biodiversite/biodiversite-la-nature-est-en-plein-burn-out-20241010_CTYTS2FN2FEBBAIITRNM6C3DSI/
Der temperatur anstieg des jahres zwanzig dreiundzwanzig lässt sich mit großer wahrscheinlichkeit auf den rückgang der albedo der erde zurückführen punkt dabei spielt der rückgang niedriger wolken die hauptrolle punkt eine neue studie zeigt komma das diese veränderungen die null komma zwei grad temperaturanstieg erklären kommen die beobachtet wurden komme aber deren ursache noch nicht bekannt ist punkt der rückgang der niedrigen wolken bedeckung lässt sich zum teil auf weniger eu sohle zurückführen komma er könnte aber auch durch die globale erhitzung selbst verursacht sein komma also ein rückblicks effekt darstellen punkt Nun nun https://www.derstandard.at/story/3000000248101/raetselhafter-temperatursprung-durch-rueckgang-von-wolken
Die New York Times über die klimapolitischen oder eher anti-klimapolitischen Positionen des Führungspersonals der kommenden Trump-Administration. Auch wenn die Positionen variieren ist deutlich, dass sie für eine weitere Expansion der US-Öl- und Gasindustrie eintreten und Bundesbehörden die Kompetenzen nehmen wollen, die Emissionen zu regulieren, https://www.nytimes.com/2024/12/04/climate/trump-cabinet-stefanik-zeldin-wright.html
Die OMV und die Adnoc fusionieren ihr petrochemiegeschäft zu dem neuen Konzern Beruf, an dem beide gleiche Anteile halten. Aus dem Artikel geht auch hervor, dass die adnock durch Indizierung ihrer Anteile mit der OMV auch über die österreichische Politik bei der OMV mitbestimmt. https://www.derstandard.at/story/3000000194416/klimaphaenomen-el-nino-soll-bis-april-zu-hoeheren-temperaturen-beitragen
Erklär-Artikel im Guardian über Carbon Dioxide Removal (CDR) und Carbon Capture and Storage (CCS). Bei der Verabschiedung der Summary for Policy Makers des letzten Teils des AR6 haben sich vor allem Vertreter Saudi-Arabiens dafür eingesetzt, Hinweise auf CDR und CCS aufzunehmen und Hinweise auf erneuerbare Energien zu streichen. https://www.theguardian.com/environment/2023/apr/25/carbon-dioxide-removal-tech-polarising-climate-science
Greenpeace klagt in Rumänien gegen das OMV-Projekt Neptun Deep, u.a.wegen einer unkorrekten Umweltverträglichkeitsprüfung und falschen Angaben über die zu erwartenden Emissionen. Neptun Deep ist das größte fossile Expansionsprojekt in der EU https://www.derstandard.at/story/3000000243470/greenpeace-klagt-gegen-milliardenteures-gasprojekt-der-omv-im-schwarzen-meer
2023 wurde mit 55,5 Milliarden Fass Öläquivalent so viel Öl und Gas gefördert wie nie zuvor. 578 Unternehmen arbeiten daran, durch zusätzliche Förderstätten weitere 240 Milliarden Fass zu produzieren, obwohl zur Einhaltung des 1,5 Grad-Ziels keine Förderkapazitäten mehr aufgebaut werden dürfen. Zu den Unternehmen mit den größten Expansionsplänen gehört die an der OMV beteiligte Adnoc. Die Zahlen sind - neben vielen weiteren z.B. zur LNG-Expansion - in der aktualisierten Global Oil & Gas Exit List (Gogel) der NGO Urgewald enthalten https://www.derstandard.at/story/3000000244513/weltweite-oel-und-gasfoerderung-erreichte-2023-ein-allzeithoch
Global Carbon Budget 2024: https://essd.copernicus.org/preprints/essd-2024-519/
Presseaussendung zum Global Carbon Budget: https://globalcarbonbudget.org/fossil-fuel-co2-emissions-increase-again-in-2024/
Am 29. Oktober fiel in einigen Gebieten in Südspanien so viel Niederschlag wie sonst in eInem Jahr. U.a. weil die Regionalbehörden nicht rechtzeitig warnten, kamen über 200 Menschen zu Tode. Am vergangenen Wochenende protestierten über 100.000 Menschen gegen die Behörden. Die rechte Regionalregierung in Valencia hatte den Katastrophenschutz abgebaut, weil sie die Bedrohung durch die Klimakrise für überschätzt hielt https://www.theguardian.com/world/2024/nov/12/spain-braces-new-storms-flooding-disaster-political-fallout-continues
TRSP Desirable Characteristics
Contact info (for the person, depositor, producer or owner, ideally with ORCID; or organisation) of the data: does the repository keep and show this information?
Russland hat durch Vetos dafür.gesorgt, dass die COP29 in dem autoritären Petrostaat Aserbaidschan stattfindet. Max Bearak berichtet in der New York Times auch über die steigenden Öl- und Gasexporte des Landes nach Europa.
https://www.nytimes.com/2024/11/11/climate/cop29-why-baku-azerbaijan.html
2024 wird – den neuesten Copernicus-Daten zufolge – mit sehr großer Wahrscheinlichkeit das erste Jahr mit einer Durchschnittstemperatur von über 1,5° über dem vorindustriellen Durchschnitt und damit das wärmste Jahr der Messgeschichte werden. Im Oktober lag die durchschnittliche Meerestemperatur über 20° – der zweithöchste Wert nach 2023.
Copernicus-Meldung: https://climate.copernicus.eu/year-2024-set-end-warmest-record
Allein das Rollback der klimapolitischen Maßnahmen der Biden-Administration durch die Trump-Regierung wird bis 2030 zu 4 Gigatonnen CO2-Äqivalenten an zusätzlicher Emissionen führen (bis 2050 zu 27GtCO2e). Die Analyse verschiedener Modellierungen der von Trump geplanten Maßnahmen durch Carbon Brief ergab im März, dass die USA damit weltweit fast eine Billiarde USD zusätzlicher „social costs of carbon“ verursachen würden. Die geplante weitere fossile Expansion unter dem Motto „drill, Baby, drill!“ ist dabei nicht einberechnet https://www.carbonbrief.org/analysis-trump-election-win-could-add-4bn-tonnes-to-us-emissions-by-2030
Die New York Times über die Pläne des Trump-Teams zur Beendigung der Klimapolitik der Biden-Administration. Dazu gehören der Austritt aus dem Pariser Abkommen, die Verlagerung (zwecks Entmachtung) der Environmental Protection Agency und die Aufhebung des Verbots von Bohrungen in bisherIgen Naturschutzgebieten und Territorien von Native Americans. https://www.nytimes.com/2024/11/08/climate/trump-transition-epa-interior-energy.html
Tim Bosch befürchtet im taz-Interview, dass die kommende Trump-Administration nicht nur aus dem Pariser Klimaabkommen, sondenr auch aus der UN-Rahmenkonvention für Klimaschutz aussteigt https://taz.de/Politologe-ueber-Klimapolitik-mit-Trump/!6047357/
Eine neue Studie bestätigt, dass die Hauptursache des immer schnelleren Anstiegs des Methan-Gehalts der Atmosphäre die Aktivität von Mikroaorganismen ist, die durch die globale Erhitzung zunimmt. Damit handelt es sich um einen Feedback-Mechanismus, durch den sich die globale Erhitzung selbst verstärkt. https://taz.de/Zu-viel-Methan-in-der-Atmosphaere/!6045201/
Studie: https://www.pnas.org/doi/10.1073/pnas.2411212121
Vorangehende Studien: https://www.nature.com/articles/s41558-023-01629-0, https://www.nature.com/articles/s41558-022-01296-7.epdf?sharing_token=CDMa5-ti34UNBqv3kfuCB9RgN0jAjWel9jnR3ZoTv0NZRKXEI-7kyXEEvNI7duu65JLcZpmhGxWTeSfYcMCqxqYk5nUrdR60izmjToMNw56RgBqIcn3JXKxSjx13vmB9ZYndGTUMt-52Vs7HT_T6K9Oth4QFRyP51eOpz8pV8l65HFDo2VSfQ6xDXklMtmvt-HGwltAINb_2xgmtAR-V4g%3D%3D&tracking_referrer=taz.de
I had hoped to somewhat emulate the success of ex-Microsoft executive Steven Sinofsky and his Substack serialization project, Hardcore Software (“personal stories and lessons from inside the rise and fall of the PC revolution”). Sinofsky’s project was an inspiration for mine, but I don’t think I’ve reached his level in terms of reader comments and reshares.
possibly due to author platform?
There’s a version of the “why writers should blog” story that is tawdry and mercenary: “Blog,” the story goes, “and you will build a brand and a platform that you can use to promote your work.” Virtually every sentence that contains the word “brand” is bullshit, and that one is no exception.
Drei Klimawissenschaftlerinnen wehren sich in einem Kommentar in Nature gegen den Vorwurf, sie betrieben keine objektive Wissenschaft, weil sie ihren Schmerz angesichts der fortschreitenden Klimakrise offen kommunizieren. Gefühle seien Teil der Realität und dürften von Forschenden nicht unterdrückt werden. Gefühle auszuschließen werde vor allem von Privilegierten zur Norm erklärt, die den Folgen der Klimakrise kaum ausgesetzt seien https://www.theguardian.com/environment/2024/oct/25/we-have-emotions-too-climate-scientists-respond-to-attacks-on-objectivity
Nature-Artikel: https://www.nature.com/articles/s41558-024-02139-3
Der neueste Emissions Gap Report der Vereinten Nationen kommt zu dem.Ergebnis, dass die Erde auf eine Temperatur-Erhöhung um 3,1° zusteuert. Würden sich alle Staaten an ihre Selbstverpflichtungen halten, käme es zu einem Anstieg um etwas weniger als 3°. Noch immer sei ein Erreichen des 1,5°-Ziels technisch möglich, verlange aber einen entschlossenen Politikwechsel und Leadership https://www.theguardian.com/environment/2024/oct/24/crunch-time-for-real-un-says-time-for-climate-delays-has-run-out
Bericht: https://www.unep.org/resources/emissions-gap-report-2024
Frankreichs dritter nationaler Plan zur Klimaanpassung enthält 51 Maßnahmen. Sie gehen von 4° Erhitzung bis 2100 aus - in Frankreich die Vorgabe für alle offiziellen Dokumente zur „ökologischen Planung“. Viele Fachleute halten diese Einschätzung für zu optimistisch. Die meisten der Massnahmen sind wenig konkret und nicht mit einer Finanzierung unterlegt. Die besondere Vulnerabilität bestimmter Gruppen wird nicht berücksichtigt https://www.liberation.fr/environnement/climat/changement-climatique-un-nouveau-plan-national-dadaptation-au-regime-sec-20241025_E5E4GSMOBJEPVGEU3EGXEJBZRY/
Die von Waldbränden außerhalb der Tropen verursachten Emissionen haben sich seit 2001 fast verdreifacht. Weltweit haben die Emissionen durch Waldbrände in dieser Zeit um 60% zugenommen. Ursache dafür ist die Kombination von heißerem und trockenerem Wetter mit dem schnelleren Wachstum der Wälder durch die höheren Temperaturen. Die Wälder können durch die Brände jahrzehntelang zu Emittenten werden. Damit ist die Funktion der Wälder als Kohlenstoffsenken gefährdet. Das bedeutet auch, dass sie andere anthropogene Emissionen weniger kompensieren und die Fähigkeit verlieren, nach einem Überschreiten der 1,5°-Grenze C0<sub>2</sub> aus der Atmosphäre zu entfernen. Außerdem müssten diese von Menschen verursachten Emissonen den C0<sub>2</sub>-Budgets der Nationalstaaten zugeordnet werden.
Überblick über die französische Rezeption des Bioregionalismus. Ich bin gestern durch den Circular Metabolism Podcast darauf gestoßen. Der Artikel ist von 2022, danach ist wohl vor allem bei der bioregionalen Architektur sehr viel passiert. Von den amerikanischen Begründern des Bioregionalismus führt eine Linie zur deep ecology und eine zur sozialen Ökologie. Eine entscheidende Figur für die französische Rezeption ist Alberto Magnaghi.
In practice, tracking all authors in all copyright notices is quite cumbersome. Instead, often only the original author is credited here even when copyright is shared with additional contributors. A more reasonable approach is to credit all authors collectively, e.g. as “the FooProject contributors” or “Original Author and others”. However, I am not sure whether that results in a valid copyright notice as the copyright holders must be clearly recognizable.
The other reason to update these notices is if there are new authors. Typically, this is done by adding a new copyright line for each set of authors, with the most recent on top. For example: Copyright 2016–2018 George Copyright 1999, 2007–2016 Fred Adding a new line is sensible since many open-source licenses require that existing copyright notices are kept intact – so you must not update them in any way. And in the above example, adding George to Fred's copyright notice would be misleading since George did not publish any of their work in 1999 and Fred didn't publish in 2018.
Did you actually fix a known issue? Let the author know about it.
Developers want to improve their project. If you find an issue, bring it up. If it's a valid concern, the author will probably want to have it fixed. In many cases, the author will consider it a valid issue, but simply not have the personal time or need to address it immediately. This is where open-source is great. Just fork the project and fix it
Not everyone has time to adhere to the specific coding styles for a project, so if you can't do a full blown pull-request, there is NOTHING wrong with opening a pull-request that only has the intention of showing the author how you solved the problem.
On behalf of all open-source developers and project maintainers, I ask you try and be polite the next time you ask for support. Try to remember that there is a real human being on the other side of the screen, and they actually want to help you.
If you feel there has been an oversight, it's okay to not give up. As long as you are being logical and open to other people's views, you will find that you might learn something new, or even teach something to the maintainer.
fed Rico new book called irreducible
for - book - Irreducible - author - Federico Faggin - to - book Irreducible
to - book - Irreducible - https://hyp.is/0J8C4lo8Ee-WxX-r7RiEHw/www.collectiveinkbooks.com/essentia-books/our-books/irreducible-consciousness-life-computers-human-nature
Vanessa Andreott
for - book - Hospicing modernity - author - Vanessa Andreotti - Dean of education - U of Victoria
per Espen Stokes is the author of what we think about when we try not to think about global warming
for - book - What we think about when we try not to think about global warming - author - Per Espen Stokes - climate crisis - psychology of - Per Espen Stokes
After publication in February 1940, How to Read a Book propelledAdler to the forefront of the Great Books Movement and into a posi-tion now referred to as a “public intellectual.”
“When I was 9 or 10,” he told The Times in 2017, “my grandmother gave me a six-volume collection of books by Robert Louis Stevenson, which inspired me to start writing stories that began with scintillating sentences like this one: ‘In the year of our Lord 1751, I found myself staggering around blindly in a raging snowstorm, trying to make my way back to my ancestral home.’”
One problem with using this extension is that the author stopped supporting their extensions years ago and has not been heard from since. You also need to bypass the version check per this article.
follow up - book - Exactly What to Say" - author - Phil. M. Jones
for - symbolosphere - language - book - Exactly What to Say - author - Phil M. Jones
Clark E. Moustakas in his delightful and seminal book Loneliness
follow up - book - Loneliness - author - Clark E. Moustakas
Emergent Strategy, Adrienne Maree Brown
follow up - book - Emergent Strategy - author - Adrienne Maree Brown
the second link is to a video made by scott shepard who's done some great work unearthing key aspects of lumens zettelcostn method that typically go unmentioned in the many videos and blog posts that are out there claiming to introduce people to the zettocauston method
Scott P. Scheper, author of "Antinet Zettelkasten," book published in 2022.
a wonderful book by kevin kian there is the story about two young fish playing off a coral reef
for - book - author - Kevin Kian
follow up - Kevin Kian? - get more info on author and book
Would it bestretching things too much, you suddenly wonder, to call yourRolodex a form of autobiography—a manuscript that you have beenwriting these �fteen years, tinkering with, revising?
Peter Pogany, Rethinking the World
for: book - Rethinking the World
book: Rethinking the World
Roland Barthes (1915-1980, France, literary critic/theorist) declared the death of the author (in English in 1967 and in French a year later). An author's intentions and biography are not the means to explain definitively what the meaning of a (fictional I think) text is. [[Observator geeft betekenis 20210417124703]] dwz de lezer bepaalt.
Barthes reduceert auteur to de scribent, die niet verder bestaat dan m.b.t. de voortbrenging van de tekst. Het werk staat geheel los van de maker. Kwam het tegen in [[Information edited by Ann Blair]] in lemma over de Reader.
Don't disagree with the notion that readers glean meaning in layers from a text that the author not intended. But thinking about the author's intent is one of those layers. Separating the author from their work entirely is cutting yourself of from one source of potential meaning.
In [[Generative AI detectie doe je met context 20230407085245]] I posit that seeing the author through the text is a neccesity as proof of human creation, not #algogen My point there is that there's only a scriptor and no author who's own meaning, intention and existence becomes visible in a text.
The story that they are telling is of a grand transition that occurred about fifty thousand years ago, when the driving force of evolution changed from biology to culture, and the direction changed from diversification to unification of species. The understanding of this story can perhaps help us to deal more wisely with our responsibilities as stewards of our planet.
Andrei VoznesenskyPermalink
Born in Moscow in 1933, Andrei Voznesensky is celebrated as “one of the most daring and popular poets of the Soviet era.” [1] While he graduated from the Moscow Architectural Institute, he abandoned his short-lived architectural career in order to work as a poet. That being said, his architectural training significantly influenced the structure of his poetry, including his famous “Videoms,” in which words are arranged visually to compose geometric shapes. [2] The centrality of structure and visuality to the reading experience is also fundamental in this poem. All indentations and line breaks reflect the form in which it was originally published.
Although his work cannot be distilled or reduced to a single literary movement, futurism significantly influenced his poetry. His work has been compared to that of Pablo Neruda, Vladimir Mayakovsky, and Boris Pasternak. [3]
While Voznesensky's poetry was censored by the Soviet press and publicly admonished by Khrushchev, he was still able to perform his work publicly [4]. He carefully navigated his position within the Soviet literary scene so as to maximize his opportunities for public expression. He is known for his performance The Poet and the Theater, which was held at the Taganka theater in 1965 and actively involved actors in the process of reading poetry from his book Antiworlds. [5] As the recipient of the 1978 Soviet State Prize, he had the opportunity to share his poetry across the world, including in the United States, which enabled him to befriend prominent politicians, artists, philosophers, and activists, such as Pablo Picasso, Robert Kennedy, and Jean-Paul Sartre. [3] He died on June 1, 2010.
 <script async src="//embedr.flickr.com/assets/client-code.js" charset="utf-8"></script>
<script async src="//embedr.flickr.com/assets/client-code.js" charset="utf-8"></script>
Voznesensky’s poem Думы о Чернобыле [Thoughts on Chernobyl] was published shortly after the Chernobyl nuclear disaster in Pravda, the official newspaper of the Soviet Union's Communist party, on June 3rd, 1986. In a LA Times interview, Voznesensky referred to the publication of this poem as illustrative of changing attitudes towards public-facing criticism in the Soviet Union:
“One year ago, it was impossible to even think these things. Maybe not arrested, but terrible time. Even now, I know the magazine director was brave guy. He didn’t even ask authorities. So it’s changing. I don’t know personally Gorbachev, but I think somebody around him read this poem and said, ‘It’s OK.’” [6]
Note: The image in the background of this project features the first page of this issue of Pravda.
Sources:
[1] Cheuse, Alan. “Remembering Poet Andrei Voznesensky.” NPR, 10 June 2010, https://www.npr.org/templates/story/story.php?storyId=127411139.
[2] "Andreĭ Voznesenskiĭ." Kul'tura.RF, https://www.culture.ru/persons/9345/andrei-voznesenskii.
[3] Polukhina, Valentina. “Andrei Voznesensky Obituary.” The Guardian, 3 June 2010, https://www.theguardian.com/books/2010/jun/03/andrei-voznesensky-obituary.
[4] Williamson, Marcus. "Andrei Voznesensky: Poet Who Fought Against Artistic Censorship in the Soviet Union." The Independent, 5 July 2010, https://www.independent.co.uk/news/obituaries/andrei-voznesensky-poet-who-fought-against-artistic-censorship-in-the-soviet-union-2018291.html.
[5] Beumers, Birgit. Yuri Lyubimov at the Taganka Theatre: 1964-1994. Taylor and Francis, 2003.
[6] Roraback, Dick. “Outspoken Soviet Poet Makes Waves in East and West.” Los Angeles Times, 8 April 1987, https://www.latimes.com/archives/la-xpm-1987-04-08-vw-23-story.html.
Image Credit:
"Andrei Voznesensky" by rdesign812 is licensed under CC BY-NC-ND 2.0. The image has not been modified in any way and falls under fair use.
Hernán Urbina JoiroPermalink
Hernán Urbina Joiro is a poet, journalist, writer, and acclaimed medical doctor from Colombia. As a member of the Academia Nacional de Medicina de Colombia, he has received national recognition for his medical research. In 2015, he received the prestigious Orden Gran Cruz de Caballero del Congreso de la República de Colombia for his artistic achievements. He has written for numerous Colombian newspapers and founded the Romanceros and Humanidad Ahora magazines. His book of poetry Canciones para el camino: Poesía escogida 1974-2019 is forthcoming from Caligrama. [1]

He traces the beginning of his poetry to his childhood, when he wrote verses about the massacres that took place between families in his town, San Juan del Cesar. [1] Urbina Joiro views his role as a doctor, who “inquires about the human condition,” as deeply tied to his poetry and journalism, in which he “reproduces in voices, meters, and images . . . the vertigo and the uncertainty of the contemporary world.” [1] Urbina Joiro utilizes poetry as a means to investigate and represent “the history of Colombia and the world,” which informs his artistic engagement with international politics and catastrophes such as Chernobyl. [1]
Sources:
[1] Urbina Joiro, Hernán. Hernán Urbina Joiro – Escritor. 2020, https://hernanurbinajoiro.com/.
Image Credit:
"Hernán Urbina Joiro" by Evelynparra19 is licensed under CC BY-SA 4.0. The image has not been modified in any way and falls under fair use.
Lina KostenkoPermalink
Lina Kostenko is one of the most renowned contemporary Ukrainian poets. She was born in 1930 in Rzhyshchiv, Kyiv Oblast and studied at the Maxim Gorky Literature Institute in Moscow. Kostenko was a leading member of the “Sixtiers” dissident movement, which consisted of "anti-totalitarian" thinkers who openly criticized political repression and corruption in their work. [1] Many of the “Sixtiers” were imprisoned, forcibly held in psychiatric hospitals, or exiled, and Kostenko herself was blacklisted in 1973 for her political activities and subversive writing by the Central Committee on Ideology of the Communist Party of Ukraine. [2] Although her work was not allowed to circulate officially, it was published in secret through samizdat, in which activists and poets transcribed and circulated each other’s forbidden works. Beginning in 1977, she was taken off the blacklist. She later received the prestigious Taras Shevchenko National Prize for her novel Marusya Churai in 1987. [2]

The Chernobyl disaster is a fundamental theme in Kostenko’s later work. [3] The untitled poem below comes from her collection The Volatile Quatrains, which deals extensively with the difficulties in bearing witness to and remembering Chernobyl.
As you read her poem, consider this excerpt from an interview between Lina Kostenko and the journalist Oksana Pakhlyovskaya:
Оксана Пахлёвская: – Ты не боишься ездить в Чернобыльскую зону?
Oksana Pakhlyovskaya: Aren’t you afraid of going into the Chernobyl zone?
Лина Костенко: – Нет. Писатель должен видеть всё.
Lina Kostenko: No. A writer must see everything.
[4].
Sources:
[1] “The Ukrainian Sixtiers Dissident Movement Museum.” Museum of Kyiv History. 2017, http://www.kyivhistorymuseum.org/en/museum-affiliates/museum-60-th.
[2] “Kostenko, Lina Vasylivna.” Virtual Museum: Dissident Movement in Ukraine. 2005, http://museum.khpg.org/en/index.php?id=1113913627.
[3] “Contributor: Lina Kostenko.” Words Without Borders: The Online Magazine for International Literature, https://www.wordswithoutborders.org/contributor/lina-kostenko.
[4] Kostenko, Lina. From an interview by Oksana Pakhlyovskaya, section IV, Al'terrnativna Barrikad. Translated by Valentina Varnovskaya. Stikhi.ru. https://stihi.ru/2014/08/06/210.
*English translation by Grace Sewell.
Image Credit:
"Lina_Kostenko_2003" by Rosiestep is licensed under CC BY-SA 4.0. The image has not been modified in any way and falls under fair use.
Author
comment
Marcia Bjornerud
Those are good points.
Reply to Dan Allosso at https://danallosso.substack.com/p/what-value-do-i-add-to-the-substack/comment/16463063
I just saw this morning that Jillian Hess, a professor/researcher/writer at a community college in New York, is also contemplating some of the same territory and trying to balance out the necessaries: https://jillianhess.substack.com/p/introducing-ps-a-new-paid-subscriber
I see that she's using both Amazon and Bookshop affiliate accounts and links in her stream. Have you delved into this for supplementation (albeit probably small)? I've done it for years and it never nets enough to even cover my hosting costs, though it makes the hobbyist portion of the outlay a bit more comfortable.
Beyond this, you might appreciate her particular Substack on note books and note taking or her new book: Hess, Jillian M. How Romantics and Victorians Organized Information: Commonplace Books, Scrapbooks, and Albums. 1st ed. Oxford: Oxford University Press, 2022. https://amzn.to/3VY4RU7
You're probably beyond needing them, but Substack has been building their writer resources and tips for helping to build paid audience. Details at https://on.substack.com/s/resources and https://on.substack.com/s/office-hours
Perhaps you're conflating too many things? Ask first, what value do I add to the world? (Arguably loads.) Then ask: How do I (best) distribute this value? To this perhaps one of your answers is Substack, which may or may not be one of many tools you use for this purpose. Then the follow on question is what value do you get back from it?
Given HCR's numbers (especially in comparison with Twitter) and her time on the platform, I suspect she may have (or at this point had) some sort of special platform deal with Substack which isn't publicly known beyond the basics of what the typical person could get. It's probably the modern digital equivalent of the sort of deal a highly visible academic might get from a magazine like The Atlantic. The pay scale may be different but we can obviously see that the daily output is wildly different too. If you're not aware, when Substack started they reached out to a wide variety of famous/semi-famous people and helped them to build a quick audience that would have taken them far more time and effort than they would otherwise have ever invested. Part of this was providing initial payment/seed money which was really their early investment for getting lots of quality content on the platform as a means of drawing the masses to come to the platform to both read and create as well. Unless you're a massive name working with them directly, you're unlikely to get this sort of deal today, and this means a tougher up hill slog for the "rest of us" as the platform doesn't need to pay for this sort of scaling/network effect now. If nothing else, knowing these early economies of Substack (and really lots of other social platforms, Medium certainly followed this script as an example) will help you to have a broader perspective and better compare your apples to others' oranges.
Ecological consequences of human niche construction: Examining long-term anthropogenic shaping of global species distributions
In the Pirandello play, "Six Characters in Search of an Author", the six characters come on stage, one after another, each of them pushing the story in a different unexpected direction. I use Pirandello's title as a metaphor for the pioneers in our understanding of the concept of evolution over the last two centuries. Here are my six characters with their six themes. 1. Charles Darwin (1809-1882): The Diversity Paradox. 2. Motoo Kimura (1924-1994): Smaller Populations Evolve Faster. 3. Ursula Goodenough (1943- ): Nature Plays a High-Risk Game. 4. Herbert Wells (1866-1946): Varieties of Human Experience. 5. Richard Dawkins (1941- ): Genes and Memes. 6. Svante Pääbo (1955- ): Cousins in the Cave. The story that they are telling is of a grand transition that occurred about fifty thousand years ago, when the driving force of evolution changed from biology to culture, and the direction changed from diversification to unification of species. The understanding of this story can perhaps help us to deal more wisely with our responsibilities as stewards of our planet.
!- Pirandello’s play Six Characters in Search of an Author : vehicle for exploring cultural evolution over the last 50,000 years
Biological and Cultural Evolution Six Characters in Search of an Author
!- Title : Biological and Cultural Evolution Six Characters in Search of an Author !- Author : Freeman Dyson !- Date : 2019
The deep AnthropoceneA revolution in archaeology has exposed the extraordinary extent of human influence over our planet’s past and its future
!- Title : The deep Anthropocene - A revolution in archaeology has exposed the extraordinary extent of human influence over our planet’s past and its future !- Author : Lucas Stephens - researcher at archaeoGLOBE project
paranoia has some surprising behaviour (like overriding ActiveRecord's delete and destroy) and is not recommended for new projects. See discard's README for more details. Paranoia will continue to accept bug fixes and support new versions of Rails but isn't accepting new features.
https://www.goodreads.com/notes/59660671-building-a-second-brain/7458926-tiago
And as if I requested it this morning, here's an example of an author using annotations to create engagement/start a conversation/start an informal book club discussion using Goodreads and annotations on their own work.
cc: @remikalir
https://www.goodreads.com/notes/57643476-annotation/3524158-markgrabe-grabe
I rarely see notifications from Goodreads about annotations (typically via Kindle) unless they're from the author of the book posting them, ostensibly to generate engagement with their readers. Interesting to see Mark Grabe sharing his annotations on @remikalir and @anterobot's book on annotation though. :)
Dear reviewer Thank you very much for the insightful suggestions, the manuscript improved a lot with the changes performed. Please find the point-by-point answer to the raised questions. In the main text, all changes are highlighted in yellow. I hope that with the changes made the new version is suitable for publication.
Best regards Valquiria Bueno
General assessment
The study is interesting and the title promises for me more than the MS finally contains.
Answer: The manuscript is part of a project aiming to study ACE1 and ACE2 expression in cells from the immune system of aging and young adults. These initial results suggest that ACE1 (and probably ACE2) plays somehow a role in the process of aging.
The background, question and the aim are relevant as explained in the introduction.
Answer: We included a piece of information in the “Introduction” trying to link ACE1 expression in tissue cells and age-related diseases, as it follows:
ACE1 has been suggested to influence age-related diseases (i.e. Alzheimer’s, sarcopenia, cancer) but the associated mechanisms are still under investigation. ACE1 polymorphisms were correlated with susceptibility to Alzheimer’s disease (AD). [15, 16] In addition, it was shown recently that in normal aging ACE1 expression is increased in brain homogenates and this expression is unchanged in early stages of AD. [17] Regarding sarcopenia, Yoshihara et al. [18] found a weak correlation between ACE polymorphism and physical function. In cancer (gastric or colorectal), patients presented higher expression of ECA1 in tumor when compared with healthy tissues. [19, 20]
The major criticism concerns the small size of the material (subjects, n=6), the small age difference (64-67 years) and the lack of younger controls.
Answer: We agree that the small number of studied subjects is a limitation of this study. In spite of the interesting results suggesting that ACE1 expression could be linked to the health status, it was not possible to perform correlation analysis due to the small sample size. Even though there is a small chronological difference between the subjects, the biological aging is very different among them and reflects the genetics, lifestyle, nutrition and comorbidities. Another limitation is the lack of younger controls to compare with the subjects studied. Our next steps are to include younger controls, to increase the number of studied subjects, and if possible to get samples from older subjects (i.e. 70-80, 80 and more years old)
Minor notes:
1)Title: Angiotensin converting enzyme (ACE) expression in leukocytes of older adults
Answer: We evaluated only ACE1 expression, and thus, title, abstract, and main text were changed to ACE1 instead of ACE. We decided to change to title for: Angiotensin converting enzyme (ACE) 1 expression in leukocytes of adults from 64 to 67 years old
2)Introduction: The last chapter, the Author should explain in more detail, how references 11-14 suggest that “ACE play an important role in the aging process”. Does this mean, that ACE is somehow regulating the aging process or in increasing age ACE -levels are changed?
Answer: References 11-14 shows that age-related diseases occurring in older adults are associated with changes in the immune system. To complete the text we added:
ACE1 has been suggested to influence age-related diseases (i.e. Alzheimer’s, sarcopenia, cancer) but the associated mechanisms are still under investigation. ACE1 polymorphisms were correlated with susceptibility to Alzheimer’s disease (AD). [15, 16] In addition, it was shown recently that in normal aging ACE1 expression is increased in brain homogenates and this expression is unchanged in early stages of AD. [17] Regarding sarcopenia, Yoshihara et al. [18] found a weak correlation between ACE polymorphism and physical function. In cancer (gastric or colorectal), patients presented higher expression of ECA1 in tumor when compared with healthy tissues. [19, 20]
Material and Methods:
The N-value of the subjects should be mentioned here, as well the relation of females/males.
Answer: Text was correct as suggested Blood was collected from adults (n=6, four females and two males) aged 64-67 years old in 2015.
Do the Authors really regard 64-67 “older age” nowadays?
Answer: Nowadays the most common term used for individuals older than 65 years is “older adults”.
Why first many years later the assays have been done in comparison to the collection of the blood? Are the samples still useable, not destroyed?
Answer: Samples are part of UNIFESP Biobank and have been kept in adequate conditions. We wanted to test cells from a period anterior to COVID-19 and those samples were the only ones that attended our purpose. We compared samples used in this study with fresh blood samples (cell viability and percentage of CD4+, CD8+ and CD19+) and the results showed good preservation of the cells.
Did the subjects have some diseases and/or drugs because the possibly were from hospital sample bank?
Answer: Samples are part of UNIFESP Biobank, but unfortunately we do not have information about diseases and medicaments.
Express the company details similarly than Amersham, cities and countries.
Answer: Changes were done as required ACE CD143 FITC (R&D Systems, Inc, Minneapolis, USA)
Results:
“Table 1 shows that older adults…..” The comparison between the present data and historical studies belongs to the Discussion.
Answer: Changes were done as required
Give also individual ages and gender of the subjects in the table 1.
Answer: The manuscript version sent to medrxiv@medrxiv.org had age and gender on tables, but due to their request, any possible variable that could identify the studied individual had to be removed. That is why in the present version these variables are not shown.
What means p-values here? Compared with which or interindividual differences in the particular variable? Should be explained
Answer: We used p-value for interindividual differences in each variable since individuals age differently (biological aging) and thus, physiological parameters could be affected by genetics, lifestyle, nutrition, and comorbidities. It is now explained in materials and methods
The numbering of tables and the text seems to me confusing. Only three tables, but in the text mentioned four. Number 4 does not exist.
Answer: For some reason table 2 is missing in the main text, please find the new version with Table 2 included
It would be good to have a list of abbreviations used in the description of the cell types for an unfamiliar reader.
Answer: In each figure and table we are now providing a description of cells evaluated.
Discussion:
A major part of the discussion deals with previous publications and not meaning or clinical significance of the present findings and comparison between the present and earlier studies.
Answer: The discussion was changed as suggested:
Our results show that for the studied population, chronological aging and biological aging don´t go at the same pace. Even individuals having a small chronological difference (64 to 67 years old), they are heterogeneous for physiological parameters such as glucose, urea, glycated hemoglobin (Hbglic), and C-reactive protein (CRP). Changes in the same functional parameters have been reported by Carlsson et al. [22] and Helmerson-Karlqvist [23] in healthy older adults. Carlsson’s study [22] found that CRP value was 2.6 with a coefficient variation of 1.4% whereas in our study, it was observed higher values of CRP in 5 out of 6 individuals. Increased CRP levels has been associated with inflammaging and our findings show that the studied population has changes in functional parameters which are likely associated with an inflammatory profile. [24] The link between RAS and inflammation has been suggested but its role is not completely clear under physiological and pathological conditions. [25, 26] In addition, the association between ACE1 altered expression in tissues (brain, muscle, heart and vessels) and the development and progression of age-related conditions such as Alzheimer’s, sarcopenia, and cardiovascular disease has been suggested but results are controversial. [17, 27, 28, 29, 30] There are few studies showing the association between ACE1 expression in cells from the immune system (monocytes, T cells) and the progression of kidney and cardiovascular disease. [9, 8, 31, 32]. Therefore, considering the lack of information on this issue, we questioned whether ACE1 (CD143) was highly expressed in cells from the immune system during the aging process. We found that ACE1 was expressed in almost 100% of T (CD4+, CD8+) and B lymphocytes and in all phenotypes of these cells. In non-lymphoid cells, ACE1 mean expression was 56,9%. In agreement with our findings, independent studies showed that T cells from healthy donors and monocytes from patients with congestive heart failure expressed ACE1, but there was no investigation on cell phenotype. [25, 26]. Our study is the first to show that either inexperienced (naive) or fully activated (memory) cells expresses ACE1. Our findings suggest a that the expression of ACE1 in lymphoid and non-lymphoid cells reflects the health status since our studied population presented changes in physiological parameters and high levels of ACE1 expression on immune cells. Previous independent studies showed that patients with unstable angina [32] or acute myocardial infarction [33] presented higher expression of ACE1 in T cells and dendritic cells than controls subjects. In addition, markers of the cell (lymphoid and non-lymphoid) functional status such as inflammatory or growth factors production could be modulated by ACE inhibitors (ACEi). Accordingly, mononuclear leukocytes from healthy subjects incubated with endotoxin exhibited high levels of tissue factor activity which was reduced in the presence of captopril in a dose-dependent pattern. This result could be related to the antithrombotic effect of ACEi. [34]. In patients with congestive heart failure, immune cells cultured with LPS secreted high levels of the pro-inflammatory TNF-alpha and these levels were significantly reduced in the presence of captopril. [35]
In those previous studies, also ACE2 has been reported, why not studied here?
Answer: Our next studies will be focused on ACE1 and ACE2 expression in cells from the immune system in both younger and older adults.
In the limitations, the Authors fairly mention the real problem: The small sample size, and I would like to say lack of younger subjects.
Answer: we agree with the limitations pointed and the text was changed as required:
This study have limitations such as the small sample size and the lack of young adults for comparison. As an example, the subject with the highest CRP and albumin also exhibited a high percentage of ACE1 expression on T (CD4+, CD8+), B and non-lymphoid cells in addition to the lowest percentage of CD4+ naive cells, and the highest percentage of CD8+ terminally differentiated (EMRA) and DN B cells. However, due to the small sample size it was not possible to associate the high expression of ACE1 on immune cells with inflammaging and immunosenescence. It would bring important information to correlate physiological parameters/health status with ACE1 expression and to find out whether age and associated chronic diseases could lead to increased ACE1 expression.
The COVID-19 point even tempting today, is too far from this study and unnecessary. Answer: Our point was to emphasize the negative impact of chronic diseases for the outcome of aging population during a viral infection and how ACE1/ACE2 expression could bring information to diagnosis and treatment. Therefore, we would like to maintain this piece of information.
Linguistic checking would improve the MS. Answer: We checked for possible linguistic mistakes
Reviewer, Heikki Vapaatalo:
I read with pleasure the very detailed answers to my comments.
I very warmly recommend acceptance of this MS for publications without any further notes.
Decision changed:
Verified manuscript: The content is scientifically sound, only minor amendments (if any) are suggested.
Dear Prof.Caruso Thank you very much for the revision of this manuscript. It is a privilege to have a manuscript reviewed by a research with high expertise on the field of ageing. Please find the answers to your questions and in the main text the changes in bold.
Sincerely yours,
Valquiria Bueno
The paper is essentially anecdotal because it studies the cells of 6 subjects without any comparison with other age groups. There is also a serious limitation because beyond the age and sex there is no information on the donors (how and why they were recruited, what drugs they took, etc.).
It is really a limitation to have only 6 individuals for the study, but they were the only ones fitting in the proposal of the manuscript. The samples were from a central bank of cells at UNIFESP and participants were considered “healthy” but there was not further information in addition to what we displayed on the tables of the manuscript. They were not living on homecares or hospitalized.
Our aim was to evaluate samples from individuals aged 60-69 years previously to COVID-19 and/or vaccination. In addition, there were no samples in the same conditions (PBMCs, -80oC) of young individuals and using fresh blood could bring a result that could not be compared mainly regarding to myeloid cells and B cells as is follows in the below reference. Braudeau C, Salabert-Le Guen N, Chevreuil J, Rimbert M, Martin JC, Josien R. An easy and reliable whole blood freezing method for flow cytometry immuno-phenotyping and functional analyses. Cytometry B Clin Cytom 2021;100(6):652-665. doi: 10.1002/cyto.b.21994.
Our goal from now on is to expand this study with young and old adults samples since it is important to understand whether ageing is associated with an increase in ACE expression on immune cells.
-To infer that chronological and biological ages do not match is inappropriate in the absence of the above information.
This piece of information regarding chronological and biological age was required by another reviewer. I agree that the concept does not match without more information on the donors. However, the information is now referenced and should be considered when older adults are studied. Vasto S, Scapagnini G, Bulati M, Candore G, Castiglia L, Colonna-Romano G, Lio D, Nuzzo D, Pellicano M, Rizzo C, Ferrara N, Caruso C. Biomarkes of aging. Front Biosci (Schol Ed) 2010;2(2):392-402. doi: 10.2741/s72. PMID: 20036955.
-However, the paper is of some interest because there are few studies on the topic.
Thanks for this positive comment. Few studies on the topic was the reason why we decided to send the manuscript for publication even though there were some important information on the donors missing and limited number of individuals.
Essential revisions that are required to verify the manuscript
1) Although we do not have data on donors, placing an age and gender column in all tables adds a minimum of useful information for the reader.
The first table submitted with age, but for requirement of MedRxiv, gender and age could no be linked to the metabolic results to preserve the anonymity of the donors.
2) Inflamm-ageing means low grade of inflammation. The value of CRP 23.1 suggests acute inflammation (also because albumin has high values, while in chronic inflammation its values decrease). Therefore the Ly averages do not have to take this subject into account.
Thank you for this comment. In a review of literature it was found an article (below) with CRP variation from 0.1 to 19.8 (Heumann Z, Youssim I, Kizony R, Friedlander Y, Shochat T, Weiss R, Hochner H, Agmon M. The Relationships of Fibrinogen and C-Reactive Protein With Gait Performance: A 20-Year Longitudinal Study. Front Aging Neurosci 2022;14:761948. doi: 10.3389/fnagi.2022.761948). There is also an article from your group showing CRPs <5g/dL and >5g/dL (Cancemi P, Aiello A, Accardi G, Caldarella R, Candore G, Caruso C, Ciaccio M, Cristaldi L, Di Gaudio F, Siino V, Vasto S. The Role of Matrix Metalloproteinases (MMP-2 and MMP-9) in Ageing and Longevity: Focus on Sicilian Long-Living Individuals (LLIs). Mediators Inflamm 2020;2020:8635158. doi: 10.1155/2020/8635158) that will be used to discuss how ageing impacts CRP levels. Considering the already small number of donors, data were maintained and statistics (mean + SD) with and without 23.1 mg/dL are now shown.
This will be the new version (discussion) about CRP Carlsson’s study [22] found that CRP value was 2.6 with a coefficient variation of 1.4% whereas in our study, it was observed higher values of CRP in 5 out of 6 individuals. In addition, it was shown by Cancemi et al. in an evaluation of individuals from 40 years to older than 95 years (long-living) that CRP increases in an age-dependent manner. Increased CRP levels has been associated with low grade of chronic inflammation (inflammaging) and our findings show that the studied population has changes in functional parameters which are likely associated with an inflammatory profile. [24] However, an individual presented CPR 23.1 mg/dL suggesting acute inflammation instead, but as all donors were not hospitalized or living on homecares, this sample was considered as part of the study. Another study evaluating gait speed found CRPs varying from 0.1 to 19.8mg/dL (Front Aging Neurosci 2022;14:761948.). Our study has an important limitation that is the lack of data on donors such as the use of continuous medicaments or sarcopenia, hypertension, cognition, among others, and thus it was not possible to correlate CRP with age-related conditions.
Table 1. Updated
Other suggestions to improve the manuscript The authors write that their findings suggest that ACE1 could play a role in several processes linked to aging including the generation and activation of autoimmune cells, due to the experimental evidence that inhibitors of ACE suppress the autoimmune process in a number of autoimmune diseases such as EAE, arthritis, autoimmune myocarditis. [49] They do not appear to have these findings in their paper. So, it needs to change the sentence.
Sentence changed to: According to experimental evidence, ACE inhibitors suppress the autoimmune process in a number of autoimmune diseases such as EAE, arthritis, autoimmune myocarditis. [49] Extrapolating these findings to our results, it is possible to suggest that ACE1 play a role in several processes linked to aging including the generation and activation of autoimmune cells.
Rviewer: Calogero Caruso
Decision changed:
Verified manuscript: The content is scientifically sound, only minor amendments (if any) are suggested.
JOHN SEELY BROWN
Pioneer in learning sciences too!
Also, the chances of breaking something are really high, because not even you remember how the code actually works.
Incidentally, I am also the author of the website you are referring to.
[author of the draft v4 validation spec here]
World Health Organization
If you want to write a book, you could dial down the scope andwrite a series of online articles outlining your main ideas. If youdon’t have time for that, you could dial it down even further andstart with a social media post explaining the essence of yourmessage.
This does make me wonder again, how much of this particular book might be found in various forms on Forte's website, much of which is behind a paywall at $10 a month or $100 a year?
It's become more common in the past decades for writers to turn their blogs into books and then use their platform to sell those books.
While the brief explanation provides information about the social context, more information about the creator and their intention would help understand more about the background of the object and its significance personally to the creator.
I second this statement. Description that sheds light on the personal relationship between the object and the creator creates an exclusive value to the object. Uncle Tom's Cabin Vase surely is not the only masterpieace created during the anti-slavery movements, so what makes it stand out can only boil down to the specific situation that inspired the creator to give birth to the it.
Gas! GAS! Quick, boys!
The author uses excitement to grab your attention
He notes that authors of such projects should consider the return on investment. It take time to go through community feedback, so one needs to determine whether the pay off will be worthwhile. Nevertheless, if his next work is suitable for community review, he’d like to do it again.
This is an apropos question. It is also somewhat contingent on what sort of platform the author "owns" to be able to do outreach and drive readers and participation.
Roland Barthes par Roland Barthes. This time, the index cards werealready there. One of the pages of illustrations of the volume reproduces three ofthem in facsimile. The text doesn’t comment on them, doesn’t even allude tothem. There is just a caption: “Reversal: of scholarly origin, the index card endsup following the twists and turns of the drive.”
In his book Roland Barthes par (by) Roland Barthes, Barthes reproduces three of his index cards in facsimile. The text doesn't comment or even allude to them, they're presented only with the captions "Reversal: of scholarly origin, the note follows the various twists and turns of movement." "...outside...", and "...or at a desk".
In this setting, the card index proves itself the most direct co-author as it physically appears in Barthes' autobiography!
the index card. This is despite the fact that itfunctions as such in a variety of different ways in relation to textualorganisation, composition and authorship. In the space that remains,I wish to tease out this idea of the index card as a creative agent inknowledge production by returning to reconsider the issue of theindex card as an archival or ‘mnemotechnical’ device.
The simple card index can serve a number of functions including as an archive, a mnemonic device, a teacher, an organizational tool, a composition device, a creativity engine, and an authorship tool.
According to Krapp, admissions like this, along with Barthes’inclusion of facsimiles of his cards in Roland Barthes by RolandBarthes, are all part of Barthes ‘outing’ his card catalogue as ‘co-author of his texts’ (Krapp, 2006: 363). The precise wording of thisformulation – designating the card index as ‘co-author’ – and theagency it ascribes to these index cards are significant in that theysuggest a usage that extends beyond mere memory aid to formsomething that is instrumental to the very organisation of Barthes’ideas and the published representations of these ideas.
Singh Chawla, D. (2022). Massive open index of scholarly papers launches. Nature. https://doi.org/10.1038/d41586-022-00138-y
Elm中的Virtual Dom是一个虚拟的Dom树,本质上是个JS的对象,里面保存了一些设定的属性。但是对象的创建过程并没有涉及到渲染。这样有效地增加了速度。在触发init和view时,就会产生一个真实的渲染好的拷贝。
所以不用担心全局的Model内容过多导致渲染过慢。
இதற்கு அப்பால் மிகச்சிலர் என்னுடைய கருத்துக்களுடன் முரண்பட்டு என் படைப்புக்களில் ஈடுபாடு கொண்டு வாசிப்பவர்களாக இருக்கிறார்கள். எவ்வாறு என இலக்கியம் எப்படி எழுதப்படுகிறதென தெரிந்துகொண்டால் அது உங்களுக்குப் புரியும். இலக்கியப் படைப்பில் ஒரு படைப்பாளி முன்வைப்பது அவன் கற்று, தெளிந்து ஏற்றுக்கொண்டவற்றை அல்ல. அவனுடைய திட்டவட்டமான நிலைபாட்டை அல்ல. இலக்கியப்படைப்பு என்பது ஒருவகை கனவு என எடுத்துக்கொள்ளுங்கள். மொழியில் நிகழும் ஒரு கனவு. ஒரு படைப்பு என்பது அந்த எழுத்தாளன் மொழிவழியாக கண்ட ஒரு கனவு. நம் கனவு நம் கட்டுப்பாட்டில் இல்லை அல்லவா? நாம் நினைப்பதே நம் கனவில் இருக்கவேண்டும் என்பதில்லை அல்லவா? எழுத்தாளன் எல்லாரையும்போல ஒரு அன்றாட உள்ளம் கொண்டவன். அவனுக்கு பல கருத்துக்கள் இருக்கும். அவை அவன் தன் வாழ்க்கையிலிருந்து பெற்றுக்கொண்டவையாக இருக்கும். அவனுக்கான ஓர் அரசியலும் இருக்கலாம். ஆனால் அவன் எழுதுபவை அப்படியே அந்த கருத்துக்களை வெளிப்படுத்துபவை அல்ல. சிலசமயம் நேர்மாறாகவும் இருக்கலாம். அவனே அறியாத நுட்பங்கள் அதில் வெளிப்படலாம். ஆகவே எழுத்தை எழுத்தாளனிடமிருந்து பிரித்தே பார்க்கவேண்டும். எழுத்து தன்னளவில் முழுமையான ஒன்று.
In an effort to mitigate these issues, some book contracts now specify the number of posts required before and after a book is published.
Perhaps better would be stipulations in the contract that incentivize authors to leverage their platforms in the form of bonuses while removing the advance money in lieu. Make the author part of the promotion, which has been part of the movement in publishing for the last decade.
“It’s become more and more important as the years went on,” said Marc Resnick, executive editor at St. Martin’s Press. “We learned some hard lessons along the way, which is that a tweet or a post is not necessarily going to sell any books, if it’s not the right person with the right book and the right followers at the right time.”
This seems like common sense to me, why hasn't the industry grokked it?
XII.
Poem XII's authorship is unknown. It was later published in a collection of Thomas Deloney's work, but Deloney's collections often contained the work of other poets. Hallett Smith has suggested that this poem out of the whole collection appears to readers as the most Shakespearean.
The author would like to acknowledge Professor Natalya Lusty, Univer-sity of Melbourne, for her guidance and support throughout the durationof the study.
This professor and the university probably helped with money and other research problems.
Lisa Heinze
There is only one author and her credentials are listed below her name. She is from Australia and studies gender and cultural studies at the University of Sydney.
Wow, Aaron himself just answered it!
answered Oct 12 '09 at 18:28
There’s a version of the “why writers should blog” story that is tawdry and mercenary: “Blog,” the story goes, “and you will build a brand and a platform that you can use to promote your work.”Virtually every sentence that contains the word “brand” is bullshit, and that one is no exception.
"Brand" is bullshit.
Some newspapers, most recently the New York Times, have forbidden writers from launching personal newsletters without permission.
Using their platform to build your own platform apparently isn't kosher any more?
We’re the ones who code. The people you talk to are the people who work on your project. When you have a question, we know what we are talking about.
Sorry to hear this Dan, but I might be able to help in terms of providing some perspective for moving forward.
These days the idea of bestseller means selling in the range of 10,000 books. The average book released these days sells only 250 copies, so if you're over that, you're doing well.
It's also incredibly uncommon for any publishers to put any serious money behind promoting their titles unless PR opportunities are falling off the trees for them. (This means that unless you've been selling a million copies of everything you write, they probably don't care.) Many publishers will assign you a pro-forma publicist to help when they can, but don't expect much from them. Most publishers will tell you to hire your own book publicist (usually for about $1,500-3,000 a month).
My guess is that the first run of your book was probably 1,000 to 2,000 books, which will bring the cost of raw printing down to $2 a copy. If you need copies of your book and they're remaindering them, you might offer the publisher $1-2 a copy plus shipping to get 50 or 100 copies for yourself for hand sales over the next decade (for speaking engagements, etc.) or selling a few copies from your own stash on platforms like Amazon, Abebooks, Alibris, etc. The cost of keeping a book in print these days is usually around $12 a year and then they print them on demand.
Some of the methods you mentioned, talks, online readings, etc. can be useful marketing for both you and your book(s). Look around your local community/state for book events, fairs, bookstores that invite authors, etc to supplement this.
Depending on your next title, it might be worth hiring a publicist if you're going the route of a text accessible to a broader public.Often this can be a reasonable risk but getting copies into reviewers' hands can be helpful, as can radio or print appearances. Another option is to pay for adds in appropriate print magazine outlets related to your material.
It's an uphill slog, but getting a publisher to take most of the risk and offering you all the free amenities of editing, proofreading, typesetting and distribution can be worth it in the end to get your material out.
When choosing your next publisher/editor, have a bit of this conversation with them at the outset to see what expectations they have for themselves. Don't tip your hand though by letting them know prior sales numbers.
Since you've got your own website/newsletter/social media presence, you should also look into affiliate accounts with the bigger online platforms. Chances are you're actually selling most of your own copies, you may as well get a 4% or larger cut of the referrals you're giving. Your link on this page alone could give you a reasonable little return on top of the boilerplate 7% you're probably getting from the publisher.
(Substack has courted a number of Times writers. I turned down an offer of an advance well above my Times salary, in part because of the editing and the platform The Times gives me, and in part because I didn’t think I’d make it back — media types often overvalue media writers.)
This is an important data point. Almost no one is putting any value on editing and other institutional support that outlets provide. Some writers can see at least a little bit of the future.
This new ability of individuals to make a living directly from their audiences isn’t just transforming journalism. It’s also been the case for adult performers on OnlyFans, musicians on Patreon, B-list celebrities on Cameo. In Hollywood, too, power has migrated toward talent, whether it’s marquee showrunners or actors. This power shift is a major headache for big institutions, from The New York Times to record labels. And Silicon Valley investors, eager to disrupt and angry at their portrayal in big media, have been gleefully backing it. Substack embodies this cultural shift, but it’s riding the wave, not creating it.
This has always been the case, especially in Hollywood. The problem becomes that everyone thinks they can become rich and famous too. Talent shows like American Idol show us that this is rarely the case. Building a platform for oneself is not an easy thing to do, even if you've got the talent.
Part I: viral entry, sensing and evasion
Ester Gea-Mallorquí is co-corresponding author, email: ester.gea-mallorqui@ndm.ox.ac.uk
Viral Immunology Unit, Nuffield Department of Medicine, University of Oxford, Oxford, UK
Corresponding email: ester.gea-mallorqui@ndm.ox.ac.uk
,
Corresponding author: ester.gea-mallorqui@ndm.ox.ac.uk
answered May 9 '13 at 15:29 alexander farkas
Posting an issue on the discussion boards for a three year old game, yesterday, I wasn't holding my breath for a reply. Earlier, this morning, a dev. responded, stating they'd look at fixing it, and it was just a few hours before it were sorted!
After being denied admission at three colleges
Stuart's elementary school was Plum Grove School, where an intense love of learning was instilled in him (his father also instilled this love of learning in him) https://etd.ohiolink.edu/apexprod/rws_olink/r/1501/10?clear=10&p10_accession_num=bgsu1554464085296459.
short stories, poetry, and novels as well as non-fiction autobiographical works
No one has requested it before so it's certainly not something we're planning to add.
To give a little more context, structures like this often come up in my work when dealing with NoSQL datastores, especially ones that rely heavily on JSON, like Firebase, where a records unique ID isn't part of the record itself, just a key that points to it. I think most Ruby/Rails projects tend towards use cases where these sort of datastores aren't appropriate/necessary, so it makes sense that this wouldn't come up as quickly as other structures.
NO support whatsoever will be given for the moment unless I gave you the program personally. This is because all of this is work in progress and I can't code while constantly writing documentation and answering questions.
DEV actively answers questions in the community.
Summary:
The strengths of the study are the findings that a single oxytocin level measured from saliva or plasma is not meaningful in the way that the field might currently be measuring. The reviewers appreciated this finding, and the careful attention to detail, but felt that the results fell short.
Reviewer #1:
This article describes the investigation of a valuable research question, given the interest in using salivary oxytocin measures as a proxy of oxytocin system activity. A strength of the study is the use of two independent datasets and the comparison between intranasal and intravenous administration. The authors report poor reliability for measuring salivary oxytocin across visits, that intravenous delivery does not increase concentrations, and that salivary and blood plasma concentrations are not correlated.
Line 77-78: While it's true that saliva collection provides logistical advantages, there are also measurement advantages (e.g., relatively clean matrix) that are summarised in the MacLean et al (2019) study, which has already been cited.
Thanks for the suggestion. We added this advantage:
Line 101 “Compared to blood sampling, saliva collection presents several logistical and measurement advantages (i.e. relatively clean matrix)(1).”
Line 86: It is important to note that the 1IU intravenous dose in this study led to equivalent concentrations in blood compared to intranasal administration.
The reviewer is right that 10 IU (over 10min) in our case increased the concentrations of plasmatic oxytocin beyond those observed for the spray or nebuliser (we reported the full time-course of variations in plasmatic oxytocin in another manuscript we published earlier this year)(2). This was an intentional aspect of our study design. We decided to use the highest intravenous dose (at the highest rate of 1IU/min) that we could get permission to administer safely in healthy volunteers as a proof of concept, so as to achieve a robust and prolonged increase in plasmatic oxytocin over the course of our full testing session. In this manner, we demonstrate that even when plasmatic levels of OT are maintained substantially increased throughout the observation interval, we cannot detect increases in salivary oxytocin. In this aspect, we believe that our manuscript goes one step beyond the important findings described in of Quintana et al. 2018(3), showing that this phenomenon is not linked to dosage (or to amount of increase in plasmatic levels of exogenous OT), as far as we can determine given the current safety standards for the administration of OT IV.
Please see also response to Reviewer 2, point 1.
Line 158: When using both ELISA and HPLC-MS, extracted and unextracted samples are correlated when measuring oxytocin concentrations in saliva, at least in dogs. (https://doi.org/10.1016/j.jneumeth.2017.08.033).
Thanks for pointing out this study. Indeed, in this specific study the authors found correlations between extracted and unextracted saliva samples. Such associations in humans have nevertheless been rare. In humans, the body of evidence suggests that the measurements obtained when comparing extracted samples to unextracted samples, or when comparing samples obtained using different methods of quantification (for instance, ELISA versus radioimmunoassay), do not correlate or show very low correlations (4, 5). Furthermore, most ELISA kits and HPLC-MS protocols to measure oxytocin have so far fallen short on sensitivity to detect the typical concentrations observed in humans at baseline (0-10pg/ml)(6). The current gold-standard method for quantifying oxytocin in biological fluids is the radioimmunoassay we used in this study(4). This method has shown superior sensitivity and specificity when compared to other quantification methods, when combined with extracted samples; therefore, it was our primary choice. We now highlight this advantage in the revised version of the manuscript more explicitly.
Line 129 “For all analyses, we followed current gold-standard practices in the field and assayed oxytocin concentrations using radioimmunoassay in extracted samples, which has shown superior sensitivity and specificity when compared to other quantification methods(7).”
Statistical reporting: I ran the article through statcheck R package (a web version is also available) and found a number of inconsistencies with the reported statistics and their p values. For example, on Line 302 the authors reported: t(123) = 1.54, p = 0.41, but this should yield a p value of 0.13. The authors should do the same and fix these errors.
Thanks very much for taking the time to check our statistical reporting thoroughly. We apologize if we were not sufficiently clear in the previous version of the manuscript, but the p-values we reported are corrected for multiple comparisons using Tukey correction. Currently, statcheck can only evaluate inconsistencies when the results are reported in the standard APA style and does not take into consideration corrections for multiple comparisons of any kind. We did check all of our statistical reporting and the p-values and correspondent statistics are correct (we only corrected an inadvertent error in reporting the degrees of freedom for these tests). In any case, we have now clarified in the manuscript when the reported p-values have been adjusted for multiple comparison to avoid any further confusion.
Line 305: The confidence intervals for these correlations should be reported.
We have now added the confidence intervals, estimated using bootstrapping, in our results section.
Line 348: This is an important point, but it's important to note that the vast majority of these studies use plasma or saliva measures. Perhaps CSF measures are more reliable, but the question wasn't assessed in the present study, and I'm not sure if anyone has looked at this question.
We are not aware of any study evaluating the stability of measurements of oxytocin in the CSF. Indeed, there are only a few studies sampling CSF to measure oxytocin in clinical patients and it is unlikely that CSF will become a widely used fluid to measure oxytocin in humans, given the invasiveness of the procedure to obtain CSF samples. Here, we wanted to refer specifically to saliva and plasma, which remain as the most popular options for measuring oxytocin in humans and which we investigated specifically in the current study. We have changed the text accordingly for clarity.
Line 466 “Our data poses questions about the interpretation of previous evidence seeking to associate single measurements of baseline oxytocin in saliva and plasma with individual differences in a range of neuro-behavioural or clinical traits.”
Line 423: I broadly agree with this conclusion, but it should be added that "single measurements of baseline levels of endogenous oxytocin in saliva and plasma are not stable under typical laboratory conditions" Perhaps these measures can be more stable using other means (i.e., better standardising collection conditions). But the fact remains, under typical conditions these measures do not demonstrate reliability.
Thanks for the suggestion. We have revised the text accordingly throughout the manuscript (examples below). Our study is a pharmacological study, which means that it is conducted in a highly controlled setting and adheres to strict protocols (i.e. we tested participants at the same time of the day, we instructed participants to abstain from alcohol and heavy exercise for 24 h and from any beverage or food for 2 h before scanning). These exclusion criteria were stricter than those applied in a large number of studies sampling saliva and plasma for measuring oxytocin for the purposes estimating possible associations with various traits associating. Most of these studies do not control, for instance, for fluid or food ingestion. Therefore, we expected our reliability calculations to represent an optimistic estimate of the reliabilities of the salivary and plasmatic oxytocin concentration used in most studies.
For now, it remains unclear to us what factors might be driving the within-subject variability in salivary and plasmatic concentrations we report in this study. Thanks to Reviewer 3, we are now confident that this is unlikely to represent measurement error (see response to Reviewer 3, point 3).
Line 117 “Here, we aimed to characterize the reliability of both salivary and plasmatic single measures of basal oxytocin in two independent datasets, to gain insight about their stability in typical laboratory conditions and their validity as trait markers for the physiology of the oxytocin system in humans.”
Line 567 “In summary, single measurements of baseline levels of endogenous oxytocin in saliva and plasma as obtained in typical laboratory conditions are not stable and therefore their validity as trait markers of the physiology of the oxytocin system is questionable.”
Reviewer #2:
Summary:
To test questions whether salivary and plasmatic oxytocin at baseline reflect the physiology of the oxytocin system, and whether salivary oxytocin index its plasma levels, the authors quantified baseline plasmatic and/or salivary oxytocin using radioimmunoassay from two independent datasets. Dataset A comprised 17 healthy men sampled on four occasions approximately at weekly intervals. In the dataset A, oxytocin was administered intravenously and intranasally in a triple dummy, within-subject, placebo-controlled design and compared baseline levels and the effects of routes of administration. With dataset A, whether salivary oxytocin can predict plasmatic oxytocin at baseline and after intranasal and intravenous administrations of oxytocin were also tested. Dataset B comprised baseline plasma oxytocin levels collected from 20 healthy men sampled on two separate occasions. In both datasets, single measurements of plasmatic and salivary oxytocin showed insufficient reliability across visits (Intra-class correlation coefficient: 0.23-0.80; mean CV: 31-63%). Salivary oxytocin was increased after intranasal administration of oxytocin (40 IU), but intravenous administration (10 IU) does not significantly change. Saliva and plasma oxytocin did not correlate at baseline or after administration of exogenous oxytocin (p>0.18). The authors suggest that the use of single measurements of baseline oxytocin concentrations in saliva and plasma as valid biomarkers of the physiology of the oxytocin system is questionable in men. Furthermore, they suggest that saliva oxytocin is a weak surrogate for plasma oxytocin and that the increases in saliva oxytocin observed after intranasal oxytocin most likely reflect unabsorbed peptide and should not be used to predict treatment effects.
General comments:
The current study tested research questions relevant for the study field. The analyses in two independent datasets with different routes of oxytocin administrations is the strength of current study. However, the limited novelty of findings and several limitations are noticed in the current report as described below.
Specific and major comments:
1) Previous study with similar results has already revealed that saliva oxytocin is a weak surrogate for plasmatic oxytocin, and increases in salivary oxytocin after the intranasal administration of exogenous oxytocin most likely represent drip-down transport from the nasal to the oral cavity and not systemic absorption (Quintana 2018 in Ref 13). Therefore, the novelty of current findings is limited. The authors should more clearly state the novelty of current results and the replication of previous findings.
We apologize for not describing the novelty and impact of our findings with sufficient clarity, and thanks for the opportunity to do so. Our study had two major goals. The first was to investigate whether single measurements of salivary and plasmatic concentrations of oxytocin can be reliably estimated within the same individual when collected at baseline conditions (i.e. without any experimental manipulation). As the reviewer highlighted, this is an important methodological question given the wide use of these measurements in a large and increasing number of studies to establish associations between the physiology of the oxytocin system and a number of brain and behavioural phenotypes in both clinical and non-clinical samples. However, to our knowledge, no previous study has appropriately conducted a thorough investigation of the reliability of these measurements (see also response to Reviewer 3, point 5). Thanks to our study, we now know that when single measurements are collected at baseline, salivary and plasmatic oxytocin cannot provide a sufficiently stable trait marker of the physiology of the oxytocin system in humans. As we highlight in the manuscript, this finding should deter the field from making strong claims based exclusively on associations of phenotypes with single measurements of peripheral oxytocin concentrations. Furthermore, our study also describes two very concrete implications of our findings which we believe are very important for the field. First, if baseline level of OT is to be used as a trait marker, future studies should, as much as possible, rely on repeated measures within the same participant but collected on different days to maximize reliability. Second, this less than perfect reliability should be taken into consideration when calculating the sizes of the samples needed to detect a certain effect, if it exists, with sufficient statistical power.
The second goal of our study was, as pointed out by the reviewer, to revisit the findings of Quintana et al. 2018(3), but this time with two major design modifications which could strengthen the conclusions from that study. The first modification was the dose of intravenous oxytocin administered, which was considerably higher (see response to Reviewer 1, point 2). The administration of a higher dose that resulted in substantial and sustained increases in plasmatic oxytocin throughout the two hours observation period can only strengthen the previous conclusion that increases in plasmatic oxytocin cannot be detected in salivary measurements, and that this is not a matter of dose (as far as we can ascertain by administering the maximum intravenous dose we could safely administer in healthy volunteers). We believe that this is an important addition to the literature.
The second modification regarded the choice of the method we used to quantify oxytocin. In this study, we used radioimmunoassay, which is superior to ELISA in sensitivity and hence more appropriate to measure the low concentrations of oxytocin in saliva and plasma typically detected in humans at baseline conditions (1-10 pg/ml; for most individuals 1-5 pg/ml)(6). For instance, in Quintana et al. 2018(3) the limitations in the sensitivity of the ELISA kit used led the authors to discard around 50% of the collected saliva samples. Hence, our study replicates and extends the previous findings from Quintana et al. 2018 in important ways, demonstrating that the lack of an association between increases plasmatic oxytocin and salivary measurements is not limited by the dose of intravenous oxytocin administered or limitations of the sensitivity of the method used to quantify oxytocin.
We have now made the novelty and contribution of our work more explicit:
*Line 77 “Currently, we lack robust evidence that single measures of endogenous oxytocin in saliva and plasma at rest are stable enough to provide a valid trait marker of the activity of the oxytocin system in healthy individuals. Indeed, previous studies have claimed within-individual stability of baseline plasmatic and salivary concentrations of oxytocin in both adults and children based on moderate-to-strong correlations between salivary and plasmatic oxytocin concentrations measured repeatedly within the same individual over time using ELISA in unextracted samples(14-16). However, these studies have a number of methodological limitations that raise questions about the validity of their main conclusion that baseline plasmatic and salivary concentrations are stable within individuals. First, measuring oxytocin in unextracted samples has been postulated as potentially erroneous, given the high risk of contamination with immunoreactive products other than oxytocin(4). It is conceivable that these non-oxytocin immunoreactive products might constitute highly stable plasma housekeeping proteins (17) that masked the true variability in oxytocin concentrations. Second, a simple correlation analysis cannot provide information about the absolute agreement of two sets of measurements – which would be a more appropriate approach to study within-subject reliability/stability. Third, it is not clear whether these findings generalize beyond the early parenting(14) or early romantic(15) periods participants were in when the studies were conducted, since these periods engage the activity of the oxytocin system in particular ways(18). Hence, establishing the validity of salivary and plasmatic oxytocin as trait markers of the activity of the oxytocin system in humans remains as an unmet need. Such evidence is urgently required, given reports that plasma and saliva levels of oxytocin are frequently altered during neuropsychiatric illness and that they co-vary with clinical aspects of disease(13).”
Line 509 “Our findings were not consistent with these expectations. We could replicate previous evidence that intravenous oxytocin does not increase salivary oxytocin(3) and extended it by showing that the lack of increase in salivary oxytocin is not limited to the specific low dose of intravenous OT that was previously used (1IU) and that it is not driven by the insufficient sensitivity of the OT measurement method (which had resulted in more than 50% of the saliva samples being discarded in the previous study(3).”*
2) As authors discussed in the limitation section of discussion, the current study has several limitations such as analyses only in male participants and non-optimized timing of collection of saliva and blood due to the other experiments. These limitations are understandable, because the current study was the second analyses on the data of the other studies with the different aims. However, these limitations significantly limit the interpretations of the findings.
Here, we would like to highlight two aspects. First, most studies in the field are indeed conducted in men to avoid potential confounding from fluctuations in oxytocin concentrations across the menstrual cycle in women. Therefore, our study is representative of the typical samples used in most human studies. Second, we did not optimize our study to collect repeated samples of saliva. Indeed, it would have been interesting to describe the full-time course of variations of oxytocin concentrations in saliva after intranasal and intravenous administration. However, this does not detract the importance of our findings in respect to our first aim (which was our main goal).
We agree with the reviewer though that it is at least theoretically possible that we could have missed the window for increases in salivary oxytocin after intravenous oxytocin if it existed, given that we only sampled one post-administration time-point. However, we believe this was unlikely for one reason. Despite the sustained increase (throughout the two-hour observation interval) in plasmatic oxytocin following the intravenous administration of oxytocin, we observed no increase in salivary oxytocin post-dosing (at ~115 min). Unless the half-life of oxytocin is shorter in saliva than in the blood (which we do not know yet), we expected the levels of salivary oxytocin to mirror the changes in plasma – potentially with a slight delay given the time that it might take for oxytocin concentrations to build up in saliva through ultrafiltration from the blood, but this was not the case. Most likely the half-life of oxytocin in the saliva is not shorter than in the blood, since a previous study found increased concentrations of oxytocin in saliva up to 7h after administration of intranasal oxytocin (as the reviewer pointed out below, in our study we no longer could detect significant increases in plasmatic oxytocin after the intranasal administration of 40 IU with two different methods at around 115 mins post-administration). Therefore, while we acknowledge these limitations we also believe they do not detract from the importance of our main findings and the potential they hold to influence the field towards a more rigorous use of these measurements. Please see below for the implemented changes in the text.
Line 554 “It is possible that we may have missed peak increases in saliva oxytocin after the intravenous administration of exogenous oxytocin if they occurred between treatment administration and post-administration sampling. This is unlikely given that the dose we administered intravenously resulted in sustained increases in plasmatic oxytocin over the course of two hours. Unless the half-life of oxytocin in saliva is much shorter than in the plasma, it would be surprising to not find any increases in salivary oxytocin after intravenous oxytocin given that concentrations of oxytocin in the plasma were still elevated at the specific time-point of our second saliva sample. Currently, we have no estimate for the half-life of oxytocin in saliva; however, given that previous studies have found evidence of increased salivary oxytocin after single intranasal administrations of 16IU and 24IU oxytocin up to seven hours post-administration(19), it is unlikely that the half-life of oxytocin is shorter in the saliva than in the plasma.”
3) As reported in page 6, the dataset A comprises administrations approximately 40 IU of intranasal oxytocin and 10 IU on intravenous. The rationale to set these doses should be described. Since the 40IU is different from 24 IU which is employed in most of the previous publications in the research field, potential influence associated with the doses should be tested and discussed.
Thank you for the opportunity to clarify this aspect of our work. With respect of our primary aims (to investigate whether single measurements of salivary and plasmatic oxytocin at baseline can be reliably measured within individuals across different days), the choice of doses is of course not relevant.
With respect to our secondary aim, namely, to investigate whether salivary oxytocin can be used to index concentrations of oxytocin in the plasma, particularly after the administration of synthetic oxytocin using the intranasal and intravenous routes, the administered doses are relevant.
The data reported here were collected as part of a larger project – which determined the choice of both intranasal and IV doses (2). As explained in our response to Reviewer 1, point 2, the selection 10IU (over 10min) was the highest intravenous dose that we could get permission to administer safely in healthy volunteers as a proof of concept, so as to achieve a robust and prolonged increase in plasmatic oxytocin over the course of our full testing session. In this manner, we demonstrate that even when plasmatic levels of OT are maintained substantially increased throughout the observation interval, we cannot detect increases in salivary oxytocin.
Regarding the intranasal OT dose, it is worth noting that the 24 IU is indeed popular in oxytocin studies, but not exclusive, and generally the selection of dose in oxytocin studies has not been informed by detailed dose-response characterizations. Our choice of 40IU was made for the purposes of matching our previous work on the pharmacodynamics of OT in healthy volunteers(20), and is a dose we (21-29) and others (e.g. (30)) have commonly used with patients.
A potentially important implication if dose variations also imply variation in the total volume of liquid administered (as is usually the case with standard nasal sprays – but not with the nebuliser), then it is likely that the potential for drip-down might increase for higher volumes and decrease for lower volumes. As far as we know, no study has ever investigated the impact of administered volume on salivary oxytocin after the intranasal administration of synthetic oxytocin, but we agree this would be an important point to look at. We have now expanded our discussion to accommodate this point.
Line 519 “We expect this phenomenon to be particularly pronounced for higher administered volumes. Further studies should examine the impact of different administered volumes on increases in salivary oxytocin.”
4) It is difficult to understand that no significant elevations in plasma oxytocin levels were observed after intranasal spray or nebuliser of oxytocin. From figure 4A, the differences between levels at baseline and post administration are similar between nebuliser, spray, and placebo. Please discuss the potential interpretation on this result.
The plasmatic concentrations of oxytocin we report in this study refer solely to the samples acquired at around 2h after the administration of intranasal oxytocin. We reported the full-time course of changes in plasmatic oxytocin in a paper published earlier this year(2) – which we now refer the reader to. We did find increases in plasmatic oxytocin after administration of oxytocin with the spray and nebuliser (around 3x the baseline concentrations) that did not differ between intranasal methods of administration. Plasmatic oxytocin reached a peak within 15 mins from the end of the intranasal administrations. Given the short half-life of oxytocin in the plasma, we believe it is not surprising that at 115 mins after the end of our last treatment administration the concentrations of oxytocin in the plasma are no longer different from the placebo condition.
Line 166 “The full time course of changes in plasmatic oxytocin after the administration of intranasal and intravenous oxytocin in this study has been reported elsewhere(2).”
5) In page 12, the reason why not to employ any correction for multiple comparisons in the statistical analyses should be clarified.
We apologize that this was not sufficiently clear, but we did correct for multiple testing using the Tukey procedure in our analyses investigating the effects of treatment on salivary and plasmatic oxytocin (this was described in page 9 – Treatment effects). If the reviewer meant something else, we would be glad to follow any further advice on multiple testing correction he/she might have.
Line 250 “Treatment effects: The effect of treatment on blood/saliva oxytocin concentration were assessed using a 4 x 2 repeated-measures two-way analysis of variance Treatment (four levels: Spray, Nebuliser, Intravenous and Placebo) x Time (two levels: Baseline and post-administration). Post-hoc comparisons to clarify a significant interaction were corrected for multiple comparisons following the Tukey procedure.”
Reviewer #3:
In the current study, baseline samples of salivary and plasma oxytocin were assessed in 13, respectively, 16 participants, to assess intra-individual reliability across four time points (separated by approximately 8 days). The main results indicate that, while as a group, average salivary and plasma samples were not significantly different across time points, within-subject coefficient of variation (CV) and intra-class correlation coefficient (ICC) showed poor absolute and relative reliability of plasma and salivary oxytocin measurements over time. Also no association was established between plasma and salivary levels, either at baseline or after administration of oxytocin (either intranasally, or intravenously). Further, salivary/ plasma oxytocin was only enhanced after intranasal, respectively intravenous administration.
The study addresses an important topic and the paper is clearly written. While the overall multi-session design seems solid, sample collections were performed in the context of larger projects and therefore there appear to be several limitations that reduce the robustness of the presented results and consequently the formulated conclusions.
General comments
1) A main conclusion of the current work is that 'single measures of baseline oxytocin concentrations in saliva and plasma are not stable within the same individual'. It seems however that the study did not adhere to a sufficiently rigorous approach to put forward this conclusion. It lacks a control for several important factors, such as timing of the day at which saliva/ plasma samples were obtained, as well as sample volume. Particularly while it is indicated that all visits were identical in structure, important information is missing with regard to whether or not sampling took place consistently at a particular point of time each day, to minimize the influence of circadian rhythm. Without this information it is not possible to draw any firm conclusions on the nature of the intra-individual variability as demonstrated in the salivary and plasma sampling.
Thanks for pointing this out. Indeed, we were not sufficiently explicit on how strict we were in controlling for some potential sources of variability that could have contributed to the lack of reliability we report here. Our data was acquired in the context of two human pharmacological studies, which by design were strict on a number of aspects to minimize unwarranted noise. All participants were tested in the same period of the day (morning) to avoid the potential contribution of circadian fluctuations of oxytocin. In dataset A, we tried, as much as possible, to match the exact time participants were tested between visits, using the start time of the first visit as a reference. With the exception of one participant, where one session was conduct 1h and 30 mins later than the other three, all the remaining participants from study A were tested within 1h of the exact start time of session 1. Further, we also instructed participants to abstain from alcohol and heavy exercise for 24 h and from any beverage or food for 2 h before scanning. Hence, we believe our sampling protocol was strict enough to discard any potential contribution of major known sources of variability in oxytocin levels.
The reviewer also inquiries about the volume of the samples. For the plasma samples, we used a standardized protocol and collected the same blood volume in all participants, visits and time-points (1 EDTA tube of approximately 4 ml). The saliva samples were collected using Salivettes. Participants were instructed to place the swab from the Salivette kit in their mouth and chew it gently for 1 min to soak as much saliva as possible. After this, the swab was then returned back to the Salivette and centrifuged. In both cases, to avoid degradation of the peptide in the collected sample, we followed a strict protocol where all samples were put immediately in iced water until centrifugation, which happened within 20 mins of sample collection. Samples were then immediately stored at -80C until analysis. Hence, differences in degradation of the peptide related to the processing of the sample are also unlikely to justify the poor reliabilities we report here.
For completeness, we have now added all of these further details to our Methods section.
Line 169 “**All visits were conducted during the morning to avoid the potential confounding of circadian variations in oxytocin levels(31, 32). In addition, we also made sure that each participant was tested at approximately the same time across all four visits (all participants were tested in sessions with less than one hour difference in their onset time, except for one participant where the difference in the onset of one session compared to the other three sessions was 1.5h). “*
Line 192 “Blood was collected in ethylenediaminetetraacetic acid vacutainers (Kabe EDTA tubes 078001), placed in iced water and centrifuged at 1300 × g for 10 minutes at 4°C within 20 minutes of collection and then immediately pipetted into Eppendorf vials. Samples were immediately stored -80C until analysis. Saliva samples were collected using a salivette (Sarstedt 51.1534.500). Participants were instructed to place the swab from the Salivette kit in their mouth and chew it gently for 1 min to soak as much saliva as possible. After this, the swab was then returned back to the Salivette, centrifuged and stored in the same manner as blood samples. For both saliva and plasma, we stored the samples in aliquots of 0.5 ml, following the RIAgnosis standard operating procedures. We followed this strict protocol, putting all samples in iced water until centrifugation with immediate storage at -80C until analysis to minimize the impact putative differences in degradation of the peptide related to differences in the processing of the samples might have on the reliability of the estimated concentrations of oxytocin.” *
Correspondingly, a deeper discussion is needed on the reason why ICC's were considerably variable across pairs of assessment sessions, with some pairs yielding good reliability, whereas others yielded (very) poor reliability.
Currently we have no insightful hypothesis on why this could have been the case. Indeed, we found higher ICCs for only 2 out of 6 pairs of visits for the plasma. However, it is plausible that this might have occurred by chance. In any case, we should note that the 95% confidence intervals for the ICCs of our different pairs of samples overlap; this suggests that there is no evidence that the ICCs we estimated for the specific two pairs where we found higher reliabilities are significantly higher than those observed in the remaining pairs.
Line 431 “If there are specific reasons explaining the higher reliability indices observed for the specific pairs of sessions, these reasons remain to be elucidated. However, it is not implausible that we might have found higher reliabilities for these specific two pairs by chance, since the 95% confidence intervals for the ICCs for all pairs of samples overlapped.”
More detailed descriptions regarding sampling procedures (timing and sampling intervals) are necessary. Also, more information is needed on the volume of saliva collected at each session, to control for possible dilution effects.
This information has been added to the revised version of the manuscript (please see response to your point number 1). As a further clarification, oxytocin concentrations were measured in plasma and saliva aliquots of 0.5 ml, following the standard operating procedures of RIAgnosis. This volume was used for all participants, sessions and time-points. Furthermore, for measuring cortisol, the salivettes were shown to allow for an almost 100% recovery, regardless of cortisol concentration, volume of the sample or method of quantification(33), suggesting that the sampling method is robust.
2) It is indicated that the initial sample would allow to detect intra-class correlation coefficients (ICC) of at least 0.70 (moderate reliability) with 80% of power. Is this still the case after the drop-outs/ outlier removals? Since the main conclusions of the work rely on negative results (conclusions drawn from failures to reject the null hypothesis) it is important to establish the risk for false negatives within a design that is possibly underpowered.
We understand the concern of the reviewer. However, according to the power calculations provided by Bujang and Baharum, 2017(34), the four repeated samples we collected in Dataset A would have allowed us to detect an ICC of 0.5 with 80% of statistical power even with only 13 subjects (which is the lowest sample size we used for the analysis on saliva in dataset A). The two samples we collected in Dataset B would allow us to detect an ICC of 0.6 with 80% of statistical power even with only 19 subjects. Hence, both datasets were powered to detect an ICC of 0.7 with acceptable power, if it existed, even after the exclusion of outliers.
3) Did the authors also assess within-session reliability? For example, by assessing ICC between pre and post-measurements in the placebo session.
Thanks for the suggestion. Indeed, we had not performed this analysis before but we agree it would be informative. We calculated the ICC and CV for the two samples acquired before any treatment administration and the intravenous infusion of saline during the placebo session. These samples where acquired with an approximate 15 min interval in between them. In this analysis, we found that the ICC was excellent 0.92 and the CV 20%. This additional analysis strengthens our findings by supporting the idea that our poor reliabilities across different days reflect true biological variability and cannot be attributed to measurement error. These new findings have now been included in the revised version of the manuscript.
Abstract
Line 44 "Results: Single measurements of plasmatic and salivary oxytocin showed poor reliability across visits in both datasets. The reliability was excellent when samples were collected within 15 minutes from each other in the placebo visit.”
Line 240 “Within-visit reliability analysis: To investigate the reliability of salivary and plasmatic oxytocin concentration within the same visit, we calculated the ICC and CV as described above for two samples acquired before any treatment administration and the intravenous infusion of saline during the placebo session. These samples where acquired with an approximate 15 minutes interval in between them.”
Line 405 “Furthermore, in a further analysis assessing the within-session stability of plasmatic oxytocin using two measurements collected 15 minutes apart from each other in the placebo visit (one sample collected at baseline and the other after the intravenous administration of saline), we found excellent within-session reliability (ICC=0.92, CV=20%). Together, this suggests that the low reliability of endogenous oxytocin measurements across visits in the current study results from true intrinsic individual biological variability and not technical variability/error in the method used for oxytocin quantification.“*
4) It is indicated that the intra-assay variability of the adopted radioimmunoassay constitutes <10%. Were analyses of the current study run on duplicate samples? Was intra-assay variability assessed directly within the current sample?
We reported the intra-assay variability determined by RIAgnosis during the development of this assay(35). This was not specifically assessed for the current study.
Introduction & Discussion
5) The introduction and discussion is missing a thorough overview of previous studies assessing intra-individual variability in oxytocin levels.
Thanks for the suggestion. We have now included in our introduction/discussion an overview of previous studies attempting to tackle this question, which unfortunately do not address this question with sufficient detail or using the appropriate methods and statistical analyses (see response to Reviewer 2, point 1). Hence, from the available evidence, it is not possible to draw robust conclusions about the validity of concentrations of oxytocin in saliva and plasma as valid trait markers of the activity of the oxytocin system. With this manuscript, we hope we can prompt further discussion and guide the field towards a more rigorous use of these measurements. A thorough discussion of this literature has now been added to the Introduction and Discussion.
Line 434 “Our observation of poor reliability questions the use of single measurements of baseline oxytocin concentrations in saliva and plasma as valid trait markers of the physiology of the oxytocin system in humans. Instead, we suggest that, at best, these measurements can provide reliable state markers within short time-intervals (5 mins in our study). Our data does not support previous claims of high stability of plasmatic and salivary oxytocin within individuals over time. For instance, in one study, Feldman et al. (2013) assessed plasmatic oxytocin in recent mothers and fathers at two time-points spaced six months apart during the postpartum period. The authors found strong correlations between the two assessments for both mothers and fathers(14). In another study, Schneiderman et al. (2012) found strong correlations between plasmatic oxytocin concentrations measured at two different instances spaced six months apart in both single and individuals recently involved in a new romantic relationship(15). Two important differences between these studies and ours are i) the method used for oxytocin quantification, and ii) the particular states participants were in when the studies were conducted. Regarding the first difference, these previous studies used ELISA without extraction, reporting concentrations of plasmatic oxytocin well above the typical physiological range of 1-10 pg/ml detected in extracted samples (in their studies, the authors report concentrations above 200 pg/ml). The inclusion of extraction has been postulated as a critical step for obtaining valid measures of oxytocin in biological fluids(4). Unextracted samples were shown to contain immunoreactive products other than oxytocin(4), which contribute largely to the concentrations of oxytocin estimated by this method. It is possible that these non-oxytocin products might represent highly stable plasma housekeeping molecules(17) that masked the true biological variability in oxytocin concentrations between assessments in these previous studies that we could detect in extracted samples in our study. Regarding the second difference, these previous studies on within-individual stability were conducted during the early parenting(14) or early romantic(15) periods, which engage the activity of the oxytocin system in particular ways(18). Instead, we used a normative sample that did not specify these inclusion criteria. Hence, we cannot exclude that during these specific periods the reliability of salivary and plasmatic oxytocin concentrations might be higher. We note though that our sample more closely resembles the samples used the vast majority of studies in the field (which sometimes even exclude participants during early parenthood(36)). Hence, our estimates of reliability are a better starter point for all studies where specific circumstances potentially affecting the activity of the oxytocin system have not been specified a priori.”
6) The paper misses a discussion of previous studies addressing links between salivary/ plasma levels and central oxytocin (e.g. in cerebrospinal fluid). I understand the claim that salivary oxytocin cannot be used to form an estimate of systemic absorption, although technically, a lack of a link between salivary and plasma levels, does not necessarily imply a lack of a relationship to e.g. central levels. The lack of effect is limited to this specific relationship.
In this study, we did not intend to investigate whether salivary and plasmatic oxytocin are valid proxies for the activity of the oxytocin system in the brain. Our data does not address that question and a thorough discussion of these studies falls, in our opinion, out of the scope of the manuscript. Instead, we focused on whether measurements of oxytocin in saliva and plasma (by far the most commonly used biological fluids to measure oxytocin) are sufficiently stable to provide valid indicators of the physiology of the oxytocin system in humans. Additionally, we also investigated whether salivary oxytocin can index plasmatic oxytocin at baseline and after the administration of synthetic oxytocin using different routes of administration.
A previous meta-analysis of studies correlating peripheral and CSF measurements of oxytocin has shown that most likely peripheral and CSF measurements do not correlate at baseline; significant correlations could be found after intranasal administration of oxytocin or specific experimental manipulations, such as stress(37). We believe that currently we still do not have a clear answer about the extent to which these peripheral fluids can actually index oxytocin concentrations in the brain (even if associations with CSF are evident in specific instances). For instance, no study has ever shown that CSF oxytocin actually predicts the concentrations of oxytocin in the extracellular fluid of the brain. Given what we currently know about the synaptic release of oxytocin in the brain(38) (in contrast with former theories of exclusive bulk diffusion in the CSF(39)), we think we have good reasons to suspect this might not be the case.
The only contribution our study can make in that respect is highlighting our current lack of understanding of how oxytocin reaches saliva if not from the blood. Currently there is no evidence of direct secretion of oxytocin to the saliva (not from acinar secretion or nerve terminals release). Hence, as it stands, the most likely mechanism for oxytocin to entry the saliva is from the blood (for instance, by ultrafiltration). If increases in plasmatic oxytocin after intravenous oxytocin cannot produce any significant increases in salivary oxytocin (shown in ours and in a previous study), how does oxytocin reach the saliva and why might it be able to predict concentrations in the CSF, if it does? In this respect, we hope our study highlights the need for further research shedding light on the mechanisms underlying these potential saliva – CSF relationships, if they exist. We would be glad to accommodate any other hypothesis the reviewer might have on this respect.
Line 522 “The lack of increase in salivary oxytocin after the intravenous administration of exogenous oxytocin that was consistently found in our study and in a previous study(3) also raises the question of how oxytocin reaches the saliva if not from the blood. Currently there is no evidence of direct acinar secretion or direct nerve terminals release of oxytocin to the saliva; therefore, transport from the blood remains as the most plausible mechanism of appearance of oxytocin in the saliva. Clarifying these mechanisms of transport is paramount, given the current hypothesis that salivary oxytocin might be superior to plasma in indexing central levels of oxytocin in the CSF(40).”
Methods
7) Related to the general comment, the variability in days between sessions is relatively high (average 8.80 days apart (SD 5.72; range 3-28). However, it appears that no explicit measures were taken to control the conducted analyses for this variability.
Thanks for point this out. Indeed, we were not sufficiently thorough in exploring the impact of this potential variability in the time gap between visits on our estimated ICCs. Thanks to the reviewer we now acknowledged this limitation of our analysis and decided to explore this further. We decided to run the following sensitivity analysis. First, we went back to our dataset A and identified all pairs of consecutive measures that were collected with an exact time interval of 7 days between visits. We could retrieve 15 examples of these pairs from 15 different participants for both saliva and plasma. Then, we recalculated the ICC and CV on this subset of our initial sample. In line with our main analysis, we found poor reliabilities for both salivary and plasmatic oxytocin; in both cases the ICCs were not significantly different from 0 and the CVs were 49% and 40%, respectively. This further analysis has been added to the revised version of the manuscript. We hope the reviewer shares our vision that our main conclusion of poor reliabilities of single measurements of baseline oxytocin in saliva and plasma cannot be simply attributed to the variability in the number of days between visits.
Line 229 “Since there was considerable variability in the time-interval between visits across participants, we conducted a sensitivity analysis where we repeated our reliability analysis focusing on 15 pairs of consecutive measures that were collected with an exact time interval of 7 days between visits in 15 participants. Here, we recalculated the ICC and CV on this subset of our initial sample, using the approach described above.”
Line 399 “These poor reliabilities are unlikely to be explained by variability in the time-interval between visits of the same individual, since we also found poor reliability indexes for both saliva and plasma when we restricted our analysis to a subset of our sample controlling for the exact number of days spacing visits.”*
8) A rationale for the adopted dosing and timing (115 min post administration) of the sample extraction is missing. Additionally, it seems that intravenous administrations were always given second, whereas intranasal administrations were given third, with a small delay of approximately 5 min. Hence, it seems that the timing of 115 min post-administration is only accurate for the intranasal administration.
We collected saliva samples before any treatment administration and after the end of our scanning session (collection of saliva samples in between was just not possible because the participants were inside the MRI machine and could not have moved their heads). For the plasma, we collected samples before any treatment administration, after each treatment administration and at other five time-points during the scanning session. Here, we only report the plasma data that was acquired concomitantly with the saliva samples (the full-time course of plasma changes in plasmatic oxytocin has been reported elsewhere(2)). In the manuscript, we report post-administration times from the end of the full treatment administration protocol. Hence, as the reviewer highlights our post-administration sample was collected at around 115 mins from the last intranasal administration and 120 mins from the end of the intravenous administration. We have now made this aspect explicit in the revised version of the manuscript.
Line 162 “For the purposes of this report, we use the plasmatic and salivary oxytocin measurements that were obtained at baseline and at 115 minutes after the end of our last treatment administration (this means that our post-administration samples were collected 115 mins after the intranasal administrations and 120 mins after the intravenous administration of oxytocin).”
9) Since the ICC of baseline samples showed poor reliability, it seems suboptimal to pool across sessions for assessing the relationship between salivary and blood measurements. It should be possible to perform e.g. partial correlations on the actual scores, thereby correcting for the repeated measure (subject ID). Further, since the sample size is relatively small (13 subjects), it might be recommended to use non-parametric (e.g. Spearmann correlations) instead of Pearson. The additional reporting of the Bayes factor is appreciated; it is very informative.
Thanks for the suggestion. In fact, for the correlation the reviewer mentions we indeed used a multilevel approach where we specified subject as a random effect (please see pages 9-10). This allowed us to deal with the dependence of measurements coming from the same subject in different visits. Furthermore, since we also had concerns about the sample size, we calculated Pearson correlations but used bootstrapping (1000 samples) to obtain the 95% confidence intervals and assess significance. Bootstrapping is a robust statistical technique which allows significance testing independently of any assumptions about the distribution of the data and is robust to outliers. Please see page 12 of the manuscript, section “Association between salivary and plasmatic oxytocin levels”.
10) Now, the authors only compared relationships between salivary and plasma levels, either at baseline or post administration. I'm wondering whether it would be interesting to explore relationships between pre-to-post change scores in salivary versus plasma measures.
Thanks for the suggestion. We have now conducted this further analysis and we could not find any significant correlation between changes from baseline to post-administration in any of our treatment conditions. As for our other correlation analyses, here we also conducted Bayesian inference, which supported the idea that the null hypothesis of no significant correlation between changes in saliva and plasma from baseline to post-administration is at least 4x more likely than the alternative hypothesis. This further analysis strengthens our confidence that changes in salivary oxytocin after administration of oxytocin using the intranasal and intravenous routes should not be used to predict systemic absorption to the plasma.
Line 260 “*As a final sanity check, we also investigated correlations between the changes from baseline to post-administration in saliva and plasma in each of our treatment conditions separately.”
Line 485 “Furthermore, we could not find any significant correlation between changes in salivary or plasmatic oxytocin from baseline to 115 mins after the end of our last treatment administration in any of our four treatment conditions. The lack of significant associations between salivary and plasmatic oxytocin (and respective changes from baseline) was further supported through our Bayesian analyses which demonstrated that given our data the null hypotheses were at least three times more likely than the alternative hypothesis.”*
11) Please provide more information on the outlier detection procedure (outlier labelling rule).
This information has now been added to the revised version of the manuscript.
Line 271 “Outliers were identified using the outlier labelling rule(41); this means that a data point was identified as an outlier if it was more than 1.5 x interquartile range above the third quartile or below the first quartile.”*
12) Please indicate how deviations from a Gaussian distribution were assessed.
We used the combined assessment of i) differences between mean and median; ii) skewness and kurtosis; iii) histogram; iv) Q-Q plots; and v) the Kolmogorov-Smirnov and Shapiro-Wilk normality tests. Deviations from a normal distribution is common in the concentration of several analytes in the saliva (42), including oxytocin (15); hence, following the current recommendations, we used log transformations of the raw concentrations but plot the raw concentrations to facilitate the interpretation of our plots.
Results
13) Please verify the degrees of freedom for the post-hoc tests performed to assess pre-post changes at each treatment level (e.g. baseline vs Post administration: Spray - t(122) = 7.06, p < 0.001) . Why is this 122? Shouldn't this be a simple paired-sample t-test with 13 subjects?
We apologize for this oversight. Indeed, we did a mistake in copying the values of the degrees of freedom from SPSS. We have now corrected these values. All the other p-values and F or T values were reported correctly and hence are not changed in the revised version of the manuscript (please see also response to Reviewer 1, question 4 regarding inconsistencies in the reported p-values).
We thank the Editor of eLife f or kindly considering our manuscript for publication and for soliciting three peer reviews. We note that the reviews were positive for the most part. We sincerely believe that the key criticisms arise regrettably from a seeming misunderstanding of the motivation and context of our work – one that we hoped was a candid presentation of available data for tarantulas and the methods used. We provide detailed responses to the reviewers’ concerns below. We further note that our manuscript has since been published with minimal changes (Foley et al. 2020 Proceedings of the Royal Society B 287: 20201688, doi:10.1098/rspb.2020.1688).
Tarantulas belong to an enigmatic and charismatic group with a nearly cosmopolitan distribution and intriguingly show vivid coloration despite being mostly nocturnal/ crepuscular. Using a robust phylogeny based on a comprehensive transcriptomic dataset that includes nearly all theraphosid subfamilies (except Selenogyrinae), we performed both discrete and continuous ancestral state reconstructions of blue and green coloration in tarantulas using modern phylogenetic methods. Using phylogenetic correlation tests, we evaluated various possible functions for blue and green coloration, for instance aposematism and crypsis. Our results suggest green coloration is likely used in crypsis, while blue (and green) coloration show no correlation with urtication, stridulation or arboreality. Our findings also support a single ancestral origin of blue in tarantulas with losses being more frequent than gains, while green color has evolved multiple independent times but never lost. We comparatively assessed opsin expression from the transcriptomic data across tarantulas to understand the functional significance of blue and green coloration. Our opsin homolog network shows that tarantulas possess a rather diverse suite of regular arthropod opsins than previously appreciated.
While color vision in (jumping) spiders is relatively well studied, to the best of our knowledge, this is the first study to comparatively consider the identity of opsin expression across tarantulas, and in relation to the evolution of coloration. Our study challenges current belief (e.g., Morehouse et al. 2017 doi: 10.1086/693977 and references therein; Hsiung et al. 2015 doi: 10.1126/sciadv.1500709) that tarantulas are incapable of perceiving colors, at least from a molecular perspective and suggests a role for sexual selection in their evolution. This also adds to the growing body of knowledge on the complexity of arthropod visual systems (e.g., see Futahashi et al. 2015 doi:10.1073/pnas.1424670112, Hill et al. 2002 doi:10.1126/science.1076196).
In short, we believe our results are timely and pertinent broadly to sensory biologists, behavioural ecologists and evolutionary biologists as it is an exhortation for sorely needed behavioural and sensory experiments to understand proximate use of vivid coloration in this enigmatic group.
Summary:
This study offers some interesting data and ideas on colour evolution in tarantulas, building upon previous work on this topic. However, the reviewers judged that the insights are too taxon-specific and that several key conclusions are too speculative. There were also concerns about the methodology for trait scoring from photographs that the authors might consider going forward.
Reviewer #1:
This study investigates the evolution of blue and green setae colouration in tarantulas using phylogenetic analyses and trait values calculated from photographs. It argues that (i) green colouration has evolved in association with arboreality, and thus crypsis, and (ii) blue colouration is an ancestral trait lost and gained several times in tarantula evolution, possibly under sexual selection. It also uses transcriptome data to identify opsin homologs, as indirect evidence that tarantulas may have colour vision.
Otherwise, a few comments:
1) Given that data is limited for the family (only 25% of genera could be included in this study), it seemed a shame not to discuss further the variation in colour and habit within genera. Based on Figure 1 and supplementary tables, the majority of "blue" genera contain a mix of blue and not-blue (and not-photographed) species. Does this mean that blue has been lost many more times in recent evolutionary history? And how often are "losses" on your tree likely to be the result of insufficient sampling for the genus (i.e. you happen not to have sampled the blue species)?
First, the taxa in our robust and well-resolved phylogeny are representative of the major lineages within Theraphosidae, i.e., we have sampled nearly all theraphosid subfamilies (except Selenogyrinae). Our ideal is also to work with a more complete genus-level molecular phylogeny and corresponding color dataset for theraphosidae. However, this group is generally not well represented in museum collections (let alone in digitized collections), while the pet trade is focussed on only a select number of taxa. While we appreciate the reviewer’s concern that adding more taxa and corresponding data could potentially change the results, we believe that with a strong backbone phylogeny recovering the major branches, the results should not change all that much (For instance, cf. Hackett et al. 2008 10.1126/science.1157704 vs. Prum et al. 2016 10.1038/nature19417, where the initial Hackett et al. backbone is robust to increased sampling). Although the way trait losses are concentrated towards the tip suggests that using a genus-level phylogeny would perhaps show a few more recent trait losses, but unlikely to contradict an ancient origin of blue coloration at the base of this group, especially given the way the outgroups are polarized (i.e., outgroups also exhibit blue).
2) A key conclusion of the study is that sexual selection should not be discarded as a possible explanation for spider colour. However, there is very little detail given in the discussion to build this case. Do these spiders have mating displays that might plausibly include visual signals? How common are sexually-selected colours in spiders generally? Where on the body is the blue coloration (in cases where it is not whole body)? I also missed whether the images used are of males or females or both, or how many species show sexual dimorphism in colouration (mentioned briefly in the Discussion, but not summarised for species or genera).
We agree with the reviewer that we should have provided more information regarding sexual dichromatism in tarantulas, and on the images we used in the study (whether male/female). However, the location of blue coloration varies wildly with species – some species have blue chelicerae, blue abdomens, or blue carapaces while others are entirely blue. We also know very little about mating (and selection, if any) strategies in tarantulas, let alone the sensory ecology of this group. However, there is intriguing anecdotal information from one species (Aphonopelma) that they can be active as early as 4pm (Shillington 2002 Canadian J. Zoology, 80: 251-259, doi: 10.1139/z01-227), while some species show an intensification of color upon maturation, often a hallmark of sexual selection. Indeed, we believe that our work will incite broad interest on these intriguing questions.
3) A quick scroll through the amazing images on Rick West's site suggests that oranges and red/pinks are not rare in tarantulas. Perhaps the data is just not available, but it would be good to mention somewhere the rationale behind the blue/green focus, rather than examining all colours.
We agree. However, in the present study, we focused on blue and green colors because the data is readily available and we wanted to build upon the previous work by Hsiung et al 2015. Given that violet/blue and likely also some green coloration are structural in origin (Saranathan et al. 2015 Nano Letters, doi: 10.1021/acs.nanolett.5b0020; Hsiung et al. 2015), these hues are unlikely to fade or vary between individuals unlike diet acquired pigmentary coloration. Hence, these colors perhaps better lend themselves to analyses using digital photographs.
I suggest defining stridulating / urticating setae for non-specialist readers. I had to look these up to understand that they were involved in defence.
We thank the reviewer for this suggestion.
I notice the Rick West website says species IDs should not be made from photos alone. Is there a risk of misidentification for any photos?
We understand the reviewer’s concern. However, Rick West is an experienced arachnologist and quite knowledgeable in tarantula systematics and taxonomy (see https://www.tarantupedia.com/researchers/rick-c-west), which is why we endeavoured to use his website as extensively as possible without resorting to photos from hobbyists. We further validated the IDs with field guides, when in doubt.
The Results section would benefit from some more clear statements of key results. For example, phrases like "AIC values to assess the relationships between greenness and arboreality are reported in Table 3" could be replaced instead with a summary statement indicating what this table shows.
We agree and thank the reviewer for this suggestion.
In the Figure 1 caption I think there is a typo: 'the proportions of species with images that possess blue colouration (grey = no available images)" but should this say "grey = not blue"?
We apologize for the confusion. This is not a typo – this is in relation to Trichopelma, for which no images of described species were available, and so we cannot conclude that none of the taxa are blue/green.
142 - the lengthy discussion here of whether there is one or more mechanisms by which blue is produced in tarantulas, and the detailed criticism of Hsuing SEMs, seems a bit out of place given that the current study does not investigate the proximate mechanism of blue colouration but merely its presence.
We respectfully disagree. The core support for Hsiung et al.’s (2015) argument against sexual selection as a driver of color evolution in tarantulas comes from their structural diagnoses of the nanostructures responsible for the violet/blue structural coloration and their subsequent argument that a diversity of divergent nanostructures rather than convergence argues against sexual selection. While it is true that we did not investigate the proximate mechanism of blue coloration here, one of us (Saranathan et al. 2015) has already done so elsewhere. It appears that in insects and spiders, the bulk of the nanostructural diversity is across families and not within.
Table S6 - It is not clear to me how the values for predicted N orthologs were calculated.
This is mentioned in line 354 of our methods – “Per the ‘moderate’ criteria from the Alliance of Genome Resources (55), hits may be considered orthologous if three or more of the twelve tools in their suite converge upon that result”.
The Table S7 caption states: "A * indicates currently undescribed species with blue or green colour that can be confidently attributed to corresponding genus. However, as the described species exhibit no blue or green colour, we conservatively scored these as 0." Is this a conservative approach though? If they have been confidently assigned to genus, I don't understand why they would not be included.
This refers to the cases where a hitherto undescribed species possesses the blue or green color. However, even though the species has not formally been described, its placement in the genus is not in question. We have not included such undescribed species in our tabulated number of species per genus, as it is difficult to express any such undescribed species as a fraction of the total number of species in that genus.
Reviewer #2:
This paper presents a broad-ranging overview of tarantula visual pigments in relationship with the color of the spiders. The paper is interesting, well-written and presented, and will inspire further study into the visual and spectral characteristics of the genus.
We thank the reviewer for her/his/their kind words.
First a minor remark, Terakita and many others distinguish between opsin, being the protein part of the visual pigment molecule and intact light-sensing, so-called opsin-based pigment, often generalized as a rhodopsin. The statement of line 65, 'convert light photons to electrochemical signals through a signalling cascade' is according to that view strictly not correct. Furthermore, the presence of opsins in transcriptomes may be telling, but it is not at all sure that they are expressed in the eyes, if at all. As the authors well know, in many animal species some of the opsins are expressed elsewhere. It may be informative to mention that.
We thank the reviewer for this clarification. As for the regions of opsin expression, we very much agree – were it not for constraints of sample availability, we would also have preferred to sequence only the eyes and brain of various tarantulas that were all exposed to similar lighting conditions. However, we encouragingly see that our “leg only” transcriptomes have far fewer (often no) opsins as compared to the whole-body data.
The blueness or greenness feature prominently in the paper, but the criteria used for determining to which class a spider belongs are not at all sure. The Color Survey and Supplementary Table S2 refer to Birdspiders.com, but that requires a donation; not very welcoming. The other used sources are also not readily giving the insight or overview which material was sampled. I therefore think that the paper would considerably gain in palatability by adding a few exemplary photographs as well as measured spectra. Of course, I am inclined to trust the authors, but I would not immediately take color photographs from the web as the best material for assessing color data with 4-digit accuracy. Furthermore, the accessible photographs do not always show nice, uniform colors, so it might be sensible to mention which body part was used to score the animals. And finally, using CIE metric might infer to many readers that the spiders are presumably trichromatic, like us. Any further evidence?
We refer to the detailed description of our method for scoring blue or green coloration in tarantulas (l. 277-303). Briefly, we calculated ΔE (CIE 1976) difference values using between the images of each taxa against a suitable reference (average of green leaves, or Haplopelma lividum, the bluest taxa in our survey based on the b value of its images). We use the ΔE Lab values to perform quantitative ancestral state reconstruction, while we use ΔE b (for blue) and ΔE a (for green) to discretize the data for understanding trait gains and losses.
BirdSpiders.com only requires one to enter names of genera as search terms in order to see photos that we used. However, we agree could have provided some photos of exemplars. We do realise that using pictures is not ideal, as opposed to reflectance spectrophotometry (our ideal as well), which is why we limited ourselves to a single reputable source (BirdSpiders.com) for consistent images, whenever possible. However, acquiring sample material and reflectance of tarantulas is challenging. This group is generally not well represented in museum collections (let along in digitized collections), while the pet trade is focussed on only a select number of taxa and doing field work to collect specimens is fraught with moral and ethical issues (e.g., see https://www.nytimes.com/2019/04/01/science/poaching-wildlife-scientists.html). This study nevertheless represents a substantial improvement upon a recent high-profile work that used the OSX “color picker” function (Hsiung et al. 2015).
Indeed, available evidence on tarantula vision (including our opsin sequences) suggests tarantulas are likely trichromats (Dahl and Granda 1989 J. Arachnol., Morehouse et al. 2017) similar to jumping spiders (e.g., Zurek et al. 2015, doi: 10.1016/j.cub.2015.03.033), so we consider CIE as an appropriate color space for a putative tristimulus system in tarantulas (see also our response to Reviewer 3). Again, this underscores the need for future studies on the sensory biology and psychophysics of this enigmatic group.
Reviewer #3:
This neat paper continues the story of structural colour evolution in a group that is rarely appreciated for their ornamentation. The study uses colour & ecological data to model their evolution in a comparative framework, and also synthesises transcriptomic data to estimate the presence and diversity of opsins in the group. The main findings are that the tarantulas are ancestrally 'blue' and that green colouration has arisen repeatedly and seems to follow transitions to arboreality, along with evidence of perhaps underappreciated opsin diversity in the group. It's well-written and engaging, and a useful addition to our understanding of this developing story. I just have a few concerns around methods and the interpretation of results, however, which I feel need some further consideration.
We thank the reviewer for his/her/their kind words.
As the authors discuss in detail, this work in many ways parallels that of Hsiung et al. (2015). The two studies seem to agree in the broad-brush conclusions, which is interesting (and promising, for our understanding of the question), though their results conflict in significant ways too. Differences in methodology are an obvious cause, and they are particularly important in studies such as this in which the starting conditions (e.g. the assumed phylogeny or decisions around mapping of traits) so significantly shape outcomes. The current study uses a more recent and robust phylogeny, which is great, and the authors also emphasise their use of quantitative methods to assign colour traits (blue/green), unlike Hsiung et al.
We thank the reviewer for his/her/their appreciation.
1) This latter point is my main area of methodological concern, and I am not currently convinced that it is as useful or objective as is suggested. One issue is that the photographs are unstandardised in several dimensions, which will render the extracted values quite unreliable. I know the authors have considered this (as discussed in their supplement), but ultimately I don't believe you can reliably compare colour estimates from such diverse sources. Issues include non-standardised lighting conditions, alternate white-balancing algorithms, artefacts introduced through image compression, differences in the spectral sensitivities of camera models, no compensation for non-linear scaling of sensor outputs (which would again differ with camera models and even lenses), and so on (the works of Martin Stevens, Jolyon Troscianko, Jair Garcia, Adrian Dyer offer good discussion of these and related challenges). Some effort is made to minimise adverse effects, such as excluding the L dimension when calculating some colour distances, but even then the consequences are overstated since the outputs of camera sensors scale non-linearly with intensity, and so non-standardised lighting will still affect chromatic channels (a & b values). So with these factors at play, it becomes very difficult to know whether identified colour differences are a consequence of genuine differences in colouration, or simply differences in white balancing or some other feature of the photographs themselves.
We thank the reviewer for his/her/their carefully considered thoughts and for drawing our attention to the work of Martin Stevens, Jolyon Troscianko, Jair Garcia, and Adrian Dyer in this regard (e.g. Stevens et al. 2007 Biol. J. Linn. Soc. Lond., doi: 10.1111/j.1095-8312.2007.00725.x). These are fair points raised by the reviewer. We are indeed aware that there are clear drawbacks in working solely with photographs from online sources as opposed to optical reflectance data (our ideal), but we are sure that the reviewer appreciates how challenging it is to source specimens of tarantulas. It is for this reason that we restricted ourselves to photographs from mostly only 1 reputable source (BirdSpiders.com). Furthermore, this is why we chose a perceptual model that permits device independent color representation, one that lets us separate chromatic variables from brightness, keeping in mind the underlying assumptions. However, some recent research suggests that CIELab space can perform reasonably well as compared to the latest algorithms for illuminant-invariant color spaces (Chong et al. 2008 ACM Transactions on Graphics, doi: 10.1145/1360612.1360660). Please also see our response below (to point #2) and also to Reviewer #2 above.
Given the dearth of tarantula specimens and in the absence of spectrometry, future work will have to try and acquire uncompressed original images (with EXIF data) and could perform image processing such as homomorphic filtering and adaptive histogram equalization (Pizer et al. 1987 Computer Vision, Graphics, and Image Processing; Gonzalez and Woods 2018 Digital Image Processing, Pearson) in order to further mitigate artefacts such as those arising from differences in illumination, especially if using images from a diversity of sources.
2) The justification for some related decisions are also unclear to me. The CIE-76 colour distance is used, and is described as 'conservative'. But it is not so much conservative as it is an inaccurate model of human colour sensation. It fails to account for perceptual non-uniformity and actually overestimates colour differences between highly chromatic colours (like saturated blues). The authors note they preferred this to CIE-2000, which is a much better measure in terms of accuracy, because the latter was too permissive (line 300). I understand the problem, and appreciate their honesty, but this decision seems very arbitrary. If the goal is to quantitatively estimate colour differences according to human viewers, then the metric which best estimates our perceptual abilities would strike me as most appropriate. Also, the fact that all species would be classified as 'blue' using the CIE-2000, when some of them are obviously not blue by simply looking at them, is consistent with the kinds of image-processing issues noted above. I only focus on this general point because it is offered as a key advance on previous work (L 40-41), but I don't think that is clearly the case (though I agree that the scoring methods of Hsiung et al. are quite vague). I'm generally in favour of this sort of quantitative approach, but here I wonder if it wouldn't be simpler and more defensible to just ask some humans to classify images of spiders as either 'blue' or 'green', since that seems to be the end-goal anyway.
We agree that CIE 1976 is an inaccurate model of “human color sensation,” but at the same time the degree of their applicability or lack thereof to non-human tristimulus visual systems is not clear. In any case, the digital photographs do not preserve UV information anyway. We hasten to add CIE 1976 is still widely used in color science and engineering research for its simplicity and perceptual uniformity, as a simple Google Scholar search would attest. We believe that the reviewer is perhaps mistaken as to our motivation for choosing the CIE 1976 and the exact nature of the shortcomings of the CIE 1976 model, which it turns out to be an unintended advantage. Our goal was not, as the reviewer suggests, to just “quantitatively estimate color differences according to human viewers,” but to do so in a device independent fashion given the constraints of working with already available digital images, and for a putative trichromat visual system. Given there are technically no limits for a and b values in the CIE 76 space, color patches with high values of chroma are computed to have too strong a difference than in actual fact (Hill et al. 1997 ACM Transactions on Graphics, 16, 109-154). This is precisely the kind of situation that we do not face here, as we are essentially comparing shades of blue rather than for instance, chromatic contrasts between saturated blue vs. green or blue vs. red. Moreover, we only use the rectilinear rather than the polar coordinate representation of the colors (in other words, we do not compute the psychometric correlates, chroma Cab, or the hue angle hab). Contrary to the reviewer’s assertion that the CIE 1976 “overestimates color differences between highly chromatic colors (like saturated blues),” a quick perusal of Table S3 affirms that a comparison of highly saturated blues such as between our “standard” H. lividum and Poecilotheria metallica reveals they are quite close in terms of chromatic contrasts (i.e., small E values). Moreover, CIE 1994 and subsequent revisions rely on a von Kries-type transformation to account for non-uniformity of the perceptual space, but as the reviewer is well aware, without an accurate idea of the illumination conditions, use of CIE 2000 is not justified.
Lastly, we are sure the reviewer appreciates that asking humans to manually score the colors of images (e.g. Hsiung et al. 2015) is neither reproducible nor enables quantitative analyses of trait evolution.
3) L26-27, 53-56, 171-176: This is a more minor point than the above, but some of the discussion and logic around hypothesised functions could be elaborated upon, given it's presented as a motivating aim of the text (52-56). The challenge with a group like this, as the authors clearly know, is that essentially none of the ecological and behavioural work necessary to identify function(s) hasn't been done yet, so there are serious limitations on what might be inferred from purely comparative analyses at this stage. The (very interesting!) link between green colouration and arboreality is hypothesised and interpreted as evidence for crypsis, for example, but the link is not so straightforward. Light in a dense forest understory is quite often greenish (e.g. see Endler's work on terrestrial light environments) including at night which, when striking a specular, structurally-coloured green could make for a highly conspicuous colour pattern - especially achromatically (which is what nocturnal visual predators would often be relying on). This is particularly true if the substrate is brown rotten leaves or dirt, in which case they could shine like a beacon. Conversely, if the blue is sufficiently saturated and spectrally offset from the substrate it could be quite achromatically cryptic at dusk or night. To really answer these questions demands information on the viewers, viewing conditions, visual environment etc. The point being that it is a bit too simplistic to observe that, to a human, spiders are green and leaves on the forest floor may be green, and so suggest crypsis as the likely function (abstract L 22-23). So inferences around visual function(s) could either be toned down in places given the evidence at hand or shored up with further detail (though I'm not sure how much is available).
We agree. Indeed, we are limited by the absence of rigorous behavioural studies. With this in mind, we have already made every effort to tone down and emphasize that our results might point towards a given function, but we do not claim it outright. It is our fervent hope that these findings will form the basis for future behavioural studies by giving researchers a starting point to test their hypotheses.
We would like to point out that the association we uncovered is actually between arboreal taxa and the presence of green coloration and not as the reviewer says “spiders are green and leaves on forest floor may be green.” These taxa live in natural crevices on trees, shrubs and essentially spend their lives arboreally. Also, green coloration in tarantulas need not be structural in origin (see e.g., Saranathan et al. 2015) and this is why to test for crypsis against foliage, we used (pigmentary) leaves as the representative model for comparison to tarantula green colors. Although, certain lycaenid butterflies (Saranathan et al. 2010 10.1073/pnas.0909616107; Michielsen et al. 2010 10.1098/rsif.2009.0352), for instance, use structural coloration to better aid in crypsis against foliage.
Minor comments:
- I'm not familiar enough with with methods for creating homolog networks to comment in detail, but the use of BLASTing existing opsin sequences against transcriptomes seems straightforward enough. As do the methods for phylogenetic reconstruction.
We agree this is straightforward.
- L48: What constitutes a 'representative' species? And how reasonable is it to assign a value for such a labile trait to an entire genus? I understand we can only do our best of course and simplifications need to be made, but I can imagine many cases among insects (e.g. among butterflies and flies) where genus-level assignments would be meaningless due to the immense diversity of structural colouration among species (including in terms of simple presence/absence).
Please see our response to Reviewer 2 above.
- Line 168: Wouldn't this speak against a sexual function? Only in a tentative way of course, but the presence of conspicuous structural colouration in juveniles, which is absent in adults, would suggest a non-sexual origin to me.
The reviewer’s inference is incorrect. We do not suggest that blue coloration is present in juveniles but absent in adults, but only that such conspicuous colors already appear in the penultimate moult right before the male creates a sperm web and is ready for mating.
Library author here. I'm always fascinated by new ways people can invalidate my assumptions. I mean that in a sincerely positive way, as it results in learning.
1) There were concerns about the normality tests and reanalysis to avoid pseudo-replication that must be addressed.
We have now checked the data by two tests for normal distribution (Shapiro-Wilk and Kolmogorov_Smirnoff) and found that flight data do not follow a normal distribution. Therefore statistical analysis of flight data have now been performed using non-parametric tests. We have used the Kruskal-Wallace test followed by Dunn’s multiple comparison test for multiple comparisons and Mann-Whitney U-Test for pair wise comparisons. This information has been included in the statistical tests section in methods. Regarding pseudo-replication, as suggested imaging data have been replotted and calculated now to include just one cell, or one lobe per brain. In addition we have included individual brain traces for every experiment as supplemental data (Figure 5 - supplement F2, Figure 6 – supplement F1, F3 and F4).
2) Discussion should be made clearer and expanded to encompass more of the literature. Specifically, the authors should expand upon the final section of the discussion to discuss more about 1) the potential context for cholinergic modulation of the PPL1-y2alpha'1 DANs (For example, consider where the acetylcholine signal onto DANs might come from. DANs may not be entirely presynaptic to Kenyon cells but might also receive input from Kenyon cells.), 2) the proposed role of these DANs (which have been studied in several contexts) and 3) modulation of innate behavior in general. The paper begins with the importance of modulating innate behavior, but the discussion on this topic is spare and focused almost entirely on research on the mushroom bodies of Drosophila. The discussion section leans heavily on summarizing the results, rather than making connections to work in other systems or networks.
As suggested we have now addressed each of these points in greater detail in the last section of the discussion which has been expanded to two paragraphs. The possibility of cholinergic inputs from KC cells to DANs stimulating the IP3R have been included in the discussion and in the final model in Figure 7. Several other references that mention the role of PPL1-y2alpha'1 DANs in modulation of behaviour are now included – see last para of the discussion. We have expanded the last section of the discussion to include possible roles for other regions of the brain in modulating flight and references to other insect brains, where relevant.
3) One common point raised by all reviewers was the need for expression of the itprDN during pupation which could have been due to either the perdurance of endogenous itpr vs. a developmental effect caused by the itprDN (the authors fully acknowledge the issue). This section raised many questions that aren't within the scope of this study, nor are easily resolved. Nevertheless, the authors must expand upon the implications of these results and suggest future studies will needed to resolve the issue.
We are indeed unable to state equivocally if adult behavioural phenotypes, arising from expression of the IP3R^DN, are only pupal or both pupal and adult. We have expanded on the implications of these results both in the results (Page 9-10) and in the discussion (page 11). One way of addressing this is to express a tagged IP3R^DN specifically in late pupae and then follow it’s perdurance in adults. This experiment has now been suggested as a way to resolve this issue in the second paragraph of the discussion.
Reviewer #1:
The authors report experiments on Drosophila to show that the proper function of an IP3 receptor in a small subset of dopaminergic neurons is required for flight behavior. Most interesting is the fact that the requirement is restricted to a time point during pupal development. Technically, the authors report a novel dominant-negative mutant for of the IP3 receptor to interfere with its function. Physiologically, the IP3 receptor-dependent impairment in the function of the dopaminergic neurons affects both synaptic vesicle release and excitability, Also, muscarinic acetylcholine receptors are required for proper development of the flight-modulating circuit during development.
The role of dopamine in the brain of Drosophila (as a model for general dopamine and brain function) is in the center of current research, and is studied by a large number of laboratories. More and more types of behavior are discovered that are modulated by dopaminergic neurons, and in particular those innervating the mushroom body. Therefore, the study is of very high interest for researchers working on Drosophila, but also to a broader readership.
The experiments are well designed. with appropriate controls at place. The conclusions drawn are highly interesting and novel (dopaminergic modulation of flight behavior, perhaps in the context of food seeking behavior, molecular mechanisms of circuit maturation).
Minor comments:
1) A test for normal distribution of data is required to determine whether parametric statistical tests are actually appropriate.
Done – please see response above.
2) It is not clear to me why the authors conclude an acute requirement of IP3R during the adult state although the phenotype can arise through a genetic intervention during earlier time points in development (Page 9, lines 297ff). This has to be outlined much clearer. My interpretation of the data is: During a certain time window after pupal formation IP3 signaling is required for a proper formation of the neuronal circuit. This is likely to be not only a cell-intrinsic (i.e., cell autonomous) effect because the mAchR is also required during this time window. This provides an excellent example (there are actually only very few!) of circuit development that requires synaptic interactions between neurons. If one keeps in mind that dopaminergic neurons have reciprocal synapses with Kenyon cells (e.g. Cervantes-Sandova, elife 2017; should be included in schematic illustration!)), and these release acetylcholine onto dopaminergic neurons, a potential circuit maturation based on the concerted activity is most interesting. I suggest that the authors point out more precisely how they think the actual phenotype comes about, of course, with all due caution.
The primary reason that we suggest an adult requirement for the IP3R in the DANs is that we see a Ca2+ response to carbachol in adult PPL1-y2alpha'1 DANs (Figure 5 – supplement 1). We put together this finding with the observation that carbachol stimulates dopamine release from PPL1-y2alpha'1 DANs (Figure 5) and that blocking vesicle release acutely in adults reduce durations of flight bouts (Figure 4) to suggest that there is likely to be an adult requirement. However, we agree that this is not conclusive and certainly does not negate a pupal requirement. As mentioned above we have addressed the pupal vs pupal+adult issue in greater detail in the results (page 9, 10) and discussion (page 11). We agree that there may be acetylcholine release from Kenyon cells at the MB synapse. This possibility has been included in the discussion and in Figure 7.
3) Statistical tests should be done across independent brains, not across different cells in the same brains.
We have done this. Thank you for pointing this out.
Additional data files and statistical comments:
A test for normal distribution of data is required to determine whether parametric statistical tests are actually appropriate.
Done.
Figure legend 5 C should be 5B. The scaling of the y-axis is not optimal.
Done.
Statistical tests should be done across independent brains, not across different cells in the same brains. This would cause a mixture of dependent and independent data. This is of importance!
Done.
Reviewer #2:
The results of the individual experiments reported by the authors are convincing. The approach is rigorous and they take full advantage of the many powerful molecular genetic tools available in Drosophila. The identification of a mechanism by which a small subset of dopaminergic cells may control behavior is significant. My concerns about the manuscript are relatively minor.
Minor comments:
I have reviewed "Modulation of flight and feeding behaviours requires presynaptic IP3Rs in dopaminergic Neurons" by Sharma and Hasan. The authors first translated to Drosophila a dominant negative (DN) strategy first tested in mammalian cells to block the function of the fly IP3 receptor. Controls using westerns to test the expression in vivo and calcium imaging to assess inhibitory activity in an ex vivo prep were generally convincing. They then show that the DNA, RNAi and a wt transgene disrupts flight as they have shown previously using both genetic mutants and RNAi. They use genetic rescue to further show that alterations in the function of itpr in dopaminergic cells are likely to mediate at least some aspects of the flight deficit. The restricted distribution of the THD' driver was used to narrow down the identity of DA cell clusters responsible for this effect to PPL1 and/or PPL3. Additional split GAL4 lines identified a deficit when the DN was expressed in the PPL1-γ2α′1 subset of DA cells that project to the mushroom bodies. This is a key finding of the paper since it localizes the requirement of the IP3R to cells that have been implicated in other behaviors. Developmental tests using TARGET/GAL80 indicate a requirement for itpr during late development. Disruption of itpr only in the adult did not have a significant effect. This seems likely to be due to perdurance of itpr as suggested by the authors. However, these data make it difficult to determine which aspects of the phenotype are due to broad developmental deficits versus disruption of IP3R in the adult (see below). The authors next test the effects of mAhR with the idea that mAChR is likely to signal through IP3R. While it was known that developmental expression of mAcHR expression is required for adult flight, the current data more specifically that the PPL1-γ2α′1 DANs are required, enhancing the impact of the paper.
To tie these results to vesicle recycling and release the authors use the shibere[ts] transgene in PPL1-γ2α′1. Flight bouts were disrupted via exposure to the non-permissive temperature both during late pupal development and the adult. The adult phenotype has been demonstrated previously but the developmental defect is novel. The demonstration of an effect in adults is important since it suggests loss of itpr during adulthood might also have an effect in adults even though this can't be tested due to perdurance. Expression of shibire[ts] in PPL1-γ2α′1 also disrupts feeding, and the authors next phenotype these effects with the itpr DN, indicating that IP3R expression in PPL1-γ2α′1 is required for both feeding and flight. However, here as with the flight experiments, it is not possible to directly demonstrate an effect in adults due to perdurance. They show that knockdown of mAChR also reduces feeding similar to its effects on flight and suggest that the deficits are due to disruption of the mAchR ->(Gq) ->IPR3 pathway. The suggestion of connections between mAchR and IPR3 within PPL1-γ2α′1 and the idea that PPL1-γ2α′1 controls two distinct behaviors are a significant finding and one of main contributions of the paper.
To help link the shibire[ts] data set with and the results of perturbing mAchR and IPR3, the authors show that carbochol induced DA release is reduced, making excellent use of the relatively new GRAB-DA lines. As a control, they show that synapse density of PPL1-γ2α′1 in the γ2α′1 MB lobes are not altered. The demonstration that DA release is altered elevates the technical strength of the paper. Moreover, although further experiments might be needed to prove their model, these data support the argument that mAchR ->(Gq) ->IPR3 pathway is disrupted in the adult. The final set of experiments in Fig 6 indicate that excitability of the PPL1-γ2α′1 DANs is also disrupted by knock down or IP3R. Is it possible that this deficit contributes to the decrease in DA release by the mAchR ->(Gq) ->IPR3 and the authors nicely explain a possible mechanism and cite relevant references in the Discussion.
The results of the individual experiments reported by the authors are convincing. The approach is rigorous and they take full advantage of the many powerful molecular genetic tools available in Drosophila. The generation of the DN transgene is a nice idea and in combination with other tools helped them to identify specific subsets of DA neurons important for the behaviors they test. However, they have previously demonstrated similar effects with mutants and RNAi, and again use them to help map the relevant cells. Since the use of the DN construct did not really go beyond the experiments using RNAi or genetic rescue, the emphasis on the importance of this reagent might be reduced in the abstract and introduction.
Flight deficits have also been seen in other experiments on these the DANs identified by the authors. Thus, the major novel finding of this section is the demonstration that itpr is required in these cells for regulating flight. While it was previously shown that feeding behavior is also required by DAN projections to the MB, the idea that overlapping cells might control both flight and feeding is interesting. Although the idea that these two phenotypes are specifically related to each other seems somewhat speculative, one major strength of the paper lies in tying together prior observations on itpr and the DANs with their current experiments. They do this again at the cellular level using GRAB to show that carbachol induced release of DA (but not synapse density) is reduced by itpr knock-down, thus tying together data on shibere, AcHR and itpr.
These connections make for an exciting story, and they have been cleverly woven together by the authors. On the other hand, they also represent a possible concern about the manuscript as a whole, since causal relationships between the deficits between the effects of blocking the effects of IP3R, mAcHR, neuronal excitability and vesicle release are not yet proven. It is therefore possible that all of these are relatively non-specific effects of disrupting the function of PPL1-γ2α′1 neurons. This modestly reduces the strength of the paper but is also a relatively minor concern. A second potential concern is that despite the interesting connections made by the authors as well as some exciting new data, some of the findings replicate previous data.
It is indeed likely that loss of the IP3R in PPL1-y2alpha'1 DANs leads to both specific (acetylcholine signaling followed by neurotransmitter release) and non-specific changes (such as loss of excitability). Both are likely to have an effect on the behavioural phenotypes modulated by PPL1-y2alpha'1 DANs. We have previously shown a role for both mAchR and the IP3R in flight. However, in this work we have addressed cell specificity and mechanism, neither of which was known earlier.
A third concern is the relationship between the effects of disrupting PPL1-γ2α′1 during development versus the adult. As the authors suggest, perdurance (of protein expression) and/or "perdurance" of previously formed tetramers could easily account for the failure of itpr and mAChR knock down in the adult to cause behavioral deficits. By the same token, it is difficult to parse out the contribution of developmental defects in the DA cells versus problems with signaling in the adult and the following issues should be addressed: the observation that synaptic bouton density is not disrupted is a good way to eliminate gross disruption of connectivity during development but does not rule out other more subtle developmental defects in neuronal function. The fact that shibire[ts] can cause effects in the adult is appreciated but does not really help us to understand what IP3R and perhaps mAcHR are doing during development.
We agree and have tried to further address this issue in the text (see above).
Additional Minor Concerns.
To validate the decrease in the overall response to carbachol in Fig 1D and E, the authors show a statistically significant difference for area under the curve. A parallel metric and statistical test might be used to support the statement that the response is delayed in 1D but not 1E.
Thank you for this suggestion. We performed the test and in fact found that both cellular and mitochondrial responses are delayed. In presence of IP3RDN. This part of the text has been modified (page 4).
"Interestingly, the mitochondrial response did not exhibit a delay in reaching peak values." Why is that? A brief explanation might be useful.
This is no longer the case. The sentence has been removed.
The second explanation of how shibire[ts] works might be shortened.
Done.
Reviewer #3:
General Assessment:
This study demonstrates that IP3R signaling (triggered by muscarinic receptor activation) affects excitability and quantal content of a subset of dopaminergic neurons to modulate flight duration and food search. I had no technical concerns and am generally supportive. My only major concern was that the narrative was fragmented. I believe this is because the perspective shifted between the IP3Rs and the dopamine neurons themselves, and was too focused. I think that streamlining the narrative and providing a broader perspective for the results will remedy this issue.
Major Comments:
-I would like the authors to expand upon their final section of the discussion to discuss more about 1) the potential context for cholinergic modulation of the PPL1-y2alpha'1 DANs, 2) the proposed role of these DANs (which have been studied in several contexts) and 3) modulation of innate behavior in general. The paper begins with the importance of modulating innate behavior, but the discussion on this topic is spare and focused almost entirely on research on the mushroom bodies of Drosophila. The discussion section leans heavily on summarizing the results, rather than making connections to work in other systems or networks.
We have expanded the last section of the discussion to include these suggestions (see above under consolidated review points).
-The developmental section seemed somewhat tangential as the authors cannot distinguish between a developmental role for the IP3R from a need to express the ItprDN transgene prior to adulthood to overcome a potential slow turnover of endogenous IP3R. In essence, it was unclear how these results contributed to the overall narrative of state modulation of behavior. Is this section informative to the development of the mushroom bodies or rigorous validation of the novel transgene?
The manuscript addresses how IP3R function impacts behaviour. In that context pupal (developmental) and adult contributions are both relevant.
We thank the editors and reviewers for taking the time to assess our paper. We note that the reviewers seemed generally supportive of the paper, including noting that the paper addressed important questions. For context, we reiterate here our main findings:
In this context, the main editorial objection that we “did not control for potential confounding of behavioral variables” is not explained in the reviews; we also note that there were no “concerns about the analytical methods used” that were pertinent to our main findings. We are thus unclear about the basis for rejection.
We respond below to the main points of each reviewer; their suggestions on terminology and of separating literature citations on rodent and primate PfC are being given due consideration.
Reviewer #1:
Maggi and Humphries examined how the coding of the present and past choices in the medial prefrontal cortex (mPFC) of the rats during a Y-maze task overlaps and whether they can be reliably distinguished. They found that the neural signals related to the animal's choice in the present and past are distinct and as a result they can be recalled separately, for example, during post-training sleep. Although these are very important questions and an interesting set of analyses have been applied, the results in this report are not entirely convincing, because the analyses did not successfully exclude some alternative hypotheses.
1) The authors analyzed the signals related to the choice, light cue, and outcome separately, and this is possible because the relationship between the animal's choices and cues were decoupled by testing the animals under at least two different rules. There were a total of 4 alternative rules and different sessions included different subsets of these rules. It is possible that at least some results reported in this paper might vary depending on which of these results were tested. For example, rules might affect how the animals learned the task. Therefore, the authors should provide more detailed information about how often different rules were used to collect the neural data reported in this paper, and whether any of the results change according to the rules used in a given session.
In the paper we did examine mPfC encoding in the trials under the two qualitatively distinct types of rule (direction-based i.e. egocentric, and cue-based i.e. allocentric), and showed that encoding of the direction, light, and outcome occurred in both rule types (figure 1e). We gave the number of sessions for those rules in the legend for Figure 1e. (We could equally decode all 3 features in direction-based and cue-based rule sessions in the inter-trial interval as well, see Maggi et al 2018, Figure 9). Thus we compared the decoding vectors across all rule-types.
Only 8 sessions contained more than 1 rule, in the sessions in which the rule was switched. In the full analysis underlying this paper, we had also separately examined the decoding in these 8 rule-switch sessions, and found equally good decoding of direction, choice, and cue. As the paper was already dense - see e.g. Reviewer 3’s comments - we elected to not show this null result in the current version of the manuscript - it is available in version 1 of this preprint - but it can be restored if desired.
2) The authors claim that the neural coding identified in this study does not depend on the signals in individual neurons by showing comparable results after removing the neurons with significant modulations. This logic is flawed, because the neurons without "significant" modulations might still include meaningful signals due to type II errors. Furthermore, if individual neurons carry absolutely no signals, how can a population of neurons still encode any signals? This might suggest some kind of joint coding, and the authors should not merely implicate such a possibility without more thorough tests.
The joint coding of information by a population of neurons is the basis for the whole paper, and is tested extensively: for example, Figure 1 is about establishing that joint coding exists in mPfC. Our point on lines 91-95 was simply to show that the decoding could not be trivially explained by one or two neurons that reliably and strongly differed in the firing rates between different labels (e.g. between left or right choice of direction). To do so, we found sessions in which there were neurons with significantly detectable tuning to the task feature, omitted those sessions, and then looked at the performance of the feature decoding in the remaining sessions - and found it was just as good. Indeed, our point is precisely that it is possible for individual neurons to carry no signals detectable by classic significance testing (potentially due to Type II errors), yet for the population to be able to perfectly encode the information.
The explanation is simply that most, and sometimes all, individual neurons do not consistently covary their firing with the changes in a feature (e.g. choose left and choose right trials) across every trial of a session. In other words, no neuron need consistently participate in encoding information. But so long as when a neuron does change its firing it does consistently vary with the feature, then across a population there are enough intermittently participating neurons on a given trial to always decode the information.
3) The authors analyzed the activity divided into 5 different epochs, where the position #3 corresponds to a choice point and #5 corresponds to the reward site. Therefore, it is surprising that the reliable outcome signals begin to emerge from the position #3 (i.e., choice point). Is this a false positive?
No, this replicated a common finding of outcome-predictive signals in prefrontal cortex; e.g. Daw, N. D., O’Doherty, J. P., Dayan, P., Seymour, B. & Dolan, R. J. Cortical substrates for exploratory decisions in humans. Nature 441, 876–879 (2006).
Fellows, L. K. Advances in understanding ventromedial prefrontal function: the accountant joins the executive. Neurology 68, 991–995 (2007).
Sul, J. H., Kim, H., Huh, N., Lee, D. & Jung, M. W. Distinct roles of rodent orbitofrontal and medial prefrontal cortex in decision making. Neuron 66, 449–460 (2010).
Kaplan, R. et al. The neural representation of prospective choice during spatial planning and decisions. PLoS Biol. 15, e1002588 (2017).
We will add these references to the next version of the manuscript.
4) The authors report that there is retrospective coding, i.e., no coding of the choice in the previous. By contrast, during the intertrial interval (while the animal's returning to the start position), the signals related to the "past" choice were still present but different from how this information was coding earlier during the trial. This is not surprising since during the intertrial interval, the animal's movement direction is opposite compared to that during the trial, so this coding change could reflect the animal's sensory environment. Whether the brain encodes the past and previous events using different coding schemes or not cannot be tested with such confounding.
We note that the reviewer’s objection here only relates to the choice of arm direction, whereas we showed independent encoding of all three features: direction, outcome, and cue position. We can thus test how the past and present are differently encoded because we showed they are both encoded in the same set of neurons. We showed at length both here (Figure 2a&c, Supplementary Figure 5a) and in Maggi et al 2018 (Figs 5-6 and accompanying supplementary figures) that we could decode the past events from the population activity during the inter-trial interval. The information of the trial and the inter-trial interval can be decoded from the same neurons, so the question is: how can the same neurons encode both the present and the past?
One interpretation of the reviewer’s comments is that they are concerned about the possible confounding of movement direction between the trial and the following inter-trial interval. Namely, that the turn directions are guaranteed to be opposite: e.g a left turn into the left-hand arm on the trial would mean a right-hand turn on the return journey of the inter-trial interval. However, that would mean the feature labels would be exactly complementary e.g. trial =[L L R L R] and ITI = [R R L R L]. So if the population was encoding the direction choice the same way in both the trial and ITI, then using the trial’s decoder of direction to decode direction choice in the ITI should result in a performance of 1-[proportion of correctly classified trials], meaning the classifier would be significantly below chance (and vice-versa for using the inter-trial interval’s decoder for the trials). However, we find the cross-decoding performs at chance (Fig 2).
5) The authors tested whether the coding of present and past events is consistent using a transfer (cross-decoding) analysis. However, this is based on simply correlation, and does not exclude the possibility that neurons changing their activity similarly according to (for example) the animal's choice might also change their baseline activity between the two periods (as revealed by the analysis of "population activity" in Figure 3) or might additionally encode different variables. In this case, decoding based on simple correlation might not reveal consistent coding that might be present.
It is unclear what the referee means by the cross-decoding analysis being “based on simple correlation”. The decoder is trained on vectors of firing rates (cf Figure 1b). The decoder assigns high weights to neurons whose activity differs most strongly between the two labels (e.g. left and right choice of direction). So a change in “baseline”, presumably meaning the average firing rate of a neuron across all trials or all ITIs, would not alter the decoder outcome. In addition to the two cross-decoding tests, we also showed the independent encoding by: (a) The angles formed by the decoding vectors trained solely on the trials and solely on the ITIs (Fig 2d-f) (b) The independence of the population rate vectors between trials and ITIs (Fig 3). Indeed, the change in population rates between trials and ITIs shown in Figure 3 is exactly those predicted by the cross-decoding results, as explained on pg 7.
Reviewer #2:
The study by Maggi and Humphries re-examines data by Peyrache et al. (2009), which the authors have themselves analysed previously (Maggi et al., 2018), recorded , in rat prelimbic/infralimbic cortex (see comment below on terminology). In particular, they look at the relationship between decoding of task events during performance of a trial, and during the subsequent intertrial interval. (n.b. in this study, unlike in many studies, the ITI is considerably longer than the trial period). They find that although task-relevant information can be decoded during these two periods, the information is encoded in orthogonal subspaces during trials ('the present') and ITIs ('the past'). They build on this to examine how information is encoded during sleep following training (vs a pre-training control period). They find that only the trial subspaces are reactivated during sleep, not the ITI subspaces, and more so if the rat received a higher rate of average reward.
On the whole, I found this an interesting paper with a clear set of findings, and well-analysed data. Although the advance in some ways an incremental one on previous studies of sleep/replay, and on the authors' previous analyses of this dataset, the study will undoubtedly be of interest to researchers who are interested in consolidation of past experience during sleep. In particular, the study benefits from being able to look for two different types of information ('past' and 'present' decoders) in the same sleep recording sessions. There were a few things that I felt the authors could address:
1) For the cross-decoding analysis in figure 2 b, it is not entirely clear from the main text which part of the trial and ITI coding is being used here. It seems to me like a more useful way of showing the cross-decoding analysis would be to show the 10x10 matrix of cross decoding accuracy for each of the 5 maze positions in both trials and ITIs. This is, I think, different from what the analysis in figure 3g is trying to show (which plots the classification error after dimensionality reduction to a 2D space).
As we strived to explain in the text, for the cross-decoding analysis we used the decoder trained on the firing rates across the entire trial and separately across the entire ITI, in order to arrive at the most stable decoding vectors. We did not show the cross-decoding for the full 5x5 matrix of positions, as the results would be quite noisy. Nevertheless, this is a constructive suggestion, and we will add this analysis. (And indeed the analysis in Figure 3 already shows that the population activity is separable in 1 or 2 dimensions between the trials and ITIs at each maze position, so we would expect the decoder weight vectors to also be independent).
2) It was surprising to me that the authors do not mention the finding in figure 4e anywhere in the abstract or introduction. It makes the reactivation story far more compelling if it can be linked to a change in behaviour during the preceding trials. I think this finding would benefit from not being buried deep in the results section.
We are happy to make this result clearer. Our main finding is of the independent coding, and this result in Fig 4e does not speak directly to the independent coding results, but rather is a lovely little result to support the hypothesis that there really is reactivation of the population vectors in sleep. Because it did not speak to the main thrust of the paper, it was omitted from the abstract given the constraints on the number of words (150).
3) The finding in figure 5 seems slightly extra-ordinary. It suggests that reactivation decoding during sleep is reliable even if very long bins of activity are used to calculate the firing rate (e.g. up to 10s). Does this relationship ever break down? Presumably with the sleep data, it would be possible to extend bins up to 1 minute, 5 minutes, etc. If there is still more reactivation at these extremely long time-bin lengths, does this mean that these neurons are essentially more persistently active? One possible way to test for this might be to project the data recorded during sleep through the classifier weights, and then calculate the autocorrelation function of this projected data (e.g. Murray et al., Nat Neuro 2014) - if this activity becomes more persistent, the shape of the ACF may change post-training.
An excellent question. Rather than persistent activity, we interpreted the consistency of reactivation across orders of magnitude time-scales as showing that the correlations between the neurons were roughly consistent; and thus when active tended to be active in roughly the same relative order. Support for this comes from the findings in Appendix Fig A4e - the correlation matrix between neurons in the trial was more consistently found in post than pre-session sleep.
Reviewer #3:
This article asks the question if within trial (present) and ITI (past) task parameters are encoded in mPFC, and how encoding during these two trial epochs are encoded. They claim that firing in mPFC reflects past and present, but population encoding of past and present are independent. Further they show that the present is reactivated during sleep, not the past.
On the face of it, this seems like an interesting paper. It is novel in that ITI encoding would be highly related to what was going on in the trial. The sleep finding is also interesting but I don't quite get the distinction between present and past for sleep. That could use some clarification.
1) I'm not an expert in regards to this type of analysis, but throughout I was left with the feeling that I would prefer at least some single neuron data and firing rate analysis to complement the highly computational analysis, which frankly, was difficult to understand or critique by somebody who is not an expert.
The goal of the paper is to assess the population coding in PfC of the same events in the past and the present. Indeed, as reported in the paper, we found 25-39 sessions which had no single neuron tuning at all to a given event in a trial (such as the choice of maze arm).
2) I would have liked to see more analysis of firing correlations with behavior. It seems to me if animals were doing different things during the trial and the ITI, then it might not be a surprise that there is independent encoding.
3) I also wonder if the finding is solely dependent on the task (which is poorly described). It seems like there should be independent coding of past and present in this circumstance because they do not feed into each other, and behavior during one is independent of behavior in the other.
4) Relatedly, the authors suggest that independent encoding can explain how the brain resolves interference between past and present, but in this task there was no interference between past and present, and the authors do not show that when there is more or less dependent encoding that there is more or less interference. Without it is unclear how to know how important this finding is as it relates to performance and general mPFC function.
We deal with these points together, as they are all on the behaviour in the trial and inter-trial interval in the task. Yes, the behaviour in the trial is independent of that in the inter-trial interval, so there is no “interference” of behaviour. But that is not of relevance to what is encoded in the PfC. The Introduction and Discussion both point out that the problem is interference of the encoding itself: the encoding of the past and present exists, as we show at length, so the question is: how can it co-exist in the same neurons? We indeed ask if there is no “interference” in the encoding simply because activity in the inter-trial interval is just a memory trace of activity in the trial, and rule that out.
We cannot address when there is “more or less dependent” encoding, because the results are what they are: there is independent encoding of the same events (Figure 2).
The task is described in detail in the Methods (pgs 20-21).
5) Could activity reflect what the animal predicts will happen on the next trial, or what they are planning to do? It wasn't clear if that was examined.
Whether activity in the inter-trial interval predicted what will happen in the next trial was examined in detail in Maggi et al 2018 (Fig 6), and shown here in Figure 2g. We found no encoding of the following trial’s choices, except for a very niche occurrence: an above chance decoding of the next trial’s direction choice when the rat had returned to the start position, during a learning session, and for a direction rule. In other words, as it turned to start the next trial, so there was decoding of the upcoming choice of arm.
Newport is an academic — he makes his primary living teaching computer science at a university, so he already has a built-in network and a self-contained world with clear moves towards achievement.
This is one of the key reasons people look to social media--for the connections and the network they don't have via non-digital means. Most of the people I've seen with large blogs or well-traveled websites have simply done a much better job of connecting and interacting with their audience and personal networks. To a great extent this is because they've built up a large email list to send people content directly. Those people then read their material and comment on their blogs.
This is something the IndieWeb can help people work toward in a better fashion, particularly with better independent functioning feed readers.
The blog was not just the venue in which I started putting together the ideas that became my second book, the one that made promotion and various subsequent jobs possible, but it was also the way that I was able to demonstrate that there might be a readership for that second book, without which it’s much less likely that a press would have been interested.
This sounds like she's used her blog as both a commonplace book as well as an author platform.
Flow is ascendant these days, for obvious reasons—but I think we neglect stock at our peril. I mean that both in terms of the health of an audience and, like, the health of a soul. Flow is a treadmill, and you can’t spend all of your time running on the treadmill. Well, you can. But then one day you’ll get off and look around and go: oh man. I’ve got nothing here. I’m not saying you should ignore flow! This is no time to hole up and work in isolation, emerging after years with your work in hand. Everybody will go: huh? Who are you? And even if they don’t—even if your exquisite opus is the talk of the tumblrs for two whole days—if you don’t have flow to plug your new fans into, you’re suffering a huge (get ready for it!) opportunity cost. You’ll have to find those fans all over again next time you emerge from your cave.
This is a great argument for having an author platform.
We would like to thank eLife editors and the reviewers for their time and effort in reviewing our manuscript, entitled: “Partial prion cross-seeding between fungal and mammalian amyloid signaling motifs” by Bardin et al. We considered carefully their comments and modified our preprint accordingly (new version posted here) and address the remarks and criticism of the reviewers in the response provided below.
The editors’ summary of the review read as follows:
Summary
Bardin and colleagues identify and characterize a third prion system in P. anserina based on a cognate innate immunity signalosome comprised of PNT1/HELLP. The authors demonstrate that the three prion pathways operate orthogonally without cross-seeding; however, the newly identified PNT1/HELLP prion can be cross-seeded by the putatively homologous human necroptosis pathway when it is reconstituted in P. anserina, which further supports an evolutionary relationship between them. The review has identified substantive concerns, which limit the novelty of the work and would require significant new studies to address the mechanistic gaps. These concerns include prior work revealing several major tenets including prion activity for PNT1/HELLP in C. globosum and evolutionary conservation to the mammalian necroptosis pathway and the absence for robust experimental support for cross-seeding, or the absence thereof, membrane disruption as the cause of incompatibility, and for the relationship among toxicity, growth, protein state, and protein interaction. Concerns were also raised about the data presented, or absent, in terms of replicates, frequency of observations, and variability.
It is our understanding that the editors and reviewers raise two types of concerns. One relates to the novelty of the work. The second type directly questions the experimental soundness of some of the presented results. We will briefly respond to the criticism regarding novelty and in detail to the methodological critique. We show the existence of a third PFD-based cell-death inducing system in Podospora, that human RHIM-motifs form prions in Podospora and that RHIM-prions partially cross-seed with PP-fungal prions. These results are nonetheless novel and do shed light on the biology of Podospora and the relation of fungal and mammalian amyloid signaling motifs. Regarding the second group of concerns, we think that by clarifying certain approaches and by giving experimental results in full detail, we are able to wave many of the criticisms. For the remaining points (essentially the question of the HELLP membrane interaction), we amend our preprint to point at the delineation of experimental results and interpretation explicitly. We gratefully acknowledge the editors and reviewers input as a mean to improve the quality of the preprint and realize in light of some of these comments that the manuscript lacked in clarity at place and that detailed results tables (that were summarized in the original preprint for the sake of conciseness) should indeed be included. But having said that, it is our intention to stand our ground regarding the central claims of the paper (as they appeared in the abstract of the preprint).
Reviewer #1
Bardin and colleagues identify and characterize a third prion system in P. anserina based on the PNT1/HELLP NLR-based signalosome based on the amyloid signaling motif PP from Chaetomium globosum. The C-terminal domain of HELLP is shown to exist in either soluble or aggregated states based on fluorescence microscopy of tagged protein in vivo, termed the [pi] state, and to form amyloid in vitro. These distinct states can be propagated independently and induce conversion of full-length HELLP upon cytoplasmic mixing, which leads to cell death. The PNT1 N-terminal domain also forms foci in vivo and can seed conversion of HELLP, also leading to cell death. The C-terminal domain of C. globosum HELLP and the RHIM regions of mammalian RIP1 and RIP3, which both contain PP motifs, can cross-seed HELLP conversion to the aggregated state but the other known P. anserina prions [Het-s] and [phi] are unable to do so.
Support for the model proposed is generally qualitative in nature, with multiple instances of data described but not presented, including the timing of conversion to the aggregated state, revision of the aggregated state in meiotic progeny, the frequencies of conversion and co-localization, and the correlations between growth and prion phenotype. For the data presented, replicates, frequency of observations, and variability are not reported.
It is unclear to us what is meant by “the model proposed”. It is not our understanding that we are proposing “a model” in this paper. The results that we claim are:
-There is a third NLR/HELL protein pair involving amyloid signaling in Podospora
-There is no cross-seeding between HELLP PFD and the two other Podospora PFDs (HET-s, HELLF)
-RHIM can form a prion in Podospora
-There is a partial prion cross-seeding between PP PFDs and mammalian RHIM in vivo in Podospora
These are the statements made in the abstract of the preprint. It is our opinion that these central claims stand in face of the reviewers criticism. We shall attempt to provide whenever possible quantitative details regarding the points raised.
Specifically:
the timing of conversion to the aggregated state
There are two types of experimental situations here. In certain sets of experiments, spontaneous conversion to the prion state is measured at different subculture durations (5, 11, 19 days of subculture) (as appears in Table 1). When induced conversion (cross-seeding) is assayed, the conversion process is measured at a single time point. Details of the timing of assay of the conversion are given in the material and methods section (and now given in Table 1).
revision of the aggregated state in meiotic progeny
Details of the progeny of a specific cross involving curing of the [π] prion are now given. Among 20 meiotic progeny containing the GFP-HELLP(214-271), 3 were cured.
the frequencies of conversion
Possibly the statement that the results are “generally qualitative” comes from the fact that several conversion experiments or barrage interaction results were presented in tables with a binary output (+ or -) in the original preprint. This presentation was chosen because the replicates of these experiments yielded only monotonous all-or-none results. All tested strains were either converted (+) or not (-). In all tables, the number of tested strains and the number of replicates per strain are now given (Table S1 to S6). This presentation results in quite boring tables but we think that this should eliminate this ambiguity.
and co-localization
For all co-localization experiments, in addition to representative micrographs, counts of independent observations for each phenotypes and of co-localizing dots are given in Tables S7 and S8.
the correlations between growth and prion phenotype.
As there is no toxic effect of prion itself in absence of HELL or HeLo containing proteins (published results for [Het-s] and [φ], and verified here for [π] and [Rhim]), this last remark appear to apply to RHIM/HELLP co-expression that results in growth defects. We observe that strains co-expressing RHIM and HELLP are affected in their growth when there are infected with [Rhim] prions. These results are presented in Table 2. We based the conclusion that the growth defect relates to acquisition of the prion phenotype because the growth defect occurs after contact with a prion infected strain. This increase in the number of strains with a growth defect requires presence of the corresponding PFD in the recipient strain. Finally, the same table presents as positive control a similar experiment with homotypic [π]/HELLP interactions.
In addition, a mechanism is proposed to explain the toxicity associated with HELLP conversion to the aggregated state - membrane localization - but this model is not supported by robust data such as a marker for the membrane in the fluorescence images or a biochemical fractionation. Moreover, the absence of functional data, such as mutations that disrupt amyloid formation, leave the model with correlative observations to support it.
We agree that we do not prove membrane association for HELLP. Considering the precedent of HET-S, it is however a plausible explanation for the documented cell-death inducing activity. We acknowledge that we do not provide experimental evidence based on biochemical fractionation or dual labeling that HELLP relocates to the membrane (this would probably require confocal microscopy). What we due claim however is that in this regard HELLP behaves analogously to HET-S, CgHELLP and HELLF. We have modified the text of the preprint to specifically make the statement that proof of membrane localization would require other approaches (in particular biochemical fractionation).
The reviewer calls for mutations that disrupt amyloid formation and that should accordingly abolish HELLP toxicity. While this type of experiment is not lacking interest (this exact type of study has been made in the case of HET-S), we feel that at the present stage the fact that toxicity of HELLP is conditional and occurs specifically in interaction with [π] (not [π*] or other Podospora prions) is a sufficient support to legitimate the suggestion that HELLP functions analogously to HET-S, HELLF and CgHELLP by activation through amyloid templating.
Finally, observations on the C. globosum system decrease the novelty of the observations.
We address this comment below (response to substantive concern 1 of the reviewer #2).
Reviewer #2
This work reports the discovery of an amyloid-based cell death signaling pathway in the filamentous fungus, Podospora anserina. This makes the third such pathway in this fungus. As for the others, the amyloid in this case has prion-like activity, is selectively nucleated by a cognate innate immunity sensor protein, and results in activation of the membrane-disrupting activity of the protein. They show that all three pathways operate orthogonally - that is without cross-seeding. In contrast, cross-seeding did occur between this pathway and the putatively homologous human necroptosis pathway when it is reconstituted in P. anserina, which further supports an evolutionary relationship between them.
Substantive concerns:
1) The novelty of this finding is somewhat dampened by this group's prior demonstration of several of the major points of interest in previous papers. They had previously discovered and characterized the homologous pathway in a different fungus, and suggested an evolutionary link between fungal amyloid signalosomes and mammalian necroptosis using strong bioinformatic and structural evidence. In addition, they had shown that the two previously known amyloid signaling pathways in P. anserina operated orthogonally. Hence the major point of novelty, as reflected in the title, is the demonstration that this particular amyloid pathway can cross-seed the human necroptosis amyloids.
We are honestly puzzled by this comment, shared indeed also by reviewer 1. At no place in the preprint do we claim that the discovery of the PP-motif is new, we build on preceding work on CgHELLP and claim novelty on distinct aspects. While argumenting on the significance of one’s work is somewhat of a vain enterprise, we shall nonetheless point the specific interest we see in these results. As part of our longstanding attention on Podospora as a model to study fungal PCD, we consider it of interest to document that this species contains three amyloid-activated HeLo/HELL-domain cell-death execution pathways. Bioinformatic surveys suggest the co-occurrence of several amyloid motifs in different fungal genomes, it is of interest we think to document this redundancy at a more functional level at least in one system. The present study is superior to the previous one on CgHELLP in the aspect that activity of the PP-motif proteins is being studied in their native context (not in a heterologous host that diverged from C. globosum tens of millions of years ago). Then, to our knowledge, RHIM-motifs have never been shown to behave as prions. There is a non-trivial relation of the concepts of amyloids and prions. The reviewer writes in a later paragraph that amyloids are inherently self-perpetuating but this does imply that all amyloids are prions (or vice versa for that matter). Showing that RHIM forms (like PP-motifs) a prion when expressed in Podospora, stresses we feel the functional similarity between the fungal and animal signaling motifs. The formation of the [Rhim] prions and their propagation in a fungal environment was not a foregone conclusion. It is our experience that not any amyloid sequence will form a prion in Podospora (Aβ, α-syn, etc..) and the reviewer is surely more than aware of the rich literature dealing with the amyloid/prion-relation in yeast models. The Podospora in vivo system might also be of use to others to study RHIM-assembly, for instance to screen for inhibitors of RHIM-assembly. As stated by the reviewer the major novelty is the demonstration of cross-seeding between fungal and human necroptosis pathways which has so far only been suggested on the basis of a sequence similarity on a minute motif of 5-10 amino acids in length. We feel that documenting cross-seeding does strengthen the hypothesis that these motifs are evolutionary related.
2) Implications of "cross-seeding". The interspecific cross-seeding observed was modest; much lower than that for intraspecific templating between proteins of the same pathway. Specifically, it failed to induce a barrage, the puncta formed at different times, and colocalization was incomplete. More importantly, cross-seeding does not imply functional or evolutionary conservation. Consider the wide range of amyloid proteins that have been reported to cross-seed each other despite in some cases very different sequences, structures, and functions - for example the type-II diabetes peptide IAPP with the Alzheimer's peptide Aβ; the yeast prion protein Rnq1 with human Huntingtin; and the yeast prion Sup35 with human transthyretin. Although a direct comparison with the present data are not possible, these cross-seeding interactions appear comparably robust. The present demonstration of limited cross-seeding therefore seems not to add much additional support for an evolutionary relationship between necroptosis and fungal amyloid cell-death pathways.
Cross-seeding is partial and not as efficient as in homotypic or intra-kingdom interactions. This is precisely our conclusion (see for instance line 470 to 473 of the original preprint). We point at this partial effect and state that it suggests both some level of structural similarity but also the existence of functionally important structural differences between RHIM and PP-amyloids. These results are in line with the fact that the consensus RHIM and PP-motifs while sharing some common position also markedly differ on others. The specificity of the cross interaction between [π] and [Rhim] prions is also supported by the absence of cross-reaction between [π] and the other Podospora prions (or between [Rhim] and [Het-s]). The same is true for the partial co-localization. These results serve as a functional context that will allow future structural data on the fold of the PP-motif to be meaningfully compared to the RHIM-structure. To insist on the partial nature of this cross-seeding underlying both relation and differences between PP and RHIM, we propose to modify the title of the manuscript to “Partial prion cross-seeding between fungal and mammalian amyloid signaling motifs”.
The reviewer states : “More importantly, cross-seeding does not imply functional or evolutionary conservation”. Absolutely so. But when two amyloid forming regions show sequence similarity (not just composition bias) and both work as functional amyloid signaling motifs leading to necroptotic cell-death then cross-seeding is a further support (not proof) of evolutionary and functional conservation.
3) Rigor of the fusion experiments. In all cases, despite having generated and validated the use of RFP- and GFP-labeled proteins, all fusion experiments to examine cell death microscopically (using Evans Blue staining) were between two GFP-expressing strains. This is frustrating because it makes it impossible to know from the images alone which of the two proteins is expressed in which cells, and in which cases of mycelia crossing paths is fusion occurring. I must therefore rely entirely on the labels provided, but they sometimes appear implausible. For example, the lower fusion event demarcated in Fig. 3C left panel would have been expected to allow GFP levels to equilibrate across the point of contact; instead there remains a sharp transition in GFP intensity between the two mycelia (third panel) indicating the cytoplasm is not being shared at the time of the image. In Fig. S8 top row, there is no apparent relationship between cell death and HELLP-GFP; moreover, cell death is seen occurring in mycelia containing either punctate or diffuse GFP-RIP3. While I appreciate that Evans Blue fluorescence may overlap with that of RFP (which should be stated) and preclude its visualization without multispectral imaging capabilities that may not be available to the authors, alternative viability stains and fluorescent proteins could in principle have been used to avoid this problem.
Evans blue shows fluorescence that does indeed overlap with RFP fluorescence, which is the reason why we used GFP labeled proteins which is indeed less convenient to distinguish strains. But Evans blue staining allow clear and rapid identification of dead cells. Even with both strain labelled with GFP, strains can be identified based on diffuse versus dot-like fluorescence. Moreover, the fusion are observed in contact zone between the two strains under the microscope where the proportion of dead cells (stained cells) is drastically increased compared to the rest of the mycelium, the relative orientation and position of the filaments allows for strain identification. As for the concerns regarding equilibration levels of GFP or HELLP presence in heterokaryotic cells, it could be explained by the fact that necroptotic cell-death due to HELLP toxic effect, as for the others HeLo or HELL domain containing proteins (Seuring et al. 2012, Mathur et al. 2012, Daskalov et al. 2016, Daskalov et al. 2020), is associated with blocking of the septa to limit the spreading of cell-death through the entire mycelium. Fungal incompatibility is associated both with cell death and compartmentation of the mycelium.
We thank the reviewer to bring to our attention the issues that may be encountered to clearly identify heterokaryotic cells on these images. Therefore, cell death imaging is presented in the new preprint using methylene blue allowing the use of RFP and GFP labeled proteins to identify unequivocally heterokaryotic cells.
Minor Comments:
1) The significance of these proteins forming "prions", as opposed to (merely) amyloids, should be articulated. This is important because prion-formation per se is irrelevant to the cell-level functions of the proteins, as nucleation of the amyloid state causes cell death and hence precludes their persistent/heritable propagation. Amyloid by nature is self-perpetuating at the molecular level and hence would seem to explain the properties of the protein. The discussion about possible exaptation of these pathways for allorecognition could be expanded or clarified in this regard.
These are interesting points. Prion and amyloids are terms with different field of application. The term prion is only meaningful in vivo. We use it preferentially here, because for the most part we document prion propagation and only indirectly amyloid formation. We feel however that it might be premature to conclude that the prion-behaviour is totally irrelevant to the function of these proteins as signaling devices. This all depends (as for other prions) on the actual balance between toxicity and infectivity. It might well be that HELLP propagates part of the amyloid signal before it actually leads to cell death. Please note that even full length HET-S can be observed in certain growth condition in the form of dots and may thus partition between a toxic and an infectious fraction.
2) Colocalization between two proteins does not imply that one has templated the other to form amyloid, even when both are capable of forming amyloid independently (see https://doi.org/10.1073/pnas.0611158104 ).
We fully agree. We have corrected the labelling of the figures that document co-localization that were previously labelled as cross-seeding experiments.
3) Statements of partial cross-seeding are supported by quantitation (Fig. 8). In contrast, the authors appear to use qualitative observations to support rather definitive statements about the "total absence of" (line 344) of cross-seeding between other pathways.
Quantitative data are now given regarding the experiment presented line 344. It is true that the statement “total absence of” relates to the absence of detectable cross-seeding in the experimental setting that was use. Here in this specific case, no prion formation of [Het-s] was detected in a total of 18x2x3 infection attempts with [Rhim] prion donor strains (18 transformants for each [Rhim]-type in triplicate).
4) Fig. S9. "Note that induction of [Rhim] in transformants leads to growth alteration to varying extent ranging from sublethal phenotype to more or less stunted growth." Can the authors suggest an explanation for this heterogeneity? From my limited perspective, it suggests the existence of amyloid polymorphisms (i.e. a prion strain phenomenon), which is quite unexpected given the lack of polymorphism among known functional amyloids in contrast to rampant polymorphism among pathological amyloids. Hence the phenomenon could be interpreted as suggesting that amyloid is not an evolved/functional state for the PP motif. In any case the phenomenon is interesting and merits further discussion.
Phenotypic variability in this experiment can be explained by variation of expression levels of the transgene and prion curing. Transformation occurs through ectopic integration in these experiments (there are no autonomous plasmids available for Podospora). As a consequence in any given experiment, the transformants will display different copy number and integration sites of the transgene and hence variability in expression level. An additional cause of variety is “escape” a due to counter-selection when strain show self-incompatibility, fungal articles in which the transgene causing incompatibility is mutated or deleted will escape cell-death and resume growth. This is very typical of self-incompatible strains and has been largely documented and used as an experimental tool for mutant selection in Podospora and other filamentous fungi. This phenomenon typically leads to sector formation. Then in the specific case of experiments involving prion proteins in addition to these mechanisms leading to genetic variability, “escape” can also occur through prion curing. If a prion causes self-incompatibility, growth recovery occurs through prion curing (this has been largely studied in the case of the [Het-s]/HET-S interaction). We do not formally exclude the possibility that part of the variability may reflect prion strain formation but other explanations should probably be considered more likely, as indeed we have no evidence for strain formation for any of the wild –type functional prion motifs we have characterized so far in fungi.
Reviewer #3
Three distinct amyloid-based cell-death pathways in fungi have been reported. The authors of the current manuscript extend their previous work of the HELLP/SBP/PNT1 pathway in Chaetomium globosum and describe a similar system in P. anserina. It is shown that the amyloid signaling domain of PTN1 can form a prion in cells deleted of HELLP, which is otherwise activated by the prion to cause cell death. Using this artificial system, the authors test whether the related RHIM motif of the human RIP1 and RIP3 protein can also form a prion in P. anserina and whether RHIM amyloids as well as other fungal amyloid-forming motifs can cross-seed PTN1.
The experiments are well executed and explained but I have a few suggestions:
1) Amyloid cross seeding is usually assayed in vitro using purified protein fragments. The artificial genetic system used here is certainly clever but the expression level of different proteins needs to be measured for better comparison of cross-seeding efficiencies.
We feel that the in vivo system presented here has important advantages, in particular is it less “artificial” than in vitro seeding in the sense that at least HELLP is in its native cellular context. Note also that the cross-seeding experiments are done with several distinct transformants which as explained above represent different expression levels of the transgene.
2) Page 16, line 333-334 and Fig 8: How were recipient strains sampled? How random was it? How many samples?
We thank the reviewer to bring this to our attention and to address these shortcomings, we added precisions on samples selection and numbers in results and in methods section.
3) Jargons/abbreviations. Page 19, line 405; Page 20, line 429: What are PAMPs, MAMPs, and PCD?
These abbreviations have been spelled out.
We would like to thank the three reviewers for their efforts and the constructive feedback. Below, we describe how we will address the reviewers’ comments in an updated manuscript.
Summary:
All of the reviewers expressed concerns about the advance that the work described in the paper represents. These issues were a focus of the consultation among the reviewers. The main concern is that the work needs to go beyond demonstrating that some ganglion cells exhibit nonlinear integration for naturalistic inputs - as that point is quite well established in the literature. The comparison between natural stimuli and gratings could help in this regard, but several issues confound that comparison (e.g. differences in dynamics of the two types of stimuli). These concerns are detailed in the individual reviews below.
Reviewer #1:
This paper investigates how retinal ganglion cells integrate inputs across space, with a focus on natural images. Nonlinear spatial integration is a well-studied property of ganglion cells, but it has been largely characterized using grating stimuli. A few studies have extended this to look at spatial integration in the context of natural images, but we certainly lack a comprehensive treatment of that issue. The current paper has a number of strengths - notably using a number of complementary stimuli and analysis tools to study a large population of ganglion cells and linking properties of responses to artificial stimuli with those to natural stimuli. It also has a few weaknesses (some detailed carefully in the paper) - such as the inability to identify ganglion cell types (aside from a few), and to pinpoint specific circuit mechanisms. These are limitations of the techniques used. This is not a request as much as setting the context of the contribution of the paper. Generally the paper was in good shape, and the data supported the conclusions well. I do think there are a number of issues that could be strengthened. Those are listed below in rough order of importance.
Statistical correlations in natural scenes:
A number of analyses in the paper rely on estimating the spatial contrast from an image and comparing the dependence of various measures of the cells' responses on spatial contrast. A danger in this analysis is that spatial contrast is likely correlated with many other statistical properties of the image, so attributing a given response property to spatial contrast has some potential confounds. This issue should be discussed as a possible caveat, unless the authors can rule it out. The paper, accurately, describes the results in terms of correlations (and not causal relationships), but some discussion of the complexity of natural image statistics would be helpful.
Spatial contrast is defined in our work via the variance of pixel intensity inside the receptive field. Indeed, spatial contrast may reflect different aspects of visual scenes, such as object boundaries, textures, or gradients in light intensity. Differences in the effects of these image features on a ganglion cell’s response will not be captured by our analysis. However, the goal of relating spatial contrast to spike count was primarily to analyze whether the spatial structure of light intensity inside the receptive field was related to the response of a given ganglion cell (beyond the mean illumination), and the pixel intensity variance provides a simple, straightforward measure of this spatial structure. To clarify this aspect and better relate it to the complexity of natural images, we will add a corresponding paragraph in the Discussion.
Comparison of grating and natural scene spatial scale:
The section starting around line 233 was confusing for several reasons. First, this section starts by measuring the spatial scale associated with the grating responses, and then comparing that to LN model performance for natural inputs. It's not clear why the spatial scale is the relevant aspect of the responses to gratings. Indeed, the next paragraph provides a measure of the relative sensitivity of the nonlinear and linear response components (via a comparison of F1 and F2 responses). It would be helpful to include some initial text to motivate the different measures of the grating responses and to anticipate that you will look at both spatial scale and sensitivity.
A related issue that bears more directly on the scientific conclusions comes up later in the blurring experiments. The issue is whether it is valid to directly compare the apparent spatial scale of nonlinear responses to images (estimated via blurring) with that of the grating responses. Natural images should have much higher power at low spatial frequencies, and this may strongly impact the spatial scale identified with the blurring experiments.
We agree that the writing may not have been entirely clear, and we will reorganize the material to discuss the extracted spatial scale and nonlinearity index in parallel as suggested. Regarding the difference in spatial scales from reversing gratings and blurred natural images: yes, it is also our interpretation that the power at low spatial frequencies plays a key role. Our main point here was to assess whether and to what degree the typical analyses of spatial nonlinearity as measured from reversing gratings translate to natural images despite the differences in spatial and temporal structure of the two stimulus classes. In a revised manuscript, we will make sure to earlier clarify the role of low spatial frequencies.
Clustering of orientation-selective cells:
An interesting suggestion in the paper is that the orientation-selective cells can be divided into two groups that differ in their spatial integration properties. Do these groups represent different orientations, as suggested in the text? That seems a simple piece of information to add. Related to this, I would suggest moving Figure S4 into the main text.
We do not have information about the absolute preferred orientations of the orientation-selective (OS) cells, as we did not keep track of retinal orientation when placing the retinas on the multielectrode array. At this point, we can therefore only rely on indirect analyses of relative preferred orientations between pairs of OS cells in the same retina. These indicate that pairs of two nonlinear OS cells tend to have aligned preferred orientation (and similarly for pairs of linear OFF OS cells), but pairs of a linear and a nonlinear OFF cell tend to have divergent preferred orientations. This is shown in Fig. S4C. For a revised manuscript, we will consider integrating Fig. S4 into the main text, as suggested.
Presentation of checkerboard stimuli and results:
The checkerboard analysis, particularly how it isolates properties of spatial integration, could get introduced more thoroughly for a reader unfamiliar with it. A related issue is how well the chosen isoresponse contour captures structure in the full distribution of responses. In some cases that looks pretty good, but in others it is less clear. Could you add a supplementary figure or something similar that characterizes how consistent the isoresponse contours are for different response levels?
These are good suggestions, and we will aim at clarifying the analysis as proposed and add information about the consistency of iso-response contours for different response levels. In the present analysis, the iso-response contours are used just for illustration, whereas the quantification of rectification and integration of preferred contrast are extracted from specific points in the stimulus-response space, which we found to work robustly for a population analysis without being strongly effected by threshold or saturation effects of the cells. We will explain this more clearly in a revised manuscript.
Drift in responses over time:
Some of the rasters - e.g. the bottom left in Figure 1C - show considerable drift over time. It is important that this drift not be interpreted as a failure of the LN model and hence indicative of nonlinear spatial integration. Can you test for drift like this across cells, and exclude any that seem potentially problematic? More generally, some assurance that the variability in the responses for a given generator signal value is real variability across images is needed.
The presentation of all 300 natural images over ten trials takes about 50 minutes and some drift over this period seems unavoidable. To minimize systematic effects of experimental drift on the measured average responses for different images, we applied randomization within trials, which assured that all images were presented once in random order in each trial before the next trial started. In addition, to quantify the real variability over images of the average response for a given generator signal, we applied a goodness-of-fit measure (CCnorm) that takes into account variability over trials.
We now also tested directly for the drift mentioned by the reviewer, but observed sizeable effects in only a small subset of cells that were included in the analysis. In most cases, drift corresponded to a global scaling that approximately affected responses to all images proportionally. This is reflected in a high correlation over images between the average responses of the first five and last five trials; 94% of analyzed cells had a correlation coefficient of at least 0.7. Such global scaling of responses does not affect the analysis of differences in average responses. In a revised manuscript, we will provide analyses of drift effects and exclude cells that contain drift effects that appear to deviate from global response scaling.
Reviewer #2:
Summary:
Understanding how retinal ganglion cells respond to natural stimuli is a central but daunting question, which retinal neurophysiologists have begun to tackle recently. Here Karamanlis and Gollisch perform large-scale multi-electrode recordings in the mouse retina and demonstrate that the responses of many ganglion cells cannot be predicted by standard linear-nonlinear models (L-LN). They go on to test a variety of clever artificial stimuli that emphasize and allow for the quantification of the non-linear aspects of RGCs responses and convincingly demonstrate that non-linear processing is associated with sensitivity to fine spatial contrasts (subunits) and local rectification. While these aspects of RGC receptive fields have been previously described, demonstrating their applicability to natural vision is a significant advancement.
Major Comments:
My first main concern is with the way the paper is written. It does not highlight the significant advancements but rather emphasizes what is already known from other studies. For example, many of the conclusions of non-linear spatial integration & signal rectification arising in bipolar cells have been well described previously. By contrast, novel aspects like the sensitivity of reversal gratings being unrelated to LN model performance for natural scenes should be explained more in detail. The authors should more clearly state the major advancements that are being made here beyond what has already been shown previously (e.g. Turner and Rieke, 2016)
It is possible that our efforts to provide context by relating our results to established findings in retinal signal integration overshadowed the novel aspects of our work. As suggested, we will aim at pointing out these aspects more clearly. For example, compared to the work of Turner and Rieke (2016), we a) focused on a different species with more diversity in accessible RGC types, b) generalized the connection of spatial integration and natural scene encoding to a wider range of cell types (e.g. including also spatially linear and nonlinear ON-OFF cells as well as cells that are inversely sensitive to spatial contrast), and c) developed methods to assess and quantitatively characterize subunit nonlinearities with multielectrode recordings of many cells in parallel, without the need for intracellular recordings or knowledge of the receptive field location.
Second, the authors never include non-linear subunits in their model to demonstrate improved performance. Testing models with filters that incorporate rectification and convexity as experimentally determined will enable them to show their utility more convincingly. Without this, the reader is left with the conclusion that there are RGCs that exhibit non-linear or linear spatial integration (already known) and that non-linear integrators cause LN models to perform poorly with natural images (Turner and Rieke, 2016).
The aim of the present work was to assess how well models with linear receptive fields account for responses to natural images in various cells of the mouse retina and whether the models’ shortcomings can be related to the cells’ spatial stimulus integration characteristics. While we agree that models with nonlinear subunits could help support the conclusions, fitting such models to recorded data is – we believe – beyond the scope of the current manuscript. The many parameters of nonlinear subunit models, such as the number, shape, and layout of subunits or their nonlinearity and weight, all likely vary considerably across the diverse population of cells in our recordings. To avoid extensive parameter fitting, simplified models with ad hoc selection of subunit layouts and nonlinearities could help assess whether spatial nonlinearities are important, as in the work by Turner and Rieke (2016). Instead, as an alternative, we chose to analyze the importance of spatial nonlinearities via the effect of spatial contrast in images with similar mean intensity in the receptive field (e.g. Fig. 2). For our data, an advantage of this approach is that it is directly applicable to cell types with diverse spatial integration characteristics, such as the cells that are inversely sensitive to spatial contrast, which wouldn’t be captured by a standard subunit model with rectifying subunit nonlinearities. In future work, however, we plan to analyze subunit models that can account for the diversity of observed response patterns.
Third, I'm not sure how 'natural' their natural images are, given static images are flashed over the cell intermittently. While such stimuli might simulate some sort of saccadic eye movements, whether this is relevant for mouse vision is not clear. Would linear models be more predictive for responses to natural movies? Some discussion on this issue would be helpful.
Rather than aiming for fully natural movie-like stimuli, we used flashed images in our work to focus on aspects of spatial integration. This indeed entails a simplification of the temporal structure of natural stimuli, which was intended, but it preserves natural spatial structure, such as the occurrence of objects, boundaries, textures, and intensity gradients, as well as continuously decreasing power for higher spatial frequencies. Nonlinear spatial integration in the presence of this natural spatial structure will likely also shape responses under natural movies. To clarify this approach, we will re-evaluate our wording regarding the application of natural stimuli in our work and discuss the simplification compared to natural movies, as suggested.
Reviewer #3:
The manuscript by Karamanlis and Gollisch examines the responses of mouse retinal ganglion cells (RGCs) to natural stimuli. The primary conclusion of the manuscript is that spatial integration of stimuli within the receptive field is nonlinear. This nonlinear integration is consistent with "local signal rectification". This results in a set of RGCs that are sensitive to spatial contrast within the RF. The Authors also note the presence of cells that are suppressed by contrast and cells that prefer uniform stimulation of the RF. To reach these conclusions the authors use multi-electrode array recordings from isolated mouse retina. Spatial RFs are estimated using white noise stimuli, which are then used to generate a null-model for linear spatial summation. They compare predictions of this null-model to the responses of the same RGCs to briefly flashed natural images. The authors find some RGCs that are consistent with this null model and many that are not consistent. The authors correlate deviations from linear spatial summation to deviations revealed by contrast reversing gratings. They also used a mixed-contrast, flashed-checkerboard paradigm to map the contrast tuning and rectification of RF subunits. Finally, the authors show that some of these results track with functionally distinct RGC types such as direction-selective and "IRS" RGCs.
The data and analyses presented in this manuscript are high quality. However, I think the study is largely consistent with many previous studies that demonstrate nonlinear spatial integration among RGCs in the mammalian (including mouse) retina. I think the Authors view the use of natural stimuli as a major departure from previous work, but I'm not convinced of this for two reasons. First, I don't see a compelling reason to think that results using contrast reversing gratings or other 'textured stimuli' (e.g. Schwartz et al Nat Neuro 2012) would fail to generalize to flashed natural scenes. Second, the implicit claim here is that a 200ms flashed natural scene interleaved with an 800ms gray screen is a natural stimulus. I think this assumes a lot about the space-time separability of the RF mechanisms, and these assumptions are not well justified.
Major Concerns:
1) I think the introduction of the manuscript is building a straw man argument, suggesting that many (or most) scientists think the retina is predominantly linear. A pubmed search of 'retinal ganglion cell' and 'nonlinear' produced more than 300 studies. Specifying subunit nonlinearity produces 28 studies. The discovery of subunit nonlinearities is roughly 50 years old and many manuscripts demonstrate Y-like receptive fields are more common across RGC types than X-like receptive fields.
The goal of our work was not to show that receptive fields of mouse retinal ganglion cell are (often) spatially nonlinear, but to test whether these nonlinearities matter for natural images. It is conceivable that spatial nonlinearities as measured with typical artificial stimuli such as spatial gratings or spatiotemporal white noise are not (as) relevant for natural images because the simultaneous occurrence of strong positive and negative contrast inside a receptive field is much rarer in natural images. Indeed, in our work we find that traditional measurements of spatial nonlinearities with reversing gratings do not provide a robust quantitative prediction of whether spatial nonlinearities matter under natural images for a given ganglion cell. As laid out in the Introduction, there is surprisingly little research yet on how spatial nonlinearities affect the encoding of natural images, and in a revised version of the manuscript, we will aim at clarifying that this is the focus of our work here.
2) The authors seem to be arguing that the spatial nonlinearities engaged by the contrast reversing gratings are not the same as those engaged by their natural scenes (Figure 3). However, I think the authors are assuming too much that the spatial and temporal components of the RFs are separable. The flashed natural scenes are interleaved with relatively long gray screens. The contrast reverse granting are reversed in a square-wave fashion with no interleaved gray screen. These distinct spatiotemporal dynamics in the stimuli seem likely to explain the difference. This would also seem likely to explain why the flashed checkerboards in Figure 4 produced results more correlated to flashed scenes in Figure 1. In summary, I don't see a strong reason to think the authors are observing anything other than subunit rectification of the sort described by Hochstein and Shapley in the 1970s and followed up in many subsequent studies.
We do not think that spatial nonlinearities as observed with reversing gratings or with natural stimuli are related to different mechanisms. The point of our analysis was rather to assess whether typical assessments of spatial nonlinearities with reversing gratings allow quantitative predictions about the relevance of spatial nonlinearities under flashed natural images, and we find that this is often not the case. We believe that this is largely due to the differences in spatial structure, in particular, the prevalence of high-contrast edges in the gratings. Yet, indeed, differences in temporal stimulus structure might also contribute. We actually tested flash-like presentations of gratings in some of our recordings, and results were quite similar to those obtained with contrast-reversing gratings and led to the same conclusions. We will describe this in the revised manuscript for clarification.
3) It is not clear to this reviewer that flashed natural images interleaved by a gray screen is qualitative more natural than white noise, sinusoidal gratings, or square-wave gratings.
The spatial structure of natural images is the focus of the present work. It is in this aspect that flashed photographs are more natural than typical artificial stimuli like spatiotemporal white noise or gratings. In particular, natural images contain a broad spectrum of spatial frequencies with relatively more power at smaller frequencies, and they combine occasional edges with intensity gradients and textures. Gratings, for example, are characterized by high power at large spatial frequencies, that is, high spatial contrast, which is well suited for triggering effects of spatial nonlinearities but occurs much more rarely in natural images. Thus, understanding whether spatial nonlinearities are important in a natural setting requires considering stimuli that match the natural spatial structure. It seems likely that nonlinear spatial integration observed under flashed presentation of natural images remains relevant when stimuli are supplemented with natural temporal structure, even though the latter may likely trigger additional effects that shape the responses (e.g. adaptation or nonlinear temporal integration).
4) The null-model constructed by the authors in Figure 1 assumes the RF follows a specific functional form (e.g. Gaussian). However, many studies show that individual RFs frequently exhibit strong deviations from a Gaussian RF. To what extent are the deviations from the null model produced by deviations from linear summation or just linear mechanisms that deviate from the specific parametric form imposed by the model?
Measuring the detailed structure of receptive fields (RFs) with high precision from time-limited experiments is a challenge, and using a fitted (elliptical) Gaussian profile is a standard procedure for limiting the effect of noise in the RF structure. We also tried using the pixel-wise spatial profile obtained from the reverse-correlation analysis as a spatial filter, but results were similar, yet often more noisy. We therefore settled on the standard procedure of using a Gaussian fit to the RF. Deviations from the Gaussian profile can indeed contribute to deviations of the model. Yet, for natural images, which have most of their power in low spatial frequencies, these deviations are likely to be small. Furthermore, our subsequent analyses show that the Gaussian RF model provides a useful baseline because it allows us to extract the relation between model deviations and image structure. In addition, the results from the model analysis were supported by the findings under presentation of blurred natural images, which did not require any assumptions about the underlying RF model. In a revised manuscript, we will point out that relying on Gaussian RFs is a choice that we make and that deviations of the receptive field structure may contribute to decreased model performance, but that the subsequent analyses support the usefulness of the applied Gaussian RF model.
5) It was unclear how the authors rule out the contribution of differences in (nonlinear) temporal integration to the effects in this study. In general, RGC RFs are not space-time separable, and it seems that the analyses in the manuscript assume they are.
Our choice of using flashed images as stimuli with no temporal structure beyond onset and offset and assessing responses via elicited spike counts was motivated by focusing on spatial stimulus integration and minimizing effects of temporal processing. Nonetheless, our extraction of receptive fields from measurements under spatiotemporal white-noise stimulation uses a space-time separation of the spike-triggered average. Thus, the lack of space-time separability of ganglion cell receptive fields can contribute to the putative underestimation of surround components, which we have discussed in the manuscript. In a revised manuscript, we will add an explicit reference to the issue of space-time separability.
6) This study overlaps significantly with Cao, Merwine and Grzywacs (2011), 'Dependence of retinal Ganglion cell's responses on local textures of natural scenes', Journal of Vision. This article is not cited here, but in my view, the major conclusions are similar.
Thank you for pointing us to this paper, which is indeed relevant for our work. Both the Cao et al. paper and our manuscript evaluate the effect of spatial contrast in natural images by relating spatial contrast to response deviations from a linear-RF model, albeit with different methods. An important difference, apart from the different species, is that our work then focuses on relating the identified effects of spatial contrast to functional characterizations of the specific nonlinear operations inside the receptive field (e.g. rectification). Furthermore, we also focus on the diversity of spatial-integration properties between cells and cell types, including the description of spatially linear cells and cells that are inversely sensitive to spatial contrast. In a revised manuscript, we will add a comparison to the methods and results from Cao et al.
7) In my experience, the strength of subunit rectification can be labile during ex vivo experiments. What controls have the author's performed to ensure the effect they are studying remain stable over the duration of their recordings?
Experimental rundown could, of course, affect subunit rectification as well as other response aspects, such as overall sensitivity. However, we observed that responses for different repeats of the same natural images were typically quite stable over the course of the hour-long stimulus. As also discussed in the response to Reviewer 1, we now analyzed how responses to late trials deviated from responses to early trials and found that only a small subset of cells displayed sizeable drift. Furthermore, those cases were mostly affected by a global drift in response size, keeping the relative responses for different images approximately constant. (For 94% of cells, the correlation of images was larger than 0.7 between average responses for the first five and for the last five trials; approximately on the level of estimated random trial-by-trial variability.) This indicates that the features of stimulus integration did not change substantially over the course of the experiment. In addition, nonlinearities as assessed with our flashed checkerboards were strongly correlated to nonlinearities under natural images, despite the fact that these stimuli were applied 1-2 hours apart. Thus, the strength of subunit rectification appears to be sufficiently stable to allow comparison over different stimuli.
We would like to thank all three reviewers for their great effort and their helpful and detailed comments on our manuscript. The reviewers noted the significance of the novel concept we present here, however, major weaknesses of the manuscript were cited in the comments from each reviewer. The criticisms can be summarized into three major categories: 1) missing key controls and analyses in the HEK293 cell models we used; 2) the HEK293 cell models being the only system used for this study; and 3) some evidences that support the mechanistic conclusion are based on correlations and lack direct demonstration for causality. We have addressed some of their concerns in the updated version of the manuscript and believe that it improved our manuscript. We would like to also briefly respond to the comments here:
First of all, we apologize for not including some key controls and analyses in our manuscript. We have now revised Figure 1 and added 5 additional Supplementary Figures to provide those controls and analyses. The mistake was caused in part by our lack of perception from an audience point of view. Our HEK293 cell system has been rigorously validated for studying TyrRS nuclear deficiency at endogenous level of expression. Those evidence were published (Wei et al., 2014, Molecular Cell, PMID: 25284223) and cited in this manuscript. But this clearly was not enough; each new experiment needs to have its independent controls and analyses, which we did preform and confirm but failed to include in the original manuscript. This mistake caused major confusion and a lack of confidence in our conclusions. Now those controls and analyses have been included in the revised manuscript as listed below:
Supplementary Figure S1 shows that 1) the ΔY/YARS and ΔY/YARS-NLSMut HEK293 cells we generated express TyrRS (WT or NLS mutant) at a level similar to endogenous TyrRS expression in the original, unmodified HEK293 cells; 2) H2O2 treatment stimulates the nuclear translocation of TyrRS; and 3) ΔY/YARS-NLSMut cells are deficient in TyrRS nuclear localization with or without H2O2 treatment.
Figure 1A is expanded to include nuclear fractionation and Western blot results as controls to show that 1) overall and cytosolic levels of TyrRS (WT or NLS mutant) do not change obviously during H2O2 treatment; and 2) ΔY/YARS-NLSMut cells are deficient in TyrRS nuclear localization with or without H2O2 treatment.
Supplementary Figure S2 shows equal expression of different transgenes in our experiments (Figure 1C and Figure 2D).
Supplementary Figure S5 is added to strengthen the evidence that co-factors are required for TyrRS to regulate target gene expression. Because HDAC1 is a shared co-factor for both TRIM28 and the NuRD complex, we used an HDAC1 inhibitor Trichostatin A (TSA) to test if it can affect the transcriptional repressor activity of TyrRS. Indeed, TSA treatment blocks the inhibition effect of overexpressed TyrRS on its target gene transcription.
Supplementary Figure S6 shows equal expression of WT and E196K TyrRS and the gain-of-function effect of the E196K mutation in suppressing target gene expression and protein synthesis.
Supplementary Figure S7 shows the quantification analysis of caspase-3 cleavage as detected by Western blot analysis in Figure 5B.
For the second major criticism which is the sole use of the engineered HEK293 cell models in the study, we agree that the main conclusions of this paper need to be confirmed in an additional cell system and ideally with the endogenous TyrRS. In fact, we have generated TyrRS nuclear deficient mice by mutating the NLS of the endogenous YARS gene and, by using the mouse fibroblasts, we have confirmed that protein synthesis is overactivated in TyrRS nuclear deficient cells. Because the study of the mouse model has not been completed and it is a separate in vivo study of nuclear TyrRS with its own objectives, we prefer not to add the mouse fibroblasts data to this manuscript but will share these data with the reviewers. However, we would like to point out that the ΔY/YARS and ΔY/YARS-NLSMut HEK293 cell lines are not stable cell lines derived from single clones but instead transient transfections that were selected for in bulk. Therefore, they originated from the same starting cell line and diverged only 1-2 passages before the experiments were performed. Genetic diversion between the NLSMut and the control cell line should therefore be limited. We apologize if that was not clear from the Material and Method section.
For the last major criticism, we acknowledge that some mechanistic aspects of nuclear TyrRS have not been unequivocally demonstrated. For example, whether the direct binding of TyrRS to its target genes and the interactions of TyrRS with TRIM28 and/or NuRD complex are responsible for the endogenous TyrRS to regulate target gene expression in cells, and whether the level of transcriptional regulation on protein synthesis genes by nuclear TyrRS is sufficient and responsible for the observed suppression in cellular protein synthesis activity. While this issue is partially addressed by the new Supplementary Figure S5 (Treatment with an inhibitor of HDAC1, the shared co-factor of TRIM28 and the NuRD complex), we acknowledge that these weaknesses are in part due to the use of ectopically expressed TyrRS in the current system and can be addressed in the future by using the mouse fibroblasts mentioned above.
Svelte will not offer a generic way to support style customizing via contextual class overrides (as we'd do it in plain HTML). Instead we'll invent something new that is entirely different. If a child component is provided and does not anticipate some contextual usage scenario (style wise) you'd need to copy it or hack around that via :global hacks.
Explicit interfaces are preferable, even if it places greater demand on library authors to design both their components and their style interfaces with these things in mind.
This has already forced me to forgo Svelte Material because I would like to add some actions to their components but I cannot and it does not make sense for them to cater to my specific use-case by baking random stuff into the library used by everyone.
The point of the feature is to not rely on the third-party author of the child component to add a prop for every action under the sun. Rather, they could just mark a recipient for actions on the component (assuming there is a viable target element), and then consumers of the library could extend the component using whatever actions they desire.
Your LazyLoad image is now inextensible. What if you want to add a class? Perhaps the author of LazyLoad thought of that and sets className onto the <img>. But will the author consider everything? Perhaps if we get {...state} attributes.
Summary:
As you will see the reviewers agreed that the premise behind this manuscript is important and timely both in the context of basic auditory science and for informing technology. However, they raised largely consistent concerns about the generalizability of your observations to other auditory stimuli and to more naturalistic listening conditions.
We appreciate the reviewers’ positive assessment underpinning the significance and timeliness of our present research endeavours. We assume generalizability of our findings to more naturalistic listening conditions because the proposed model framework successfully explained the outcomes of experiments that were conducted under listening conditions differing in reverberation and source stimuli. Those differences, however, only occurred across but not within experiments and thus were not considered in the model explicitly. The set of experiments and relevant cues was chosen such that the investigation of decision strategies for the combination or selection of cues in the context of perceptual externalization could be conducted on a limited but still divers set of cues. The proposed framework allows to easily extend the set of cues. For example, in another work (see Li et al., in press), we successfully modelled the impact of situational changes of the amount of reverberation on externalization perception by extending the framework to reverberation-related cues. This further strengthens our assumption that our findings can be generalized. Nevertheless, we understand that more direct evidence for this generalizability would further increase the confidence in the conclusions we draw.
Reviewer #1:
I agree with the authors that the question at the basis of this work is timely and important both from the point of view of understanding auditory perception and for informing technology. However I am not convinced that the findings here will necessarily generalize to other stimuli/listening situations.
I think the biggest limiting factor here is that the primary data on which the modelling is based are drawn from many different studies which used different stimuli, different tasks, different presentation environments and different equipment). I can see how testing the model on existing data is an important first step, but I would think that a critical next step is to form a set of (contrasting) predictions to be tested on a single stimulus set, within a single group of participants, as a way of confirming model validity. In this experiment I would also avoid using static non-reverberant environments since we know that these factors greatly affect spatial perception.
We do not follow the reasoning why the above mentioned diversity of experimental paradigms is a limitation. On the contrary, in our opinion, the diversity of the considered experiments demonstrates robustness of our findings for a variety of experimental procedures. We agree that an additional validation experiment would further strengthen our study, but we question its necessity and still believe that the present modelling work is extensive and compelling enough to warrant publication.
Other comments:
1) The title greatly overstates the main findings, it would be toned down.
In the title, we aimed at describing the research topic in general terms accessible to a broad readership. We take your comment as an advice to state the main findings instead.
2) Intro, line 30-33 this statement is misleading. As written it appears to claim temporal aspects of auditory perception are based on short term regularity, whilst spatial perception is based on long term effects. This is not correct see e,g Ulanovsky 2004.
Agreed. We will remove the sentence or rephrase it in more general terms because the misleading distinction is actually irrelevant to our study.
3) As a reader not highly familiar with the auditory spatial processing literature I found the results section very dense and hard to follow. If you are targeting a general audience it is important to clarify concepts, avoid using abbreviations where possible etc.
Thank you for your advice. We will aim to increase the level of abstraction within the results section.
4) When discussing the various decision strategies which you tested, consider explaining how they might be implemented by the auditory system, at which stage of processing etc.
Our study approached the problem from an algorithmic point of view and did not touch upon the more detailed level of neural implementation. While the cue processing has a clear neurophysiological basis in the subcortical layers of the auditory system, we will include some speculation about the involved cortical networks in a revised version of the manuscript.
5) It is very difficult to evaluate your results without more information about the stimuli and studies from which they were taken. Whilst you do provide references, I think the paper would be much clearer if you provide a more complete description of the stimuli (even in table form; paradigms etc).
We appreciate your advice and will provide more details about the simulated experiments in a table.
Reviewer #2:
The current study compares four decision rules, factoring in seven potential acoustic cues, for predicting perceived sound externalization for single-source binaural sound with stationary interaural cues. Test stimuli included a harmonic vowel complex, noise and speech. Results show that monaural and binaural cues shape externalization. However, how listeners weighted these cues varied across the tested conditions. The authors consider the fact that some of these cues covary acoustically, by additionally testing their model on subsets of two of these cues only. No single externalization cue emerged as a clear predictor for perceived externalization. However, overall, a static cue weighting strategy tended to outperform dynamic cue weighting for predicting externalization.
Major concerns dampen enthusiasm for the current work.
1) It is unclear what neural mechanism is being tested. A premise of the current approach is that perceived sound externalization is primarily driven by acoustic cues. However, we know this not to be true. Context matters. As pointed out by the authors (l370-372), when listening to sounds processed with head related transfer functions (HRTFs) over headphones, listeners can externalize sound better when the context of the test room matches the room where HRTFs were recorded (Werner and Klein 2014).
Sound externalization is an auditory percept and as such primarily driven by acoustic cues. How those cues are used for perceptual inference is certainly context dependent. From the present study, we conclude that the auditory system evaluates deviations from a small set of expected acoustic cues in a fixed weighted (and not selective) manner. We further explain that these expectations, which are represented as templates in the model, must be adaptive to the context. This is well in line with your example of room divergence (Werner and Klein, 2004): listeners are thought to establish expectations about reverberation-related acoustic cues and evaluate incoming sensory information against those expectations with a fixed weighting between cues. If expectations are not met (i.e., acoustic cues deviate from their templates), perceptual externalization degrades.
2) Most external sounds are neither anechoic nor stationary. Therefore, any neural decision metric on externalization must have been shaped by lifelong experience with dynamic, reverberant cues for interpreting externalization. The current work mostly models stationary single source sound that was either anechoic or mildly reverberant, providing pristine spatial cues. I do not follow the author's point that this would not matter (l498-502): "While the constant reverberation and visual information may or may not have stabilized auditory externalization, they certainly did not prevent the tested signal modifications to be effective within the tested condition. In our study, we thus assumed that such differences in experimental procedures do not modulate our effects of interest." That is an untested assumption.
Others showed that the type of spectral manipulations we considered remain effective also if reverberation is present (e.g. Hassager et al., 2013) and if listeners are exposed to dynamic cues by moving their heads or the sound source (Brimijoin et al., 2013). We used the above-mentioned argument in order to motivate why we ignored certain differences across studies in the first place and the high explanatory power obtained with the proposed model framework suggests that this simplification was adequate. We agree that the above-mentioned sentence can be easily misunderstood and we will modify it by including the explanation stated here.
3) Many of the current test stimuli are perceived as ambiguous - providing 50% externalization ratings - and thus do not provide a sensitive test of brain mechanisms of sound externalization.
The field mostly agrees that auditory externalization is not a binary phenomenon but a matter of degree – we very recently published a review article that discusses this issue in detail (Best, et al., 2020). Hence, the experimental outcomes, denoted as externalization scores, ranging from 0 to 1 indicate the degree of externalization that is considered to mediate perceived egocentric distance. The externalization scores do not indicate the level of perceptual ambiguity.
We will include this explanation in the manuscript in order to prevent further misunderstanding.
4) Reverberation enhances perceived externalization, but this cannot be predicted by any of the tested decision metrics which only consider stationary monaural or binaural cues.
True, there are also other cues potentially affecting the degree of auditory externalization. Reverberation-related acoustic cues are one of them. The main purpose of our study was to identify the basic functional mechanisms that integrates or selects between various cues – the purpose was not the identification of all possible cues that may affect auditory externalization. Thus, we chose a set of experiments that can be narrowed down a priori, particularly allowing to ignore reverberation-related cues.
For the effect of reverberation-related cues, we point interested readers to another modelling study (Li et al., in press) that we conducted in parallel, in which we applied the here proposed framework also to reverberation-related cues and obtained good predictions.
On balance, this reviewer is unconvinced that the current work will generalize to realistic dynamic and reverberant conditions.
We agree with the reviewer that our study does not address dynamic and variable reverberant conditions. It was by-design limited to static conditions with fixed reverberation because we had no reason to believe that the targeted decision strategies applied to combine or select cues would be fundamentally different in more complex conditions.
S. Werner and F. Klein, "Influence of Context Dependent Quality Parameters on the Perception of Externalization and Direction of an Auditory Event," presented at the AES 55th International Conference: Spatial Audio (2014 Aug.), conference paper 6-4.
Reviewer #3:
The manuscript "Decision making in auditory externalization perception" aims to identify cues that create/hinder an auditory externalization percept by using a template-based modeling approach. The approach as well as the findings are very interesting, and the study is thoroughly conducted. However, the manuscript adds little new knowledge to the field. Furthermore, a critical discussion is missing. The authors use a template-based model, but do not discuss the possible problems with such an approach. Particularly as each condition uses another model fit. This potentially allows the model to use cues that the auditory system cannot or does not consider. Nevertheless, the approach can still teach us which cues are potentially important for auditory externalization.
1) The title seems inappropriate as the main work seems to be on the identification and combination of cues for externalization but not on the decision making.
In combination with Reviewer #1’s first comment, we understand that the title could have been more specific. We will change the title accordingly.
2) The model needs a more detailed explanation in the introduction. Otherwise the result section is not understandable without consulting the methods section.
We will carefully re-evaluate which methodological details are necessary to understand the results section on a more abstract level.
3) Add a Discussion on template-based models and fitting conditions. The risk of mathematical inspired models is that features are exploited that the auditory system cannot access. A more sophisticated front-end than a gammatone filterbank might reduce this risk. Alternatively, the use of physiologically inspired front-ends as in Scheidiger et al. (2018) might be interesting to consider. Nevertheless, I acknowledge that some of the features used in this study are backed by physiological and psychoacoustical studies.
We agree with the concern behind the use of efficient functional approximations of the auditory periphery. Interestingly, however, we are very confident that this particular approximation does not provide spurious cues, especially in the context of monaural spectral shapes, because we did cross-validate the effectiveness of those cues with a physiologically more accurate model (Zilany et al., 2014) in previous work (Baumgartner et al., 2016).
We will incorporate a corresponding explanation in the manuscript.
4) It is known that the monaural spectral shape is important for externalization, for example from the studies that you have used. Thus, I partly question the novelty of the findings.
We partly agree. It has also been suggested that interaural spectral cues are important for externalization perception. Further, it is also known that other cues contribute (e.g., reverberation-related cues as already discussed in response to the comments of Reviewer #2). Now, which cues contribute to which degree and how are they integrated? This is the main research question behind our study, with the ultimate goal to better understand the mechanisms of cue integration in the context of a perceptual inference task.
5) I am not too familiar with template based models but I wonder if there is a problem if you use your models to fit and test with the same datasets?
Cross-validation (i.e., using separate data sets for fitting/training, validating, and testing) is particularly important for complex models that allow overfitting. Such models can often be very closely fit to comparably small sets of data and thus the goodness of fit is not discriminative between those models. Here, in contrast, we compared the goodness of fit for models that contained a rather small and equal number of model parameters and this goodness of fit did strongly differ across models and was therefore informative for model selection in itself. If we separated the data sets, we would need to jointly assess the differences in initial model fits (to training data) together with the differences in predictive power (for testing data).
References:
Baumgartner, R., Majdak, P., & Laback, B. (2016). Modeling the effects of sensorineural hearing loss on sound localization in the median plane. Trends in Hearing, 20, 2331216516662003.
Best, V., Baumgartner, R., Lavandier, M., Majdak, P., & Kopčo, N. (2020). Sound Externalization: A Review of Recent Research. Trends in Hearing, 24, 2331216520948390.
Brimijoin, W. O., Boyd, A. W., & Akeroyd, M. A. (2013). The contribution of head movement to the externalization and internalization of sounds. PloS one, 8(12), e83068.
Li, S., Baumgartner, R., & Peissig, J. (in press). Modeling perceived externalization of a static, lateral sound image. Acta Acustica.
Zilany, M. S., Bruce, I. C., & Carney, L. H. (2014). Updated parameters and expanded simulation options for a model of the auditory periphery. The Journal of the Acoustical Society of America, 135(1), 283-286.
Reviewer #1:
This manuscript provides evidence that drug administration during a reconsolidation window does not necessarily prevent memory recall, as has been shown by many groups. The authors attempted to replicate several published experiments and despite demonstrating that the drugs had other effects on the animals' behavior and physiology (e.g. weight gain), no effects on memory were observed.
The paper is nicely prepared.
We sincerely thank the reviewer for these kind words and the support to publish our replication efforts.
Reviewer #2:
General assessment:
In this study, Luyten et al. aimed to replicate post-retrieval amnesia of auditory fear memories reported numerous times in the literature. They used a variety of behavioural approaches combined with systemic pharmacological treatments (propranolol, rapamycin, anisomycin, cycloheximide) after reactivation of fear memories. Interestingly, none of the treatments induced a significant decrease of freezing responses during subsequent retrieval tests. Authors strengthened their null results by using Bayesian statistics, confirming the absence of drug-induced amnesia.
Overall, the study is really interesting. Experiments and analyses are very well designed and bring some important findings to the debated topic of post-retrieval amnesia and its clinical relevance.
We are grateful that the reviewer appreciates our work and recognizes the general importance of our null findings. We genuinely thank them for the time that they took to evaluate our paper in detail and hope to provide some clarifications in our responses below.
I have nevertheless several comments for the authors to consider.
-Despite being very detailed, the authors should clarify and uniformize their Methods section and Supplemental information (e.g. number of CS, contexts used...) to improve the understanding of the different approaches. Similarly, methods for the reinstatement protocol (Exp 2) are missing.
We understand that the information in the main text is quite dense, but we explicitly chose to focus on the central message here, i.e., that we applied standard procedures that should have allowed us to detect amnestic effects in consideration of most of the published literature. In addition, the crucial overview of the number of training and test trials, as well as the context that was used for each session is depicted in Fig. 1-3, immediately above the results of the respective experiments.
In the Supplement, we provide a more extensive (and repetitive) report of the experimental procedures. The idea is that the reader can find the most important information in the main text, and all additional details in the Supplement (or in our preregistrations on the Open Science Framework: https://osf.io/j5dgx ). For example, in the main text, it is mentioned that reinstatement in Experiment 2 consisted of two US presentations in context A, one day before the final test (see p. 6 and Fig. 1C). The Supplement (p. 1) adds that the reinstatement session started with 300 s of acclimation, followed by the first US and 180 s later by the second US, and that the rat was removed from the context 120 s after last US onset. For all phases of Experiment 2, the US was a 0.7-mA, 1-s shock.
- In exp 5, tests 1 and 2 are supposed to have 12 CS each. However, only 8 dots are represented on the graph. Did the authors average some freezing values after the initial 4 first CS presentations?
Thank you for noticing this. We did not average freezing values, but just did not measure freezing on all trials, as we were not specifically interested in the concrete freezing levels on each trial, but rather in the overall extinction curve. As mentioned in the legend of Fig. 2, freezing during CS5-7-9-11 was not measured (and hence also not shown). In other words, the 8 dots on the graph represent CS1-2-3-4-6-8-10-12.
-There is an obvious difference in baseline freezing response before the test in Exp 7 (Figure 5A-B). Discussion of these differences is an important point and was thoroughly discussed by the authors in the Supplement.
Thank you for pointing this out.
-Ln 384-387: "... additional Bayesian analyses were carried out that collectively suggested substantial evidence for the absence of an amnestic effect". Despite the "substantial effect" given by the meta-analysis, I am a bit confused by the meaning of an "anecdotal evidence against drug < control" reported in half of the experiments. How do the authors interpret these results?
In short, Bayesian analyses provide evidence that is categorized starting from ‘no evidence’, to ‘anecdotal’, ‘substantial’, ‘strong’, etc. depending on the obtained Bayes factor. Grouping studies with anecdotal and substantial evidence in a meta-analysis can result in overall substantial evidence, which is what we observed here.
Addressing this remark in more detail, we want to point out that the use of frequentist analyses (ANOVAs and t-tests) allowed us to conclude that we could not replicate the amnestic effects of previously published studies – we did not obtain a statistically significant amnestic effect although we had sufficient power to detect the effect sizes that had been previously reported. However, those analyses do not permit us to make inferences about the evidence against an amnestic effect. Bayesian analyses, on the other hand, do allow us to quantify the obtained evidence against an amnestic effect (i.e., the null hypothesis) for each single experiment or by combining the results of several studies. When a single study suggests only anecdotal evidence against an amnestic effect, this implies that we cannot conclude based on that study alone that we have proper evidence for the absence of an effect. Rather, we can only conclude that we have no evidence for the presence of an amnestic effect and weak (‘anecdotal’) evidence for its absence. However, a collective analysis of our studies does lead to the conclusion of substantial evidence for the absence of an amnestic effect overall.
-The effect of cycloheximide on memory consolidation is indeed unexpected. Even if beyond the scope of the current study, what is the authors' hypothesis to explain that cycloheximide in their conditions induced a pro-mnesic effects on the consolidation of fear memories but altered the consolidation of extinction?
As indicated by the reviewer, this is beyond the scope of the current study. We have no additional data on this effect and can only guess at its meaning. Also note that the effect was rather small and disappeared quickly during the test under extinction.
One purely speculative hypothesis is that the injection with cycloheximide was more arousing than the vehicle injection, either due to sensations caused by the substance during injection or due to the rapidly emerging malaise it induced (or a combination of both), which we have documented in the Supplement (p. 5).
In line with work by McGaugh, Roozendaal and colleagues, such arousal around the time of training could, in theory, enhance consolidation of a fearful memory, and thus explain greater fear memory during test (see e.g., Roozendaal & McGaugh (2011), https://doi.org/10.1037/a0026187 ). Then again, a similar argument could be made for improved consolidation of the extinction memory (de Quervain et al. (2019), https://doi.org/10.1007/s00213-018-5116-0 ), which we did not observe. One could suggest that – assuming that we have observed ‘true’ effects here – the arousal component had the upper hand during the consolidation of the fear memory, while the protein synthesis inhibition overruled such effects during consolidation of the extinction memory. As this is all highly speculative, we prefer to not add this to the Discussion.
-Cycloheximide seemed to induced post reconsolidation amnesia of fear memory after extinction training (Exp 8, Fig 3G) but not after single CS reactivation. Can the authors please develop this point? Is it possible that several presentations of the CS is required to destabilise the initial memory trace?
First of all, it is important to emphasize that cycloheximide-treated rats in Experiment 8 (Fig. 3G) froze more during the CSs of Test 2 than control animals, arguing against a drug-induced reconsolidation blockade of the initial fear memory. Furthermore, the obvious within-session extinction during Test 1 in Experiment 8 suggests that it did not function as a typical reactivation-without-extinction session (Merlo et al. (2014), https://doi.org/10.1523/JNEUROSCI.4001-13.2014 ).
In light of the current literature, reactivation with a single CS is by far the most common way to destabilize a memory trace that was formed with one or three CS-US pairings. As mentioned in our paper, this should provide an appropriate degree of prediction error for the memory to become malleable (p. 12).
Theoretically, it is indeed possible that more than one (e.g., two) CS presentations could allow for destabilization of the memory trace, although others who have used reactivation sessions with more than one CS presentation did not find the amnestic effects that they did observe with a single CS (Merlo et al. (2014); Sevenster et al. (2014), https://doi.org/10.1101/lm.035493.114 ).
Reviewer #3:
Luyten et al's study examines the phenomenon of drug-induced post-retrieval amnesia for auditory fear memories in rats, and report that after several experiments using Propranolol, Rapamycin, Anisomycin or Cycloheximide that they essentially observe no disruption of reconsolidation, (i.e., no amnesia). This is a well-executed, written and meticulous study examining an important phenomenon. The author's lack of observing amnesia using these "reconsolidation blockers" highlights an important fact that systemic administration of these drugs at the time of memory retrieval may not robustly influence reconsolidation processes despite what the existing literature may collectively indicate. The author's data clearly indicate this point and it is important the scientific community be made aware of these difficulties in blocking reconsolidation using systemic administration of these drugs.
We are thankful for these generous comments and value the reviewer’s thorough and thoughtful assessment of our work. We also appreciate the reviewer’s position that it is important to get this message across to the scientific community.
This group has previously published similar studies disputing similar phenomena. First highlighting a lack of amnesia following the reconsolidation-extinction paradigm and then more recently demonstrating a lack of amnesia attempting to block the reconsolidation of context fear memories. This is now their third installment focusing on Cued fear memories. Certainly, these findings are important, but arguably the novelty of such findings may be diminished a bit.
We appreciate that the reviewer is well aware of some of our other work in this domain that supports a more general and widespread reproducibility crisis in this field.
Regarding the novelty, one key point to stress here, which is also articulated in the paper (p. 3, 13), is that the current rodent findings (which we could not replicate) are the ones that provide the most direct basis for the clinical translations that have been proposed (e.g., by giving patients a propranolol pill after retrieval of a traumatic or phobic memory, see e.g., https://kindtclinics.com/en/ or Kindt & van Emmerik (2016), https://doi.org/10.1177/2045125316644541 ), and are therefore critical in their own right, not only because of their fundamental scientific relevance, but certainly also in light of their clinical reach.
In one of the "control" experiments where the experimenters administer anisomycin immediately post training, they observe a paradoxical result - they observe memory strengthening instead of the expected blockade of consolidation and amnesia. This result highlights a number of things to consider when we interpret these overall results. For one protein synthesis inhibitors(PSIs) are toxic and when administered systemically usually result in inducing the animals to have diarrhea and generally just makes them sick. This of course will make the animals stressed and agitated and result in increasing their stress and likely amygdala activity. All of this could likely be the reason why the animals exhibited memory strengthening or no impairment in consolidation even with a PSI on board. See PMCID: PMC7147976. Figure 6. In this study, they could rescue the impairment of PSI on consolidation by increasing BLA principal neuron firing. Thus an important take away is something like this could easily be happening in the reconsolidation experiments - that there is no blockade because the animals are stressed either due to PSI on board or because some issues with experimenter/animal interactions, etc lead to higher BLA neural activity and rescue of the reconsolidation process.
We agree that (systemic) protein synthesis inhibitors can induce signs of sickness in the animals (particularly in the first hours after injection) and have provided a detailed description of our relevant observations in the Supplement (p. 4-5). The reviewer is completely correct in stating that this may cause some amygdala activation which could interfere with the amnestic effects that we expected to see, as described in the paper by Shrestha, Ayata et al. (2020), and in line with our reply to Reviewer #2’s first comment regarding our cycloheximide experiment. Yet, effective induction of amnesia with these drugs has repeatedly been reported in the literature.
Nevertheless, although relevant, the current remark has relatively little implications for our findings. In the large majority of our experiments, we did not use these toxic protein synthesis inhibitors (PSIs) (such as cycloheximide and anisomycin), but drugs that have generally been administered systemically throughout the literature (with successful amnestic effects). Furthermore, in the experiments where we did administer systemic cycloheximide or anisomycin, we observed no differences compared to vehicle-treated rats in contextual freezing (e.g., 9% on average in Experiment 7) immediately prior to the crucial test tones (Test 1, 24h after injection) – which argues against high levels of stress or agitation. Moreover, a blinded experimenter could not tell the difference between PSI-treated versus vehicle-treated animals while handling the animals for the test session, and observed no behavioral abnormalities, nor signs of pain or distress, as mentioned in the Supplement. We acknowledge that these experimenter observations may not entirely reflect what is happening in the animals’ amygdala, but they at least go against the notion that PSI-treated animals would be too sick to be tested properly.
I don't think the authors go far enough articulating the important differences between systemic and intra-cranial administration of these drugs. Time is a potential factor. Immediate administration of the drug at high concentration in the target brain region (BLA) versus many minutes until the drug gets to the target region with uncertain concentration levels that may not mirror levels reached with intracranial administration. It's unfortunate the authors were not able to include intra-BLA administration of these drugs in this study. I do not necessarily expect them to do such experiments, since they have already done so much and it is not clear the laboratory has the appropriate expertise to conduct such experiments, but this comparison would be helpful.
We fully agree that our results do not provide any information about the replicability of intracranial administration of drugs to induce post-retrieval amnesia of cued fear memories. We had already clearly acknowledged this in the first version of the paper (p. 11), but have now added an extra section to the Discussion (p. 13) to highlight this point in the new version posted on BioRxiv (Version 2). Notwithstanding the expertise of our laboratory to carry out intracranial infusions, we agree with the reviewer that such experiments are beyond the scope of this article.
It is, however, noteworthy that the drugs that we used in 6 experiments did not necessarily rely on intracranial administration in prior successful studies. Rapamycin, for example, has generally been used systemically (not intracranially). Propranolol has been used either systemically or intracranially in rodents and always systemically in human subjects (healthy and patients). Bearing in mind the timing issue that was raised by the reviewer, we moreover included an experiment with pre-reactivation administration of propranolol (Experiment 4), where the drug was injected 5-8 minutes before the rats heard the reactivation tone.
I think it is important that the authors make some statement of training conditions on cannulated versus cannulated rats. For example, every animal in Nader's 2000 study was bilaterally cannulated targeting the BLA. In contrast every animal in this study underwent no such surgery. I think this is relevant. In my experience non cannulated animals are a bit smarter than cannulated animals and the training conditions across these two differing groups may not equate to the same level of learning. And of course, differences in learning levels can lead to differences in the ability of the retrieved memory to destabilize.
Thank you for pointing this out. We are aware that there may be differences between operated and non-operated animals and already briefly discussed this matter in the Supplement (p. 4). We have now also added this issue to the Discussion in the new section (p. 13) where we emphasize the differences between systemic and intracranial drug administration in relation to the previous comment.
That being said, the comment regarding (non-)cannulated rats only really applies to Experiment 7 where we tested the effects of systemic anisomycin or cycloheximide. Prior cued fear conditioning studies indeed used intracranial administration of these drugs. The argument does not hold for Experiments 1-6, as systemic propranolol and rapamycin have repeatedly been reported to have amnestic effects in non-operated rats, with procedures identical to or closely resembling ours.
The authors mention possibly examining markers of memory destabilization. GluR1 phosphorylation, Glur2 surface levels, protein degradation/ubiquitination have all been used to assess if destabilization has occurred. I do not fully agree with their reasons for not performing such experiments. They could examine some or one of these phenomena across differing training conditions between retrieval, no-retrieval animals. This likely could be informative. However, the authors may not possess the necessary expertise to conduct such experiments, so I'm not stating these experiments need to be completed, but certainly the study could be strengthened with such data.
We agree that including yet more control experiments, using different experimental approaches could further strengthen the study. Nevertheless, the main conclusion of our paper – i.e., reconsolidation blockade using systemic administration of several drugs is considerably more difficult to reproduce than what the literature collectively indicates – is strongly and sufficiently supported by the data that we already report here. Overall, we believe that our conclusion does not require such additional controls. Moreover, even though the comparisons suggested by the reviewer could indeed be scientifically interesting, it is still unclear whether such experiments would provide sufficiently clear cut-offs as to which experimental condition would then allow for adequate memory destabilization and interference.
Experiment 3E - Propranolol without reactivation. I don't see any data for this on the graphs. Am I missing something?
Our apologies for the confusion. The legend shown next to Fig. 1F applies to all panels of Fig. 1, but only Experiment 1 (shown in Fig. 1A-B) contained a no-reactivation group as an additional control. Experiment 3 (shown in Fig. 1E-F) did not. We have moved the legend to the bottom of Fig. 1 to clarify this.
The authors should probably cite this paper too, PMID: 21688892. The authors in this study find no evidence that propranolol inhibits cued fear memory reconsolidation.
Thank you for bringing this to our attention. We were aware of this paper, but it had slipped through the cracks. We have cited it in the new version of the paper (p. 11).
We thank the editors for considering our manuscript for publication in eLife and the reviewers for their work. However, we would like to discuss several of their comments.
The key issue seems to be a lack of novelty of our work, which is not correct in our opinion.
We would like to quickly reiterate why we think that our findings are novel and have very broad implications.
The importance of polygenic adaptation is becoming increasingly clear. Unfortunately, it is widely assumed that polygenic adaptation is very difficult, if not impossible, to study in natural populations, because the associated allele frequency shifts are too small to be experimentally characterized (Pritchard et al., 2010). Hence, typically the collective response of many loci are considered, which frequently results in wrong results due to population stratification (Berg et al., 2019; Sohail et al., 2019).
Therefore, we have used experimental evolution to characterize polygenic adaptation. Experimental evolution is widely recognized as a powerful tool because of the possibility to replicate experiments. Here, we expand the power of experimental evolution by an hitherto unrecognized aspect: the impact of linkage disequilibrium - we demonstrate that two founder populations with different levels of linkage disequilibrium (LD) result in entirely different selection responses. The consequence of different LD structures is shown by our observation that the same population (i.e. identical LD structure) evolving in two different environments shows the same selection response, but a different population with different LD structure in the same environment shows different selection responses.
This result has important implications for all studies of polygenic adaptation in natural populations because LD is not accounted for in studies of polygenic adaptation, but like in our study, haplotype blocks with multiple loci could result in a strongly selected allele. Hence, LD will determine the likelihood of this to occur. Furthermore, accounting for linkage provides the opportunity to study polygenic adaptation also in natural populations - a substantial change to the current testing paradigms.
The second key result of our study is that we demonstrate that selection in hot and cold environments does not fit the simple model of polygenic adaptation, where the same set of loci is responding in different directions, when opposing selection regimes are applied. As pointed out by reviewer #2, this is particularly important as it shows that current models of polygenic adaptation are not well-suited to understand adaptation imposed by contrasting ecological factors. We show that there is almost no overlap between the haplotype blocks selected in the hot and cold environment. Most importantly, this is not a matter of power as we show that the blocks responding in one selection regime are not changing their frequency in the opposite direction in the other selection regime. We anticipate that this insight will have a profound impact on theoretical models of polygenic adaptation. Furthermore, as we studied temperature adaptation, our results will have also important consequences for the battery of ongoing studies aiming to link selection signatures to response to climate change.
In brief, we think that very minor clarifications in our manuscript can solve the technical issues identified by the reviewers and will provide a clearer picture about the general implications of our findings.
A detailed response to the comments of the reviewers is given below.
Reviewer #1:
Otte et al. used an evolve and re-sequence strategy to explore "the genetic architecture of adaptive phenotypes". The authors previously found different genetic architectures across different founder populations evolving in a common hot environment. The authors chose one of these founder populations for replicated experimental evolution (5 replicate populations) in a cold environment for 50 generations. The authors were surprised to discover the same number of loci evolve under strong selection between the hot-evolved and cold-evolved replicate populations, though the 20-ish loci are largely non-overlapping. The distribution of selection coefficients was also similar. They interpret this commonality as evidence that the founder population history has a larger effect on adaptive architecture than the selection regime.
The study demonstrates a comprehensive effort to discover the number of genome regions and distribution of selection coefficients that emerge from a highly controlled experimental evolution project. The experienced team applies a sophisticated toolkit to this powerful experimental design - a toolkit that grows ever more sophisticated with each new experimental run that they perform. However, the authors set me up to learn why such different adaptive architectures emerge from different founder populations. Ultimately, the researchers acknowledge that they "cannot pinpoint the cause for the differences in the inferred adaptive architecture..."
Here, the reviewer correctly identified one of the main new questions that arose from the new experiment we performed in this study. In a large part of the discussion and the associated analyses we are providing answers to this question, i.e. possible alternative explanations for the different observed architectures in the Portugal vs. the Florida population. We can indeed not pinpoint "the" cause for the differences that the reviewer seems to request here as a definite answer, but we favour one of the explanations that has not yet been discussed in literature previously (LD).
Some results simply recapitulated the previous Portugal E&R study and other results recapitulated a D. melanogaster E&R study.
This statement about "some results" is ignoring the main new experiment of this study, which is the Portugal population evolving in a cold temperature. For this, we carried out a new selection experiment in a new environment, which finds different selection targets than the previously published experiments. This new experiment therefore does not recapitulate the previous results. We then compare this new experiment to a previous one, and this comparison raises a set of new questions that we address in this manuscript. Only for the purpose of making that comparison, we indeed "simply recapitulated" "some results" of the previous study. The statement is therefore misleading in the way it is put here. Furthermore, the D. melanogaster study is also not recapitulated: in that study, it was not possible to identify selected haplotypes. The D. melanogaster study was therefore unable to determine how many selection targets were shared between the hot and cold selection regimes. The identification of selected haplotypes was a major improvement in this study, which made it possible only now to determine how many targets are shared and to evaluate whether selection targets behave as predicted by the trait optimum model.
I did not find the "common adaptive architecture" across different selection regimes to be a particularly compelling discovery of sufficiently broad interest.
This is a very subjective opinion and it would be good if the reviewer had explained why this is no interesting discovery to her/him. We feel that this statement simply reflects that the reviewer does not fully appreciate the complexity of polygenic adaptation. We would like to point out again, that this result has important implications for the interpretation of selection signatures in natural populations.
Other concerns and questions can be found below:
Major concerns:
1) Pg. 4: It is my understanding that the power of multiple populations from a single founder evolving in parallel allows for more rigorous identification of loci targeted by selection. I found it surprising to discover that if a lack of replication emerges from an experimental evolution study, this outcome is interpreted as "genetic redundancy." First, genetic redundancy has a precise definition in genetics that muddles the author's meaning. And second this interpretation seems rather post-hoc.
This statement shows that the reviewer is disregarding the work of Barghi et al (2019, PLoS Biology) and the definition of redundancy in the context of polygenic adaptation as discussed by Laruson et al. (2020) or Barghi et al 2020 (Nature Reviews Genetics). In any case, this is a semantic issue and should not be considered as a major issue with our manuscript.
2) To "shed more light on the different selection responses" is a weak motivation. The introduction sets me up to understand why selection responses are so different but no major insights into the "why" emerge from the cold-adaptation experiment.
We modestly disagree - we clearly discuss different explanations of “why” and favor one of them (LD)
3) More explanation of figure 1 in the main text is needed. Does each point correspond to a SNP that consistently changes across all five populations? Or is this the union?
The reviewer does not seem to be familiar with the statistical analyses that have been used in our study in the same way as it is common practice in the field. Despite the common use of this test, we still provided a detailed explanation in M&M and explicitly mentioned the test in the figure legend. But this can easily be detailed even further and should not be a major issue with this manuscript.
4) Line 210: How did the researchers define "stress" and determine that the degree of stress is equivalent across two temperature regimes? The absence of these data undermine the potency of the comparison.
It is not clear why the reviewer requires a more elaborate definition of temperature stress - the concept of extreme temperatures imposing stress is well established and we cite the relevant literature for Drosophila in the text. Furthermore, it is not apparent why the reviewer requests the degree of stress to be equivalent between the two temperature regimes.
5) How can the authors be sure that the only difference between the hot and cold populations was temperature? Was competition/population size/etc held constant? Might the lack of overlap between hot and cold adapted loci stem from one such regime selecting for a different phenotype? (i.e., not temperature tolerance)
As clearly stated in M&M, the culture conditions were the same with the exception of temperature.
6) Line 237: The authors assert that most alleles show a temperature-specific response - a discovery with precedent in the literature, including from this team of researchers. The authors attribute the absence of common loci between temperature regimes to the high number of generations (50) compared to the number across seasons cited in Bergland et al. The researcher could easily look for common targets at earlier time points of experimental evolution to test this idea.
This is an interesting suggestion, but the reviewer fails to explain why the analysis of early generations should be more informative than the analysis of later generations. Several studies have already documented the opposite.
7) Line 292-293: This section reads as disingenuous - the researchers could have explored overlap between Portugal and Florida founders using only the selected loci coordinates and look for non-random overlap using simulations/resampling tests.
The reviewer seems to assume that we could easily apply the same test for overlap that we used for the hot vs. cold comparison within the Portugal population to the Portugal hot vs. Florida hot comparison. But this is not feasible, and we clearly explain why the comparison of selected haplotype blocks between different founder populations is not helpful (low LD results in different haplotype blocks - even with the same target)
8) Discussion: The speculation about why such different architectures emerged across Portugal and Florida was diluted by the absence of initial fitness estimation upon subjection to a cold environment (which would have offered evidence for different initial "optima" across founder populations) as well as the change in fitness from generation 0 to generation 50.
It is not apparent why the reviewer requests a fitness estimate at the cold environment. Our analysis only included a single population in the cold environment. Hence, the only informative comparison is the one in the hot environment which has been done for both populations and is referenced in the manuscript.
9) The simulations and corresponding discussion would make for an interesting review/opinion piece but not as new results for this manuscript.
Unlike the reviewer, we think that a good discussion puts the results into perspective with different hypotheses on how to explain it and link this to the current literature.
Minor Comments:
1) Pg. 3. The recurrent citation of Barghi et al. in the Introduction undermined the reader's impression that fundamental questions are being addressed in this article.
Maybe it escaped the reviewer’s attention that we cited three different Barghi et al. papers and only one reports experimental data (cited only once), while the others are required to describe the theoretical framework, including the concept of "redundancy" which the reviewer misunderstood. New fundamental questions in this current manuscript are addressed using the Portugal population, which was selected in a cold temperature regime (not hot-evolved Florida, which was the topic of Barghi et al. 2019).
2) Lines 33-39: The argument that parallel signatures of selection across distinct natural populations are insufficient to address the polygenic basis of adaptive phenotypes, and so comparatively more contrived E&R studies are required, was unconvincing.
Unfortunately, the reviewer does not provide support for this strong statement. In fact, we find the statement of “contrived E&R studies” not as objective as we would have liked to see in a scientific discourse.
3) Line 158: Confusing. Should "among" actually be "within"?
The reviewer is not right - the correct wording is "among" not within: multiple different haplotypes can carry the actual target of selection, and they can differ by additional variants which themselves are not selected for. Multiple haplotypes with the selection target are also experiencing more pronounced frequency changes than expected under neutrality. The correlation of their allele frequency trajectories depends, however, on the extent that hitchhiking SNPs are shared among these haplotypes. To account for this, we used a less stringent correlation cutoff.
4) Line 486: I believe that the authors would be hard-pressed to find in the literature a paper declaring that "single population...[is] sufficient to understand the genetic basis of adaptive traits".
In fact, many selection tests are targeting only a single population and most studies only apply them to a single population.
Reviewer #2:
This reviewer mainly asks us to discuss some of his/her ideas - this can be done, but since reviewer#1 felt already that there is too much discussion in our manuscript this is a bit of a mixed message.
Overall Review: This is another commendable study from the Schloterer lab that features next generation genome-wide sequencing of multiple evolving populations. It compares results obtained with two different selection regimes, one hot and one cold, and two different founding populations of Drosophila simulans, one from Portugal and one from Florida. The results reveal a lack of consistency among selection regimes and founding populations. Temperature-dependent adaptation is shown to be "local" or "contingent," rather than globally consistent. My chief recommendations concern the experimental and theoretical contexts within which this study should be interpreted.
Major points:
1) I do not require any additional data collection or statistical revision. My comments are organized in terms of experimental paradigm (A) and theoretical significance (B).
A.
2) The typical paradigm for experimental evolution in this and many other labs is the use of hybrid populations created from isofemale lines. This method for founding experimental populations can be expected to generate some degree of random "historicity" as the isofemale lines approach fixation of specific genotypes with high stochasticity. Then there are further stochastic and historical effects which arise when such lines are hybridized. The strengths and limitations of this paradigm should be addressed. Most importantly, such stochastic historical effects might be the source of the discrepancy between the replicate lines derived from Portugal and Florida.
We would like to emphasize that we were using freshly established isofemale lines kept in the laboratory for at most 10 generations, as stated in the M&M section.
3) As the authors themselves point out, there is a comparative difficulty arising from the different scales of replication used for the Florida versus Portugal experiments.
The reviewer is correct, and since we were aware of this, we performed statistical tests to account for this.
A further question for large-scale experimentation is whether a larger and uniform level of replication might produce more similar results, such as 20 evolving populations from each source. Or indeed, three sets of ten evolving populations from three distinct founders from the two sources, with a total of 60 evolving experimental lineages. The authors should discuss whether they believe that their findings would hold up with such an expanded experimental protocol.
This is an interesting thought of its own, but we feel that it does not contribute much to our current study.
4) The authors themselves point out at one point that their experiments might have benefitted from some phenotypic characterization of the presumed temperature adaptation. That raises the more general question of how the field of experimental evolution can progress with some labs just doing phenotypes and other labs just doing genome-wide sequencing. Surely this and other studies would be strengthened by combining the two types of assay. Furthermore, genomic evolution might be usefully analyzed in terms of the degree to which specific genomic changes can be associated with specific phenotypic changes, as that is the foundation for adaptation itself.
We would like to draw the attention to the fact that we performed a laboratory natural selection experiment, for which the environmental factor is known, but not the actually selected phenotype - hence the phenotyping is not as trivial as implied by the reviewer.
B.
5) This is yet another study that finds difficulties with the invocation of noroptimal selection along a one-dimensional functional gradient. Such models have been long-standing favorites of evolutionary theorists, such as Kimura and Lande. But that preference may arise more from the ease with which these models can be formulated and analyzed by theoreticians. Actual evolving populations don't seem to embody the precepts of such theory, whether the issue is the maintenance of genetic variation (see the work of Turelli, for example) or the evolution of closely studied populations, as illustrated by this study. An alternative point of view that the authors should discuss is that such models are indeed NOT usually correct.
It is very interesting that this reviewer feels that our data demonstrate that the prevailing model of polygenic adaptation is wrong, but our manuscript is still considered to be of insufficient novelty.
6) There are alternative theoretical frameworks that address the maintenance of genetic variation and the response to selection. Among these are schemes of protected polymorphism arising from overdominance, epistasis, and frequency-dependent selection. If the thrust of the preceding point 4 is accepted, then it would be theoretically salient for the authors to suggest what type of underlying population genetic machinery would best account for their findings, in place of the noroptimal selection-mutation balance model.
We thank the reviewer for these interesting suggestions. However, their predictions are not at all trivial to test. For this reason, generations of population geneticists tried to test them, so we feel that this task is well beyond the scope of this manuscript.
Reviewer #3:
In their manuscript 'The adaptive architecture is shaped by population ancestry and not by selection regime,' Otte and colleagues use an evolve and resequence strategy to examine the response of a Portugal population of D. simulans responds to cold temperature. The authors identify putative targets of selection and compare the number of targets, their location, and the distribution of selection coefficients to previous work on the same population exposed to hot temperatures as well as a different population exposed to hot temperatures. The topic is of general interest, the work is sound and the writing is clear and concise.
1) It is not clear what the novel contribution of this manuscript is. The title indicates that the key finding is that population of origin mediates response to selection rather than the selection regime. However, the authors fail to provide compelling data to support that. The data are from 1 population under two selection regimes and a second population under one of those regimes. There simply aren't enough comparisons to infer that population ancestry plays a bigger role than selection regime in adaptive evolution.
We disagree with the reviewer and would like to repeat the logic of our experiment:
Comparison 1: contrast of different populations in the same environment -> different architecture
Comparison 2: contrast of the same population in different environments -> same architecture
With this simple design it is possible to reach the conclusion that the architecture is affected by population history more than by selection regime and no more populations are needed to reach this conclusion. This insight has not been reported before.
2) The authors also seem to argue that a contribution of this paper is that it illustrates that temperature adaptation is not a single trait. This was the major finding of a 2014 paper from the same group in D. melanogaster- a single founder population was exposed to hot and cold temperatures and the authors found almost no overlap between the putatively selected variants in the two different temperature regimes.
We would like to point out that the analysis of Tobler et al. (2014) is on the basis of individual SNPs, which is difficult to interpret because of the many segregating inversions in D. melanogaster. All the complications of these data and the implications for the interpretation can be found in the discussion of Tobler et al. (2014). In the current study, we are identifying selected haplotype blocks, which is mandatory to compare the architectures and selection responses.
3) Beyond the limited impact of the current work, there are some additional specific issues. The authors note that it was 'remarkable' that the distribution of selection coefficients and the number of inferred selection targets between the hot and cold experiments was 'highly similar.' What is the null expectation? Where does the null come from?
This is a minor semantic issue. Naturally, there is no null model for the number of selection targets, but if two populations selected for the same trait provide different architectures, different selection regimes should be even more likely to generate different architectures.
4) The discussion is somewhat unsatisfying and largely speculative. The 'different trait optima' section reads as straw man; this could be reframed to better guide the reader.
Naturally, the discussion intends to put the results in a broader context. It would have been helpful to read how s/he envisions a reframing that would improve the manuscript.
There is little support for the 'differences in adaptive variation' hypothesis.
It would have been helpful to read which kind of support the reviewer would have expected beyond the evidence we have already provided.
The section on LD was interesting, but the simulation findings should reside in the results section.
This could be easily moved, but we feel that it is well-placed in the discussion as we use the simulations to compensate for the lack of literature on this field (again demonstrating the novelty of our manuscript).
References:
Barghi, N., R. Tobler, V. Nolte, A. M. Jakšić, F. Mallard, K. A. Otte, M. Dolezal, T. Taus, R. Kofler, & C. Schlötterer (2019). Genetic redundancy fuels polygenic adaptation in Drosophila. PLOS Biology 17: e3000128.
Barghi, N., J. Hermisson, & C. Schlötterer (2020). Polygenic adaptation: a unifying framework to understand positive selection. Nature Reviews Genetics . Berg, J.J., Harpak, A., Sinnott-Armstrong, N., Joergensen, A.M., Mostafavi, H., Field, Y., Boyle, E.A., Zhang, X., Racimo, F., Pritchard, J.K., et al. (2019). Reduced signal for polygenic adaptation of height in UK Biobank. Elife 8.
Bergland, A. O., E. L. Behrman, K. R. O’Brien, P. S. Schmidt, & D. A. Petrov (2014). Genomic Evidence of Rapid and Stable Adaptive Oscillations over Seasonal Time Scales in Drosophila. PLoS Genetics 10, e1004775.
Láruson, Á. J., S. Yeaman, & K. E. Lotterhos (2020). The Importance of Genetic Redundancy in Evolution. Trends in Ecology and Evolution 35: 809–822. Pritchard, J.K., Pickrell, J.K., and Coop, G. (2010). The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Current biology : CB 20, R208-215.
Sohail, M., Maier, R.M., Ganna, A., Bloemendal, A., Martin, A.R., Turchin, M.C., Chiang, C.W., Hirschhorn, J., Daly, M.J., Patterson, N., et al. (2019). Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. Elife 8.
Note from the authors:
This is the authors' response to the reviewers' comments for the manuscript “Perceptual gating of a brainstem reflex facilitates speech understanding in humans” submitted to eLife via Preprint Review. We appreciate the time and effort the reviewers took to carefully revise our work. We believe all comments and suggestions will improve the manuscript for future publication. All the authors’ comments detailed in this response will be implemented in the next version of this manuscript.
Reviewer #1: [...] Reviewer 1-Comment 1: 1) An important aspect of assessing the efferent feedback through the CEOAEs and ABRs is to ensure that different stimuli have equal intensity. The authors write in the methodology that the speech stimuli were presented at 75 dB SPL. However, it is not stated if this applies to the speech stimuli only, such that the stimuli that include background noise would have a higher intensity, or to the net stimuli. If the intensity of the speech signals alone had been kept at 75 dB SPL while the background noise had been increased, this would render the net signal louder and influence the MOCR. In addition, it would have been better to determine the loudness of the signals according to frequency weighting of the human auditory system, especially regarding the vocoded speech, to ensure equal loudness. If that was not done, how can the authors control for differences in perceived loudness resulting from the different stimuli?
Response to Reviewer 1-Comment 1:
Controlling the stimulus level is a critical step when recording any type of OAE due to the potential activation of the middle ear muscle reflex (MEMR). High intensity sounds delivered to an ear can evoke contractions of both the stapedius and the tensor tympani muscles causing the ossicular chain to stiffen and the impedance of middle ear sound transmission to increase (Murata et al.,1986; Liberman & Guinan,1998). As a result, retrograde middle ear transmission of OAE magnitude can be reduced due to MEMR and not MOCR activation (Lee et al., 2006). For this reason, we were particularly careful to determine the presentation level of our stimuli.
As pointed out by the reviewer and stated in the Methods section: Experimental Protocol: “The speech tokens were presented at 75 dB SPL and the click stimulus at 75 dB p-p, therefore no MEMR contribution was expected given a minimum of 10 dB difference between MEMR thresholds and stimulus levels (ANSI S3.6-1996 standards for the conversion of dB SPL to dB HL)”. 75 dB SPL was indeed selected as the presentation level for all natural, noise vocoded and speech-in-noise tokens. All tokens were root-mean-square normalized and the calibration system (sound level meter (B&K G4) and microphone IEC 60711 Ear Simulator RA 0045 563 (BS EN 60645-3:2007), (see CEOAEs acquisition and analysis section)) was set to “A-Weighting” which matches the human auditory range. Therefore, the net signal was never above 75 dBA. We acknowledge the lack of details about the calibration procedure in the current manuscript and will consequently add them in a future Methods section.
Reviewer 1-Comment 2: 2) Many of the p-values that show statistical significance are actually near the threshold of 0.05 (such as in the paragraph lines 147-181). This is particularly concerning due to the large number of statistical tests that were carried out. The authors state in the Methods section that they used the Bonferroni correction to account for multiple comparisons. This is in principle adequate, but the authors do not detail what number of multiple comparisons they used for the correction for each of the tests. This should be spelled out, so that the correction for multiple comparisons can be properly verified.
Response to Reviewer 1-Comment 2:
Bonferroni corrections were explicitly chosen as the multiple comparisons adjustment across our post-hoc statistical analyses because they are a highly conservative test that protect from Type I error. All the p-values reported in our study are corrected p-values for post-hoc comparisons. However, we agree that for verification purposes, the number of comparisons for each statistical analysis should be clarified in the Methods section and will be added to a future version of the manuscript.
Reviewer 1-Comment 3: 3) Line 184-203: It is not clear what speech material is being discussed. Is it the noise vocoded speech, the speech in either type of background noise, or these data taken together?
Response to Reviewer 1-Comment 3:
Lines 184-203 correspond to “Auditory brainstem activity reflects changes in cochlear gain” in the Results section. Line 186 describes changes in ABR components during noise-vocoded speech: “Click-evoked ABRs—measured during simultaneous presentation of vocoded speech—showed task-engagement-specific effects similar to the effects observed for CEOAE measurements.” The subsequent 3 sentences refer to the same (noise-vocoded) condition, whereas the remaining sentences in the section refer to the speech-in-noise conditions. As pointed out by the reviewer we did not specify a specific masked condition in the sentence: “Conversely, although wave III was unchanged in both masked conditions for active vs. passive listening, wave V was significantly enhanced: [F (1, 26) = 5.67, p = 0.025 and F (1, 25) = 8.91, p = 0.006] when a lexical decision was required.” Here the rANOVAs correspond to masked conditions: speech in babble noise and speech-shaped noise respectively. This will be rectified in a future version of the manuscript.
Reviewer 1-Comment 4: 4) Line 202-203: The authors write that "the ABR data suggest different brain mechanisms are tapped across the different speech manipulations in order to maintain iso-performance levels". It is not clear what evidence supports this conclusion. In particular, from Figure 1D, it appears plausible that the effects seen in the auditory brainstem may be entirely driven by the MOCR effect. To see this, please note that absence of statistical significance does not imply that there is no effect. In particular, although some differences between active and passive listening conditions are non-significant, this may be due to noise, which may mask significant effects. Importantly, where there are significant differences between the active and the passive scenario, they are in the same direction for the different measures (CEOAEs, Wave III, Wave V). Of course, that does not mean that nothing else might happen at the brainstem level, but the evidence for this is lacking.
Response to Reviewer 1-Comment 4:
Lines 202-203 also correspond to “Auditory brainstem activity reflects changes in cochlear gain” in the Results section. As suggested by the reviewer, the effects observed in the ABRs may be driven by the MOCR. We agree with this observation in lines 195-197, explaining that the decreased magnitude of ABR components is consistent with reduced magnitude of CEOAEs measured during active listening in the vocoded condition, since a reduction in cochlear gain can reduce the activity of auditory nerve (AN) afferents synapsing in the cochlear nucleus (CN). However, we did not explain that this trend is also observed during the passive listening of speech-in-noise, therefore demonstrating that vocoded and speech-in-noise are differently processed at the level of the brainstem and midbrain. In a future version of the manuscript, we will restrict our interpretation to statistical comparisons in the Results and leave potential mechanisms for the Discussion section.
Reviewer 1-Comment 5: 5) The way the output from the computational model is analyzed appears to bias the results towards the author's preferred conclusion. In particular, the authors use the correlation between the simulated neural output for a degraded speech signal, say speech in noise, and the neural output to the speech signal in quiet with the efferent feedback activated. They then compute how this correlation changes when the degraded speech signal is processed by the computational model with or without efferent feedback. However, the way the correlation is computed clearly biases the results to favor processing by a model with efferent feedback.
The result that the noise-vocoded speech has a higher correlation when processed with the efferent feedback on is therefore entirely expected, and not a revelation of the computational model. More surprising is the observation that, for speech in noise, the correlation value is larger without the efferent feedback. This could due to the scaling of loudness of the acoustic input (see point 1), but more detail is needed to pin this down. In summary, the computational model unfortunately does not allow for a meaningful conclusion.
Response to Reviewer 1-Comment 5:
While claims of bias would be understandable had we used shuffled auto-correlograms (SACs) to compare the expression of temporal fine structure (TFS) cues for natural speech versus vocoded stimuli (TFS cues reconstructed from the envelope of our vocoded stimuli would have differed dramatically from those original TFS cues in natural speech) (Shamma and Lorenzi, 2013), there is no inherent reason for SAC analysis of envelopes cues being biased towards either vocoded or speech-in-noise conditions as both stimuli retain the original envelope cues from natural speech. Indeed, since the purpose of our simulations was to compare the relative effects of adding efferent feedback on the reconstruction of the stimulus’ envelope cues in the AN for the two degraded stimuli, SACs offered a targeted analysis tool to extract the relevant information with fewer intermediate steps and presumptions than either encoder models or automatic speech recognition systems.
We do agree with the reviewer that results of our simulations for the vocoded condition may have been less unexpected than those of speech-in-noise, as the envelopes of vocoded stimuli closely resemble those of natural speech in the absence of a masking noise. However, our results also demonstrate that adding efferent feedback could generate negative correlation changes for a number of vocoded words: either at individual frequencies (low and high spontaneous rate AN fibres (see raw data)) or on average across all frequencies tested [high spontaneous rate AN fibres only (Fig Supplement 3)]. This suggests that noise-vocoding speech (i.e. implementing the envelope from broader channel bandwidths while also scrambling spectrotemporal information in said channels) can disrupt envelope representation in the 1-2kHz range of certain words enough that efferent feedback should not be automatically presumed able to rectify their envelope cue reconstruction in AN fibres.
As for the speech-in-noise conditions, our intuition for the negative correlation changes observed is that the signal-to-noise ratios (SNRs) tested were not large enough to allow for the isolated extraction of the target signal’s envelope by expanding the dynamic range of AN fibres. As the test stimuli and their SNRs were directly acquired by finding iso-performance in the psychophysical portion of this study (and appropriately normalized as input for the MAP_BS model), we consider the results of the simulation to be indicative of the actual benefit/disadvantage that activating efferent feedback might have on envelope representation of vocoded or speech-in-noise tasks in the AN [and not artefacts of poorly calibrated stimulus presentation level (see Responses to Reviewer1-Comment 1 and 6 for more details about methodology)]. Although this result may be surprising when viewed in the context of physiological and modelling studies demonstrating efferent feedback’s masking effect, our results may help to explain why MOCR anti-masking appears SNR- and stimulus- specific in numerous human studies (de Boer et al., 2012; Mertes et al., 2019).
Reviewer 1-Comment 6: 6) The experiment on the ERPs in relation to the speech onsets is not properly controlled. In particular, the different acoustics of the considered speech signals -- speech in quiet, vocoded speech, speech in background noise -- will cause differences in excitation within the cochlea which will then affect every subsequent processing stage, from the brainstem and on to the cortex, thereby leading to different ERPs. As an example, babble noise allows for 'dip listening', while with its flat envelope speech-shaped noise does not. Analyzing differences in the ERPs with the goal of relating these to something different than the purely acoustic differences, such as to attention, would require these acoustic differences to be controlled, which is not the case in the current results.
Response to Reviewer 1-Comment 6:
Our fundamental methodological strategy was not to compare or even control the acoustics of the signals (although we did this to some extent by normalizing the presentation level and long-term spectrum across all signals), but instead to maintain iso-performance across conditions and, in doing so, allow the identification of brain mechanisms underlaying performance in a lexical decision task where speech intelligibility was manipulated.
We do acknowledge the reviewer’s comment regarding acoustic differences across our speech signals. This is why in the Results section we describe that: “Early auditory cortical responses (P1 and N1) are largely driven by acoustic features of the stimulus (Getzmann et al., 2015; Grunwald et al., 2003)”. Therefore, our ERP analysis instead focuses on later, less stimulus-driven components such as P2, N400 and LPC: “Later ERP components, such as P2, N400 and the Late Positivity Complex (LPC), have been linked to speech- and task-specific, top-down (context-dependent) processes (Getzmann et al., 2015; Potts, 2004).”
With regards to the reviewer’s example: “…babble noise allows for 'dip listening', while with its flat envelope speech-shaped noise does not”. We could argue that in our specific listening conditions “dip listening” did not offer a perceptual advantage over speech in speech shaped noise because:
1) Higher SNR was required in the babble noise conditions to achieve the same level of performance than for the speech-shaped noise manipulations.
2) Listeners have fewer chances to use the spectral and temporal dips compared to sentences(Rosen 2013) when listening to monosyllabic words (used in our study)
3) The dips in the signal are expected to decrease both in depth and frequency with the number of talkers in a babble noise masker (8-talker babble used in our study), with no differences in masking effectiveness for more than 4-talker babble noise (Rosen et al., 2012).
Overall, we believe that having modulated maskers effectively impaired speech intelligibility (Kwon and Turner 2001), but the most effective one was babble noise confirming that the best speech is its own best masker (Miller, 1947).
Reviewer #2: [...] Reviewer 2-Comment 1: 1) A core premise of the experiment is that the non-invasive measures recorded in response to click sounds in one ear provide a direct measure of top-down modulation of responses to the speech sounds presented to the opposite ear. This is not acknowledged anywhere in the paper, and is simply not justifiable. The click and speech stimuli in the different ears will activate different frequency ranges and neural sources in the auditory pathway, as will the various noises added to the speech sounds. Furthermore, the click and speech sounds play completely different roles in the task, which makes identical top-down modulation illogical. The situation is further complicated by the fact that the clicks, speech and noise will each elicit MOCR activation in both ipsi- and contralateral ears via different crossed and uncrossed pathways, which implies different MOCR activation in the two ears.
Response to Reviewer 2-Comment 1:
We employed broadband clicks across all stimulus manipulations and listening conditions to activate the entire cochlea so that resulting OAEs could be used to measure modulation of cochlear gain by olivocochlear efferents.
Historically, studies have applied clicks in one ear (to evoke OAEs) and a broadband noise suppressor in the other to monitor contralateral MOCR activation, demonstrating that clicks are suppressed consistently when subjects actively perform either an auditory (Froehlich et al., 1993, Maison et al., 2001; Garinis et al., 2011) or visual tasks (Puel et al., 1988; Froehlich et al., 1990; Avan & Bonfils 1992; Meric & Collet 1994). Therefore, while we acknowledge that the presence of clicks may have made the task of discriminating vocoded and words-in-noise more difficult, we would have expected to observe suppression of click-evoked OAEs for all stimulus manipulations whether subjects were actively or passively listening to speech stimuli in order to minimize the impact of the irrelevant clicks. In contrast, we observed that contralateral suppression of CEOAEs was both stimulus- and task-dependent. Unlike natural and vocoded speech, active listening of speech-in-noise did not produce significant MOCR activation; while passive listening (equivalent to visual attention) generated an MOCR effect in the opposite direction to their active-listening analogues for all 3 speech manipulations.
Despite spectrotemporal, level and task-difficulty similarities between noise-vocoded speech and speech-in-noise manipulations, the stimulus-dependence of these results suggests that MOCR activation was controlled in a top-down manner according to the auditory scene presented. We speculated that this arises from improved peripheral processing of specific speech cues during active listening, whereas the opposite effects in passive listening are associated with attenuating auditory inputs to prioritize visual information. In line with this, we observed that introducing efferent feedback to our auditory periphery model differentially affected the auditory nerve output for the 3 most challenging speech manipulations: the resulting enhancement or deterioration of envelope cue representation offering an explanation for divergent patterns of MOCR gating for noise-vocoded and speech-in-noise.
In summary, we predict that observed changes in CEOAE amplitudes in the contralateral ear will mirror cochlear gain inhibition in the ear processing speech. Bilateral descending control of the MOCR despite speech being presented monaurally is not unexpected for two reasons:
1) Unlike simple pure tone stimuli, speech activates both left and right auditory cortices even when presented unilaterally to either ear (Heggdal et al., 2019)
2) Cortical gating of the MOCR in humans does not appear restricted to direct ipsilaterally descending processes that impact cortical gain control in the opposite ear instead likely incorporating polysynaptic, decussating processes to affect both cochlear gain in both ears (Khalfa et al., 2001).
Together this evidence makes it difficult to envisage a case where unilaterally-presented speech does not influence top-down control of cochlear gain bilaterally.
Reviewer 2-Comment 2: 2) The vocoded conditions were recorded from a different group of participants than the masked speech conditions. Comparing between these two, which forms the essential point in this paper, is therefore highly confounded by inter-individual differences, which we know are substantial for these measures. More generally, the high variability of results in this research field should caution any strong conclusions based on comparing just these two experiments. A more useful approach would have been to perform the exact same task in the two experiments, to examine the reproducibility.
Response to Reviewer 2-Comment 2:
We ensured that the two populations tested across the three experiments were all normal hearing adults assessed using the same criteria. They were also age- and gender- matched and were recruited from undergraduate courses at Macquarie University (therefore presumably possessed similar literacy); however, we acknowledge this as an important issue and controlled for these issues, as far as we could, by:
1) Ensuring that CEOAE SNRs were above a 6 dB minimum which allowed for more reliable and replicable recordings within and between subjects (Goodman et al., 2013).
2) Carefully analysing and selecting ABR waveforms above the residual noise. Residual noise was calculated by applying a weighted average method based on Bayesian inference that weighs individual sweeps proportionally to their estimated precision (Box & Tiao, 1973). This helped preserve all trials without any rejection required for artefacts. ABR waveforms with residual noise equal to or higher than the averaged signal were discarded.
3) Ensuring that individual ERP components represented a reliable individual average by: a) removing noisy trials (trials between -200 ms and 1.2 sec from sound onset which had absolute amplitude values higher than 75 μV) and b) maintaining between 60-80% of total trials per condition.
In addition, we assessed potential differences across common variables between experiments such as, lexical performance during natural speech (see Results section), ABR components and CEOAE magnitude changes relative to the baseline during the Active and Passive listening of natural speech (as part of the 1st author’s thesis dissertation: Hernandez Perez, H., & Macquarie University. Department of Linguistics, degree granting institution. (2018). Disentangling the Influence of Attention in the Auditory Efferent System during Speech Processing / Heivet Hernandez Perez): “During active or passive listening of natural speech, no statistical differences between the populations assessed in the noise-vocoded and speech-in-noise experiments for: wave V-III amplitude ratio- Active listening [t (12) = 0.90, p=0.39], Passive listening: [t (23) = 1.58, p=0.13]; wave V-Active listening: [t (23) = 0.09, p=0.93]; Passive listening: [t (24) = -0.24, p=0.81]; CEOAE magnitude changes-Active listening [t (23) = -0.21, p=0.83; Passive listening [t (24) = -0.36, p=0.72].”
These results ruled out the possibility that the effects observed across the three experiments were due to intrinsic differences between the populations tested. This would be discussed in a future version of the manuscript and added as supplemental material.
Reviewer 2-Comment 3: 3) The interpretation presented here is essentially incompatible with the anti-masking model for the MOCR that first started of this field of research, in which the noise response is suppressed more than the signal, which is contradictory to the findings and model presented here, which suggest no role for the MOCR in improving speech in noise perception.
Response to Reviewer 2-Comment 3:
Physiological evidence for the MOCR anti-masking effect in animal models (Wiederhold, 1970; Winslow & Sachs 1987; Guinan & Gifford 1988; Kawase et al., 1993) has led to the hypothesis that the MOCR may play an important role in aiding humans to perceive speech in noise (Giraud et al., 1997; Liberman & Guinan 1998). The strictly non-invasive nature of human experiments has made measuring MOCR effects on OAE amplitudes the main technique for testing this anti-masking hypothesis. However, OAE inhibition (the MOCR-mediated reduction in OAE amplitude) has been reported as either increased (Giraud et al., 1997; Mishra and Lutman, 2014), reduced (de Boer et al., 2012; Harkrider and Bowers, 2009) or being unaffected (Stuart and Butler, 2012; Wagner et al., 2008) in participants with improved speech-in-noise perception. More recently, Mertes et al. (2019) suggested that the SNR used to explore speech-in-noise abilities might explain the contradicting results in the literature. The authors found that the MOCR only contributed to perception at the lowest SNR they tested (-12 dB), suggesting that the role of the MOCR for listening-in-noise may be highly dependent on the SNR, which in turns influences the extent to which the MOCR does or does not provide a benefit for hearing in noise. Therefore, our human and modelling data not only expands but also challenges the classical MOCR anti-masking effect by suggesting that, in humans, this effect is not only SNR-specific (which we controlled) but it is also task-specific (i.e whether participants are attending to the contralateral masker or not) and stimuli-dependent (i.e masker intrinsically noisy Vs signal-in-noise). We acknowledge that we can discuss further how our data advances the current state of the MOCR anti-masking effect in a future version of the manuscript.
Reviewer 2-Comment 4: 4) The analysis of measures becomes increasingly selective and lacking in detail as the paper progresses: numerous 'outliers' are removed from the ABR recordings, with very uneven numbers of outliers between conditions. ABRs were averaged across conditions with no explicit justification. The statistical analysis of the ABRs is flawed as it does not compare across conditions (vocoded vs masked) but only within each condition separately (active v passive) - from which no across-condition difference can be inferred. The model simulation includes only 3 out of 9 active conditions. For the cortical responses, again only 3 conditions are discussed, with little apparent relevance.
Response to Reviewer 2-Comment 4:
In regard to the reviewer’s comment “The analysis of measures becomes increasingly selective and lacking in detail as the paper progresses: numerous 'outliers' are removed from the ABR recordings, with very uneven numbers of outliers between conditions. ABRs were averaged across conditions with no explicit justification.” During the analysis of the ABR measurements, we not only dealt with outliers but also with several missing data points (ABR components below the residual noise). The statistical analysis used to assess potential differences within ABR components was rANOVAs. This type of analysis is particularly restrictive when dealing with missing data points, because it will only include participants with all data available: (2 Conditions X 4 Stimuli manipulations for the noise vocoded experiment). This is why, ABR components’ sample sizes across experiments appeared uneven.
Regarding the reviewer’s comment: “ABRs were averaged across conditions with no explicit justification.” Our rANOVA had the following design: Factor 1 (Conditions: Active Vs Passive); Factor 2 (Stimuli: natural, 8 channels noise vocoded (Voc8) …etc) and finally the Interaction (Conditions x Stimuli). ABR conditions were not simply averaged together; we only found a significant Conditions effect in the rANOVA that collapses all stimuli manipulations into Active Vs Passive conditions. Therefore, it was only statistically valid, to make inferences and potential interpretations about the Conditions main effect. This would be clarified in both the statistical design and in the Results section of a future version of this manuscript.
In regard to the reviewer’s comment: “The statistical analysis of the ABRs is flawed as it does not compare across conditions (vocoded vs masked) but only within each condition separately (active v passive) - from which no across-condition difference can be inferred”. Up to this point in our data analysis, we were only interested in within-speech-manipulations comparisons (similar to the CEOAE analysis i.e, within noise-vocoded manipulations). We agree with the reviewer that a simple comparison between speech manipulations (noise-vocoded Vs masked speech) for the variables that are reflecting attentional changes (Active Vs Passive listening) could be useful to infer differences across experiments (noise-vocoded Vs speech-in-noise). This analysis will be added in a future version of the paper.
Finally, regarding the comment:” The model simulation includes only 3 out of 9 active conditions. For the cortical responses, again only 3 conditions are discussed, with little apparent relevance”. At this stage of our analysis, we wanted to understand the potential reasons why the control of the cochlear gain appeared to be dependent on the way speech was being degraded i.e, noise vocoding the speech signal Vs speech-in-noise. Iso-performance being achieved in 3 task-difficulty levels, we thought to test how both the biophysical model and the auditory cortex (ERP components) would respond to the hardest and most challenging speech degradations (noise vocoded 8 channels, speech in babble noise +5 dB snr and speech in speech-shaped noise +3 dB snr) (see Figure 1B in Results section), where differences in the cochlear gain are most evident across experiments (see Figure 1B in Results section). In these extreme conditions we hypothesized that both the model and the auditory cortex activity would display the most obvious differences in the processing of the different speech degradations. We acknowledge the reviewer’s comment and in a future version of this manuscript, this line of thought will be more clearly described.
Reviewer 2-Comment 5: 5) The assumption that changes in non-invasive measures, which represent a selective, random, mixed and jumbled by-product of underlying physiological processes, can be linked causally to auditory function, i.e. that changes in these responses necessarily have a definable and directional functional correlate in perception, is very tenuous and needs to be treated with much more caution.
Response to Reviewer 2-Comment 5:
We acknowledge the reviewer’s view about being cautious when interpreting non-invasive measures associated with human perception. However, the physiological measurements used in this study are not new in the field of auditory or speech perception, they are gold-standard methods to assess auditory function in both animal and human models. The novelty of our approach lays in imposing attentional states (Active listening) and (Passive listening) while concurrently probing along the auditory pathway in order to gain a holistic understanding of MOCR-mediated changes during a speech comprehension task. The strength of our methodology arises from extensively and continuously monitoring both the attentional states and the quality of our physiological measurements.
Reviewer #3: [...] Reviewer 3-Comment 1: 1) However, I have several substantial concerns with the design, conceptualization, data analysis and interpretation of the results. I have had challenges to understand the hypotheses and rationale behind this study. A number of experimental paradigms have been employed, including peripheral/brainstem physiological measure, as well as cortical auditory responses during active versus 'passive' listening. Different noise conditions were tested but it is not clear to me what rationale was behind these stimulus choices. The authors claim that "our data comparing active and passive listening conditions highlight a categorical distinction between speech manipulation, a difference between processing a single, but degraded, auditory stream (vocoded speech) and parsing a complex acoustic scene to hear out a stream from multiple competing and spectrally similarly sounds" (lines 401-403). This seems like too much of a mouthful. I cannot see that the data support this pretty broad interpretation.
Response to Reviewer 3-Comment 1:
The main objective of this study is to examine the role of the auditory efferent system in active vs. passive listening tasks for three commonly employed speech manipulations. To address this, speech intelligibility was degraded in three ways: 1) noise vocoding the speech signal; 2) adding babble noise (BN) to the speech signal at different SNRs or 3) adding speech-shaped noise (SSN) to the speech signal at different SNRs. The reason for using noise-vocoded speech while contralaterally recording CEOAEs is that it allowed speech intelligibility to be manipulated without increasing noise levels (a classical way of evoking the MOCR (Berlin et al., 1993; Norman & Thornton 1993; Kalaiah et al., 2017b)). This avoided confounding CEOAE magnitude changes due to purely stimulus-driven MOCR activation with attention-driven MOCR on CEOAE magnitudes. Moreover, because the level of the speech spectrum decreases with increasing frequency, white noise (which is the most commonly used stimulus to evoke MOCR in the literature) predominantly masks only the high frequency component of the speech signal, therefore it is not considered an efficient speech masker. However, BN (besides representing a more ethological auditory type of noise) and SSN (which is the spectrally matched long-term averaged of the speech signal) have the same long-term average spectrum as speech. Therefore, these noises were able to mask the speech signal equally across frequencies.
Reviewer 3-Comment 2: 2) Despite maintaining iso-difficulty between vocoded vs speech-in-noise (SIN) conditions, the authors neither address (a) the fundamental differences in understanding vocoded vs. SIN speech nor (b) any theoretical basis for how the noise manifests in vocoded speech. If the tasks are indeed so obviously 'categorically' different - then it should not be surprising they engage different processing (the 'denoising' may not be comparable). I would prefer much more clearly defined and targeted hypotheses and a justification of the specific stimulus and paradigm choices to test such hypotheses. It appears to me that numerous measures have been obtained (reflecting in fact very different processes along the auditory pathway) and then it has been attempted to make up some coherent conclusions from these data - but the assumptions are not clear, the data are very complex and many aspects of the discussion are speculative. To me, the most interesting element is the reversal of the MOCR behavior in the attended vs ignored conditions. However, ignoring a stimulus is not a passive task! It would have been interesting to also see cortical unattended results.
Response to Reviewer 3-Comment 2:
The motivation behind this study arises from controversy in the literature regarding attentional effects at both the level of the cochlear (via MOCR) and the brainstem. Previous studies of attentional effects on CEOAEs have not only prevented direct comparison among them but have also distorted the interpretation of their results. Most have implemented paradigms with large differences in their arousal state [or alertness levels (Eysenck, 2012)] and stimulus type between the active auditory task (e.g. speech stimuli presented while CEOAEs are recorded) and passive listening conditions (no task, CEOAEs recorded during no-noise conditions or with-noise conditions) (Froehlich et al., 1990; Meric et al., 1994; Srinivasan et al., 2012). Our experimental paradigm addressed these issues in three main ways: 1) using the same stimuli for both active and passive listening conditions; 2) using a controlled visual scene across the experimental sessions; and 3) attempting to control for differences in alertness during the passive condition by asking subjects to watch an engaging cartoon movie. The homogeneity of visual and auditory scenes across the experiments allowed the effects of attending to the speech on CEOAE magnitude to be disentangled from the stimulus-driven effects.
In addition, it was never assumed that the “Passive listening” or the “auditory-ignored” condition was a passive task. In this condition subjects were asked to ignore the auditory stimuli and to watch a non-subtitled, stop-motion movie. To ensure participants’ attention during this condition, they were monitored with a video camera and were asked questions at the end of this session (e.g. What happened in the movie? How many characters were present?) (See Methods section). The aim of a passive or an auditory-ignoring condition is to shift attentional resources away from the auditory scene and towards the visual scene. As shown in (Figure supplement 4) all ERP components were also obtained in the Passive listening condition and they are of a smaller magnitude than ERP components observed in the active listening conditions, demonstrating that cortical representation of the speech-onset was enhanced in all active listening conditions.
Reviewer 3-Comment 3: 2) Overall, I'm struggling with this study that touches upon various concepts and paradigms (efferent feedback, active vs. passive listening, neural representation of listening effort, modeling of efferent signal processing, stream segregation, speech-in-noise coding, peripheral vs cortical representations...) where each of them in isolation already provides a number of challenges and has been discussed controversially. In my view, it would be more valuable to specify and clarify the research question and focus on those paradigms that can help verify or falsify the research hypotheses.
Response to Reviewer 3-Comment 3:
In our study, we sought to explore how active listening of degraded speech modulates CEOAE magnitudes (as a proxy for efferent-MOCR effects). With the specific Research question: Does auditory attention modulate cochlear gain, via the auditory efferent system, in a task-dependent manner? and Hypothesis: Decreases in speech intelligibility raise auditory attention and this reduces cochlear gain (measured using CEOAEs).
In particular, unlike previously published studies, we assessed auditory changes objectively and subjectively as part of a highly controlled experimental paradigm, maintaining a constant performance across three experimental manipulations of speech intelligibility as well as minimizing influences of MEMR activation and controlling for homogeneity of both visual and auditory scenes across conditions. We agree with the reviewer that due to the complexity of our study, each section should be more explicit in its hypothesis and aims. This will be clarified in a future version of this manuscript.